")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
A Case Study and Literature Review of the Diagnosis of Danon Disease in Patients Presenting Only with Severe Cardiac Symptoms
Authors Sun YQ, Lv Q, Chen D, Da Y, Zhao XY, Dong JZ
Received 11 October 2022
Accepted for publication 1 August 2023
Published 17 August 2023 Volume 2023:16 Pages 767—775
DOI https://doi.org/10.2147/PGPM.S392800
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Yu-Qing Sun,1 Qiang Lv,1 Dong Chen,2 Yuwei Da,3 Xiao-Yan Zhao,4 Jian-Zeng Dong1
1Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart Lung & Blood Vessel Diseases, Beijing, People’s Republic of China; 2Department of Pathology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart Lung & Blood Vessel Diseases, Beijing, People’s Republic of China; 3Department of Neurology, Beijing Xuanwu Hospital, Capital Medical University, Beijing, People’s Republic of China; 4Department of Cardiology, The First Affiliated Hospital, Zhengzhou University, Zhengzhou, Henan, People’s Republic of China
Correspondence: Jian-Zeng Dong, Email [email protected]
Abstract: The clinical manifestations of Danon disease, which result from the primary deficiency of the lysosome-associated membrane protein 2 gene, include cardiomyopathy, skeletal myopathy, and different degrees of intellectual disability that vary greatly among patients. The present study reports on two cases of Danon disease in two patients who only presented cardiac symptoms. Cardiac symptoms usually occur in adolescence and during a patient’s twenties, and most patients die from heart failure. However, the lab results from these cases suggested that other systems were involved, despite no other clinical symptoms. Significantly, the two patients had elevated serum cardiac troponin I, which often manifests in the acute cardiac phase, especially in severely affected patients with rapidly fatal outcomes. Danon disease is a multi-system involvement disease. Therefore, clinicians must be aware of its complexity when evaluating newly diagnosed patients due to its vastly different clinical course and prognosis and the importance of multidisciplinary management.
Keywords: Danon disease, cardiomyopathy, serum cardiac troponin I, management
Introduction
Danon disease is an X-chromosome-linked dominant disorder caused by mutations in the lysosome-associated membrane protein 2 (LAMP2) gene.1 The typical symptoms are cardiomyopathy, skeletal myopathy, and intellectual disability.2 Other less prevalent manifestations include retinal, hepatic, and pulmonary diseases.3 There is prominent cardiac involvement in patients with Danon disease, and almost all patients have the manifestation of cardiomyopathy, which can sometimes be the only manifestation of Danon disease. Male present earlier than female and have a higher morbidity rate. Male predominantly express the hypertrophic cardiomyopathy (HCM) phenotype, while female are seen to express the HCM or dilated cardiomyopathy (DCM) phenotypes equally.4 Symptoms can vary, from being asymptomatic for several years to sudden cardiac death.5 This paper analyses the case studies of two patients presenting only cardiac symptoms but where tests indicated that other systems were involved. The final diagnosis was based on the genetic analysis of the LAMP2 gene. The disease is rare and lacks specificity in clinical manifestations, and patients are often seen in different departments (pediatrics, neurology, cardiology), which leads to diagnostic difficulties and missed diagnoses. Therefore, this paper discusses management strategies that may provide options for any practicing cardiologists who may encounter this diagnosis. This study was conducted with approval from the Ethics Committee of Beijing Anzhen Hospital. This study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from all participants.
Case Studies
Patient 1
A twenty-one-year-old Asian male presented with palpitations for 7 months in Beijing Anzhen Hospital. An electrocardiogram showed atrial fibrillation (AF) and a pre-excitation pattern (Figure 1). Echocardiography and magnetic resonance imaging (MRI) of the heart showed extensive symmetrical hypertrophy of the left ventricle and interventricular septum. The left ventricle ejection fraction (LVEF) displayed in the echocardiography and MRI was 34% and 14.3%, respectively. In the MRI, subendocardial lamella mixed with high signals could be seen in the anterior septum, the anterior and posterior wall of the left ventricle from basal segment to central segment, and the lateral wall from basal segment to apical segment. A patchy midmyocardial delayed enhancement was observed in the MRI, reflecting the development of myocardial fibrosis (Figure 2). No skeletal muscle strength or mental abnormalities were found, and no family history was elicited. However, blood test results showed increased serum enzyme levels of transaminase, creatase, and cardiac troponin I (cTnI), which remained elevated at follow-up (Table 1). Biopsy specimens were taken from the patient’s left bicep, and the results of muscle pathology were as follows: (1) hematoxylin and eosin staining showed there were vacuoles in the muscle fibers, and some vacuoles had basophilic granular material deposition; (2) nestin staining indicated there were dark granular stains in the muscle fibers; (3) desmin staining revealed there were desmin-positive substances in the muscle fibers; and (4) periodic acid–Schiff staining showed glycogen and glycoprotein deposits. The pathological diagnosis was glycogen storage disease or desminopathy (Figure 3). The genetic analysis detected a pathogenic heterozygous mutation (c.973delC) in the LAMP2 gene, which confirmed a diagnosis of Danon disease. However, the patient’s parents’ genetic tests showed no abnormalities (Figure 4).
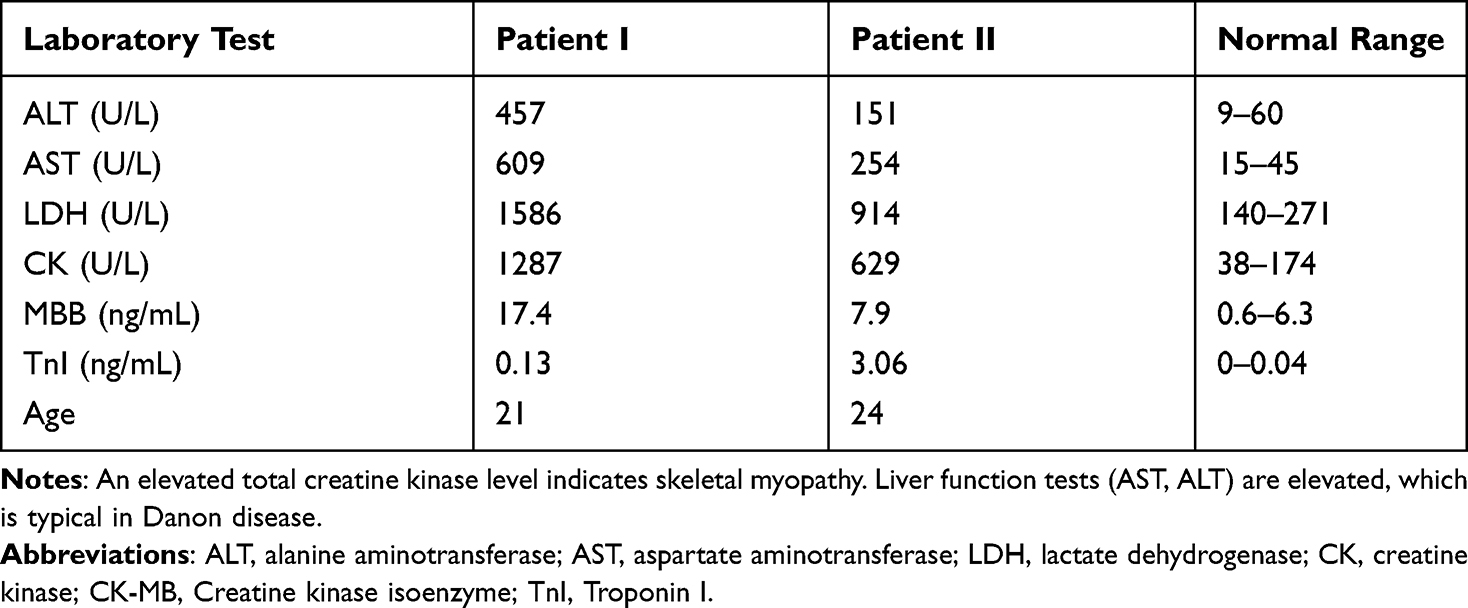 |
Table 1 Clinical Blood Tests |
 |
Figure 1 The electrocardiogram of patient I showed atrial fibrillation and pre-excitation pattern (The absence of variability of the pre-excited QRS complex in the setting of atrial fibrillation is highly suggestive of a fasciculoventricular pathway). |
 |
Figure 2 (A) A 21-year-old male patient’s two-chamber PSIR image shows LGE located at the sub-epicardium. (B and C) Same patient’s short-axis and four-chamber PSIR image show LGE located at the sub-epicardium of lateral wall, mid-myocardium of septal wall. Abbreviations: PSIR, Phase-Sensitive Inversion-Recovery Image; LGE, Late Gadolinium Enhancement. |
 |
Figure 3 (A) HE staining showed there were vacuoles in muscle fibers, and some vacuoles had basophilic granular material deposition. (B) NES staining indicated there were granular dark stains in the muscle fibers. (C) Desmin staining revealed there were desmin positive substances in the muscle fibers. (D) PAS staining showed glycogen and glycoprotein were deposition. The pathological diagnosis was glycogen storage disease or desminopathy. Abbreviations: NES staining, Nestin Staining; PAS staining, Periodic Acid-Schiff staining. |
 |
Figure 4 Patient I: LAMP2:NM_002294:exon8:c.973delC: p.Leu325fs (The red arrow represents the patient’s genetic mutation site). The father of Patient I: LAMP2:c.973delC chrx:119575705 (The red circle represents the site without genetic mutations). The mother of Patient I: LAMP2:c.973delC chrx:119575705 (The red circle represents the site without genetic mutations). Patient II: LAMP2:c.35C>A chrX-119602990 p.S12* (The red circle represents the patient’s genetic mutation site). |
The hemizygous c.973delC (GRCh37:ChrX:119575705) mutation in the LAMP2 gene (NM_002294.2) has not been reported as a disease-causing mutation or as a benign polymorphism. The c.973delC mutation causes a shift in the reading frame, beginning with leucine codon 325, changing it to tryptophan, and creating a premature stop codon at position 21 of the new reading frame (p.Leu325TrpfsX21). It is expected to result in an abnormal, truncated protein or an absence of protein from this allele due to nonsense-mediated messenger RNA decay. Loss-of-function variants in LAMP2 are known to be pathogenic (PubMed ID [PMID]: 21415759). The c.973delC mutation is assumed to be de novo in this case, and is absent from controls in the Exome Sequencing Project, the 1000 Genomes Project, and the Exome Aggregation Consortium. Based on this evidence, this mutation has been classified as pathogenic according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) 2015 guidelines.6
Patient 2
Patient 2 was a 24-year-old male presenting with a distinctly different clinical course and initial manifestation to Patient 1. The patient was initially referred to a local hospital due to an unexplained elevation of transaminase 9 years ago, 2 years before being referred to the Heart Failure Center, Beijing Anzhen Hospital of Capital Medical University. Dyspnea and weakness followed the admission. Clinical examinations revealed hepatomegaly with obvious edema. The patient reported severe fatigue during physical activity. The laboratory results were similar to those of Patient 1 (Table 1). The echocardiographic examination revealed right ventricular hypertrophy (interventricular septum, 13 mm; right ventricle wall, 11 mm) and dilated heart (left atrium, 41×37 × 58 mm; right atrium, 46 × 56mm; right ventricle, 50 mm; left ventricle at end-diastole, 57 mm). The LVEF was 23%. In the MRI, most of the walls of the left ventricle and the anteroseptal, anterior, and lateral walls of the right ventricle were observed with delayed enhancement, which reflected myocardial fibrosis (Figure 5). For the sequence analysis, DNA was extracted from the blood. A hemizygous nonsense mutation (c.35C>A) was identified in the LAMP2 gene of the patient (Figure 4). Only the relatives on the mother’s side of the family. The patient’s mother died of DCM in her 40s, and their grandfather died at 36 in unexplained circumstances. Nevertheless, no genetic testing was performed. The c.35C>A mutation creates a premature translational stop signal at codon 12 (p.Ser12*) of the LAMP2 gene. It is expected to result in an absent or disrupted protein product. While this variant has not been reported in the literature, truncating variants in LAMP2 are known to be pathogenic (PMID: 21415759). The c.35C>A mutation is absent from controls in the Exome Sequencing Project, the 1000 Genomes Project, and the Exome Aggregation Consortium. Based on this evidence, this mutation has been classified as likely pathogenic according to ACMG/AMP 2015 guidelines.6
 |
Figure 5 (A and B) 24-year-old male patient’s two-chamber and four-chamber PSIR image shows LGE diffusely distributed at the sub-epicardium. |
Patient II showed elevated levels of cardiac biomarker troponin I (TnI) upon admission, which is indicative of myocardial damage. This finding suggests that the patient may have underlying cardiac disease, which could be the cause of their presenting symptoms. Elevated TnI levels are commonly associated with cardiac events such as acute myocardial infarction (AMI) or unstable angina. However, it is also important to note that other non-cardiac conditions such as sepsis, pulmonary embolism, and renal failure can also cause increased TnI levels. Further diagnostic tests such as electrocardiogram (ECG) and echocardiogram should be performed to confirm the diagnosis of cardiac disease. If a cardiac origin is confirmed, appropriate treatment such as antiplatelet therapy, revascularization, or other interventions should be initiated. In addition, monitoring TnI levels over time can provide valuable information about the patient’s cardiac status and response to treatment. TnI is a useful marker for determining the extent and severity of myocardial injury and can inform ongoing management decisions. Overall, the elevated TnI levels in patient II highlight the potential importance of cardiac disease in their clinical presentation and the need for prompt and thorough cardiac evaluation and management.
Literature Review and Discussion
The present study describes two cases of Danon disease in patients only presenting with severe cardiac symptoms but with lab results that suggested other systems were involved and there are many literature studies on its characteristics (Table S1). Notably, elevated serum cTnI, as was the case with the two patients in the present study, has not been widely described in previous cases to develop after the progression of the disease. Furthermore, this paper also focuses on management strategies for Danon disease.
First, the epidemiology of Danon disease was reviewed. The exact prevalence of Danon disease is unknown. However, according to current case reports, it may affect any ethnic group. According to a 2019 report, an analysis of previous related articles found that of the 146 Danon disease cases identified, there were 90 male (61.6%) and 56 female (38.4%) cases. The ethnicity of the patients was reported in the minority of cases (n = 66, 45.2%), and in most cases, the patients were Caucasian (72.7%). Danon disease cases were mostly reported by hospitals in Europe (54.1% of cases), followed by North America (21.9%) and Asia (20.6%).7 Meanwhile, with the development and wide application of LAMP2 gene testing technology, the diagnosis rate of Danon disease may increase yearly.8 A recent report suggested that speculative incidence rate was higher than previous estimates and could be as high as 1% to 6% of patients with HCM of an unknown etiology.9 One study identified Danon disease in 2 out of 50 pediatric patients with HCM (incidence of 4%),10 while the prevalence could reach 17% if expanding the consideration to patients in who HCM is accompanied by other features.9 In addition, the onset of Danon disease in male patients is generally earlier with a more serious clinical presentation, with disease symptoms appearing during childhood or adolescence.11
In 1940, Antopol et al12 reported that two brothers suffered from myocardial hypertrophy caused by excess glycogen storage in the heart and skeletal muscle, which may have been the earliest description of Danon disease reported in 1981.2 In 2000, it was first reported that the mutation of the LAMP2 gene caused Danon disease.1 Although the exact function of LAMP2 is still controversial, the LAMP2 protein plays a key role in mediating autophagosomal and lysosomal fusion during autophagy.13 A LAMP2 protein deficiency, caused by gene mutations, is an essential factor in the pathogenesis of Danon disease and is associated with a variety of clinical phenotypes.
Symptoms of Danon disease can vary from being asymptomatic for several years to sudden cardiac death. The main clinical manifestations are cardiomyopathy, skeletal myopathy, and intellectual disability. Cardiac symptoms occur in almost all patients with Danon disease. To this paper’s knowledge, as a consequence of X-chromosome inactivation, female heterozygotes for Danon disease are somatic mosaics; thus, while the condition is highly penetrant, clinical expression is attenuated compared with that of the hemizygous male.14 As a result of their hemizygosity, male patients are severely affected. Among previously reported cases, female patients had a predominant presentation pattern consisting of isolated cardiomyopathy (without additional disease hallmarks), while male patients more commonly presented a triad of cardiomyopathy, skeletal myopathy, and intellectual disability.7
The most common cardiac symptoms reported include chest pain,15 heart murmurs,16 palpitations, and fatigue.17 Palpitations and chest pain were the first symptoms in most patients.13 In male patients, Danon-related cardiomyopathy was progressive and typically manifested as a hypertrophic phenotype during the course of the disease and later progressed to dilated features in 11% to 12% of cases.11 One case report demonstrated that the explanted heart of one patient with Danon disease had severe cardiomegaly (weight 872 g [normal range 253–504 g, predicted for height]) and severely symmetric left ventricular hypertrophy (left ventricular free wall width 25 mm [normal 15 mm]).18 In contrast to male patients, affected women patients show an approximately equal prevalence of DCM (28%) and HCM (33%).11 However, unlike other HCM with preserved systolic function, patients with Danon disease with an HCM pattern and preserved systolic function have worse prognoses.19 Male patients are unlikely to reach the age of 25 years without cardiac transplantation.4
Arrhythmias were very common in patients with reported Danon disease, with an incidence of 80% to 100%. The pre-excitation syndrome, Wolff–Parkinson–White (WPW) syndrome, was the most common form of arrhythmia in patients with Danon disease, in the setting of PRKAG2 mutation there is a variable incidence of ventricular pre-excitation, which may vary with specific mutations. In the most prevalent variant R302Q it was reported from 70–100%.20 Pre-excitation has been shown to develop post-natal in murines models of PRKAG2. It has been associated with fasciculoventricular pathways in most cases in humans.21 Fasciculoventricular pathways has been shown to be ubiquitous in humans,22 and the likely mechanism of ventricular pre-excitation in those glycogen storage disease (Prkag2 and Lamp2) is the accumulation of glycogen leading to electrocardiographic manifestation in otherwise ubiquitous concealed fasciculoventricular pathways.22 Boucek et al11 concluded that among 145 patients with Danon disease, WPW syndrome was found in 68% of male and 27% of female patients. The presence of WPW syndrome has been described in different types of cardiac hypertrophy, eg, Andersons–Fabry disease, Pompe disease, protein kinase adenosine monophosphate-activated non-catalytic subunit gamma 2 cardiomyopathy, and Danon disease. However, the exact mechanism of WPW syndrome is unclear.23 An analysis of the Spanish multicenter Danon Registry showed that only 11 of the 27 patients with Danon disease had WPW syndrome. Presumably, previous studies may have overestimated the incidence of WPW syndrome in patients with Danon disease.24 Data on the incidence of AF in Danon disease is scarce. However, one study revealed that five of the seven patients suffered from AF, with one of them experiencing a thromboembolic stroke at the age of 30 years, and the authors speculated that patients with Danon disease harbored a significantly increased risk of prematurely occurring AF and thromboembolic events.23
Finally, malignant arrhythmia was found to be one of the leading causes of death. One study showed that four out of six patients with Danon disease developed ventricular arrhythmias and suddenly died.25 AF, atrial flutter, and life-threatening ventricular arrhythmia were common, with a prevalence of up to 60%.26 Other electrocardiogram abnormalities included complete atrioventricular blocks, supraventricular tachycardia, delta waves, and high voltages.27,28
It is noted in this study that elevated serum cTnI was observed in two patients. So far, only a few determinations of serum cardiac troponins have been conducted in Danon disease. It is generally known that cardiac troponins are commonly used in the diagnosis and prognosis of ischemic cardiomyopathy and that cardiac troponins could have a predictive value for adverse outcomes in DCM and HCM.29,30 In one case report, notably elevated serum cTnIs were found in the severely affected family Danon members (three brothers and their mother).14 Elevated serum cardiac troponins were also found during the acute cardiac phase.31 Therefore, although elevated serum cardiac troponin is not specific, it can be considered to be related to Danon disease. Clinicians should be alert to this to prevent unnecessary catheter treatment and to merit exploration for clinical decisions during the entire diagnostic process.
Among reported cases, skeletal myopathy manifested as general weakness, exercise intolerance, and elevated serum creatine kinase.24 Learning difficulties were detected in patients, but disease progression remained relatively stable.32 Moreover, cognitive and psychiatric changes were uncommon.33 Additionally, in a Japanese survey, two patients (10%) had retinopathy.34
Whether or not family history indicates the presence of Danon disease, all sporadic HCM patients should undergo clinical examinations, such as muscular, plasma creatine kinase concentration, transaminase concentration, electrocardiogram, and nervous system tests. Laboratory abnormalities, such as detectable elevations in hepatic and skeletal muscle enzymes, are common, with values greater than three times the normal limit. Echocardiography is commonly used in diagnosing patients. Cardiac MRIs are applied to determine the extent of myocardial fibrosis. Laboratory diagnostic tools include finding normal acid maltase activity, vacuolar features in skeletal and/or endomyocardial biopsy, LAMP2 protein deficiency in various tissues (including leukocytes), and mutation detection in the LAMP2 gene.35 Ultimately, the LAMP2 gene test is considered the best for diagnosing and excluding Danon disease. Once the diagnosis is confirmed, it is recommended that cascade assessment.36 Patients with Danon disease may have multiple medical needs, and the corresponding treatment teams may include a combination of primary-care physicians, cardiologists, geneticists, neurologists, ophthalmologists, rehabilitation medicine specialists, and physical therapists.3 Due to the limited understanding of this disease, no treatment guidelines have been published. In patients with Danon disease who have HCM, clinicians may refer to the 2011 American College of Cardiology Foundation/American Heart Association and 2014 European Society of Cardiology guidelines for the diagnosis and treatment of HCM.37,38
However, the prognosis of Danon disease is entirely different from that of HCM, especially in men. Male patients generally do not live more than 30 years, but female patients can live up to 40 or 50 years.11 Malignant ventricular arrhythmias and heart failure are the most important causes of shortening patients’ life spans. Therapeutic interventions in Danon disease are implantable cardioverter-defibrillator (ICD) implantation and cardiac transplantation. One study showed that there was a high incidence of failure to terminate ventricular arrhythmias in patients that underwent implantation of an ICD, as was the case with five out of seven patients.23 Cardiac transplantation appears to be the only effective treatment.39 Survival after transplant is similar in patients with Danon disease as compared with patients undergoing cardiac transplantation for other causes.36 Meanwhile, applying a left ventricular assisting device at the appropriate time before heart transplantation is a crucial management method.40 However, current cardiac transplantation criteria focusing on the acquired heart disease are inappropriate for inherited diseases without further evidence, such as Danon disease.41 And the assessment of candidacy for cardiac transplantation should occur promptly following the development of a functional decline, particularly in young men. The timing of cardiac transplantation in patients with this disease is a major issue in clinical management and needs further study.
Abbreviations
LAMP2, lysosome-associated membrane protein 2; HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy; AF, atrial fibrillation; MRI, magnetic resonance imaging; LVEF, left ventricle ejection fraction; cTnI, cardiac troponin I; ICD, implantable cardioverter-defibrillator.
Data Sharing Statement
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was conducted with approval from the Ethics Committee of Beijing Anzhen Hospital. This study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from all participants.
Consent for Publication
All participants signed a document of informed consent.
Funding
No external funding received to conduct this study.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Nishino I, Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406(6798):906–910.
2. Danon MJ, Oh SJ, Dimauro S, et al. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31(1):51–57. doi:10.1212/WNL.31.1.51
3. D’Souza RS, Levandowski C, Slavov D, et al. Danon disease: clinical features, evaluation, and management. Circ Heart Fail. 2014;7(5):843–849. doi:10.1161/CIRCHEARTFAILURE.114.001105
4. Maron BJ, Roberts WC, Arad M, et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301:1253–1259.
5. Thakur R, Afzal A, Carey SA, et al. Repeat cardiac transplant indicated by severe cardiac allograft vasculopathy in a patient with Danon disease. Rev Cardiovasc Med. 2018;19(2):69–71. doi:10.31083/j.rcm.2018.02.903
6. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–423. doi:10.1038/gim.2015.30
7. Brambatti M, Caspi O, Maolo A, et al. Danon disease: gender differences in presentation and outcome. Int J Cardiol. 2019;286:92–98.
8. Nishino I. Autophagic vacuolar myopathy. Semin Pediatr Neurol. 2006;13(2):90–95. doi:10.1016/j.spen.2006.06.004
9. Arad M, Maron BJ, Gorham JM, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. New Engl J Med. 2005;352(4):362–372. doi:10.1056/NEJMoa033349
10. Charron P, Villard E, Sebillon P, et al. Danon’s disease as a cause of hypertrophic cardiomyopathy: a systematic survey. J Med Genet. 2004;41(10):751.
11. Boucek D, Jirikowic J, Taylor M. Natural history of Danon disease. Genet Med. 2011;13(6):563–568. doi:10.1097/GIM.0b013e31820ad795
12. Antopol W, Boas EP, Levison W, Tuchman LR Cardiac hypertrophy caused by glycogen storage disease in a fifteen-year-old boy.Am. Heart J. 1940;20:546–556. doi:10.1016/S0002-8703(40)90933-4
13. Cheng Z, Fang Q. Danon disease: focusing on heart. J Hum Genet. 2012;57(7):407–410. doi:10.1038/jhg.2012.72
14. Roos JCP, Daniels MJ, Morris E, et al. Heterogeneity in a large pedigree with Danon disease: implications for pathogenesis and management. Mol Genet Metab. 2018;123(2):177–183. doi:10.1016/j.ymgme.2017.06.008
15. Marino M, Musumeci O, Paleologo G, et al. Ischemic stroke due to hypoperfusion in a patient with a previously unrecognized Danon disease. Neuromuscular Disord. 2016;26(12):890–894. doi:10.1016/j.nmd.2016.09.025
16. Sabourdy F, Michelakakis H, Anastasakis A, et al. Danon disease: further clinical and molecular heterogeneity. Muscle Nerve. 2009;39(6):837–844.
17. Csányi B, Popoiu A, Hategan L, et al. Identification of two novel LAMP2 gene mutations in Danon disease. Can J Cardio. 2016;32(11):1355. e23–1355. e30. doi:10.1016/j.cjca.2016.02.071
18. Yu L, Wan K, Han Y, et al. A rare phenotype of heterozygous Danon disease mimicking apical hypertrophic cardiomyopathy. Eur Heart J. 2018;39(34):3263–3264. doi:10.1093/eurheartj/ehx722
19. Guo S, Zhou L, Wang R, et al. Danon disease: two patients with atrial fibrillation in a single family and review of the literature. Exp Ther Med. 2019;18(3):1527–1532.
20. Sternick EB, Oliva A, Gerken LM, et al. Clinical, electrocardiographic, and electrophysiologic characteristics of patients with a fasciculoventricular pathway: the role of PRKAG2 mutation. Heart Rhythm. 2011;8(1):58–64. doi:10.1016/j.hrthm.2010.09.081
21. Patel VV, Arad M, Moskowitz IP, et al. Electrophysiologic characterization and postnatal development of ventricular pre-excitation in a mouse model of cardiac hypertrophy and Wolff-Parkinson-White syndrome. J Am Coll Cardiol. 2003;42(5):942–951. doi:10.1016/S0735-1097(03)00850-7
22. Macías Y, Tretter JT, Anderson RH, et al. Miniseries 1-Part IV: how frequent are fasciculo-ventricular connections in the normal heart? Europace. 2022;24(3):464–472. doi:10.1093/europace/euab286
23. Konrad T, Sonnenschein S, Schmidt FP, et al. Cardiac arrhythmias in patients with Danon disease. Ep Europace. 2017;19(7):1204–1210. doi:10.1093/europace/euw215
24. López-Sainz Á, Salazar-Mendiguchía J, García-álvarez A, et al. Clinical findings and prognosis of Danon disease. an analysis of the Spanish Multicenter Danon Registry. Revista Española de Cardiología. 2019;72(6):479–486. doi:10.1016/j.recesp.2018.04.027
25. Spinazzi M, Fanin M, Melacini P, et al. Cardioembolic stroke in Danon disease. Clin Genet. 2008;73(4):388–390. doi:10.1111/j.1399-0004.2008.00971.x
26. Cheng Z, Cui Q, Tian Z, et al. Danon disease as a cause of concentric left ventricular hypertrophy in patients who underwent endomyocardial biopsy. Eur Heart J. 2012;33(5):649–656.
27. Jhaveri S, Herber J, Zahka K, et al. Arrhythmias and fasciculoventricular pathways in patients with Danon disease: a single center experience. J Cardiovasc Electrophysiol. 2019;30(10):1932–1938. doi:10.1111/jce.14049
28. Kim J, Parikh P, Mahboob M, et al. Asymptomatic young man with Danon disease. Tex Heart Inst J. 2014;41(3):332–334. doi:10.14503/THIJ-13-3279
29. Kawahara C, Tsutamoto T, Nishiyama K, et al. Prognostic role of high-sensitivity cardiac troponin T in patients with nonischemic dilated cardiomyopathy. Circ J. 2011;75(3):656–661. doi:10.1253/circj.CJ-10-0837
30. Kubo T, Kitaoka H, Yamanaka S, et al. Significance of high-sensitivity cardiac troponin T in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;62(14):1252–1259. doi:10.1016/j.jacc.2013.03.055
31. Braksator W, Chybowska B, Kuch M, et al. Noninvasive characterization of a case of Danon disease. Revista Española de Cardiología. 2010;63(4):493–495.
32. Sugie K, Yoshizawa H, Onoue K, et al. Early onset of cardiomyopathy and intellectual disability in a girl with Danon disease associated with a de novo novel mutation of the LAMP2 gene. Neuropathology. 2016;36(6):561–565. doi:10.1111/neup.12307
33. Yardeni M, Weisman O, Mandel H, et al. Psychiatric and cognitive characteristics of individuals with Danon disease (LAMP2 gene mutation). Am J Med Genet A. 2017;173(9):2461–2466. doi:10.1002/ajmg.a.38320
34. Sugie K, Komaki H, Eura N, et al. A nationwide survey on Danon disease in Japan. Int J Mol Sci. 2018;19(11):3507. doi:10.3390/ijms19113507
35. Cenacchi G, Papa V, Pegoraro V, et al. Danon disease: review of natural history and recent advances. Neuropath Appl Neuro. 2019;46(4):303–322.
36. Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm. 2022;19(7):e1–e60. doi:10.1016/j.hrthm.2022.03.1225
37. Gersh BJ, Maron BJ, Bonow RO, et al.; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124(24):2761–2796. PMID: 22068435. doi:10.1161/CIR.0b013e318223e230
38. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–2779. PMID: 25173338. doi:10.1093/eurheartj/ehu284
39. Echaniz-Laguna A, Mohr M, Epailly E, et al. Novel Lamp-2 gene mutation and successful treatment with heart transplantation in a large family with Danon disease. Muscle Nerve. 2006;33(3):393–397. PMID: 16372318. doi:10.1002/mus.20471
40. Kitahara H, Nawata K, Kinoshita O, et al. Implantation of a left ventricular assist device for Danon cardiomyopathy. Ann Thorac Surg. 2017;103(1):e39–e41. doi:10.1016/j.athoracsur.2016.07.022
41. Banner NR, Bonser RS, Clark AL, et al. UK guidelines for referral and assessment of adults for heart transplantation. Heart. 2011;97(18):1520–1527. doi:10.1136/heartjnl-2011-300048
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.