")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
A Case Report of Dyschromatosis Symmetrica Hereditaria with Glucose-6-Phosphate Dehydrogenase Deficiency
Authors Wang P, Tang C, Zhao Y, Wang P
Received 17 February 2023
Accepted for publication 4 April 2023
Published 19 April 2023 Volume 2023:16 Pages 1047—1050
DOI https://doi.org/10.2147/CCID.S407052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Panpan Wang,1 Chenyu Tang,1 Yige Zhao,1 Ping Wang2
1Department of Dermatology, Hangzhou Third People’s Hospital, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 2Department of Dermatology, Hangzhou Third People’s Hospital, Affiliated Hangzhou Dermatology Hospital of Zhejiang University School of Medicine, Hangzhou, People’s Republic of China
Correspondence: Ping Wang, Department of Dermatology, Hangzhou Third People’s Hospital, Affiliated Hangzhou Dermatology Hospital of Zhejiang University School of Medicine, Westlake Ave 38, Hangzhou, People’s Republic of China, Tel +8613588812862, Email [email protected]
Abstract: Dyschromatosis symmetrica hereditaria (DSH) is a pigmented genetic skin disorder with an incompletely understood pathogenesis characterized by reticular hyper- and hypopigmented skin patches on the dorsal aspect of the extremities, freckle-like patches on the face, and unaffected palms and feet. There is no effective treatment available. Glucose-6-phosphate dehydrogenase (G6PD) deficiency has not been reported in the literature of DSH. We describe for the first time a case of DSH with G6PD deficiency and a family history of psychosis.
Keywords: dyschromatosis symmetrica hereditaria, glucose-6-phosphate dehydrogenase deficiency, psychosis, ADAR1
Introduction
DSH is a rare autosomal dominant disorder associated with mutations in the ADAR1 gene.1 Currently, DSH has been reported in combination with Aicardi-Goutie`res syndrome 6 (AGS6),2 psoriasis,3 acromegaly,4 eyelid hemangioma,5 intracranial hemangioma,6 and dystonia,7 while we report a case with G6PD deficiency, a comorbidity that has not been reported yet. In addition to this, the patient had a family history of psychiatric disorders, which requires further study of the pathogenesis of DSH.
Case Presentation
The patient is a 7-year-old male. The patient presented with skin hypopigmentation interspersed with punctate pigmentation on the fingertips of both hands 4 years ago and freckle-like pigmentation on both cheekbones of the face without treatment, and then the lesions gradually increased in size with age, showing characteristics of heavy summer and light winter and aggravated by sun exposure. The patient came to our dermatology outpatient clinic on June 2, 2022. Past history: the patient was diagnosed with glucose-6-phosphate dehydrogenase (G6PD) deficiency 7 years ago and was untreated.
Dermatologic examination revealed symmetrical distribution of hypopigmented patches interspersed with punctate pigmentation on the dorsal skin of both fingers and freckle-like pigmentation on both cheekbones of the face, no blisters, papules, scales or other damage at the lesions, no pain, itching or other discomfort (Figure 1A and B). Skin CT showed: Compared to the surrounding normal skin, the pigment ring in the basal layer of the leukoplakic area was completely absent, and no inflammatory cell infiltration was seen in the dermal papillae, with clear boundaries to the surrounding normal skin (Figure 2A and B). Laboratory tests revealed glucose-6-phosphate dehydrogenase 176↓. Family investigation: parents were not consanguineously married, and in the patient’s II generation III line, only the patient’s mother had the same skin lesions as the patient on both hands and both cheekbones, with no other history of G6PD deficiency, except for a family history of psychiatric disorders in the patient’s paternal line (Figure 3). He was ultimately diagnosed with “1. dyschromatosis symmetrica hereditaria; 2. glucose-6-phosphate dehydrogenase deficiency” based on the above clinical manifestations, examination results and family history. Considering the young age of the patient, he was instructed to apply high-frequency sunscreen and wear sun-protective clothing for sun protection and to avoid exposure to the sun.
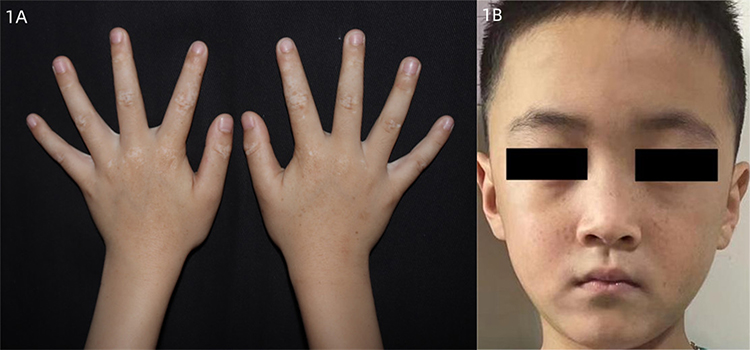 |
Figure 1 (A and B) Symmetrical distribution of hypopigmented patches interspersed with punctate pigmentation on the dorsal skin of both fingers and freckle-like pigmentation on both cheekbones of the face, no blisters, papules, scales or other damage at the lesions, no pain, itching or other discomfort. |
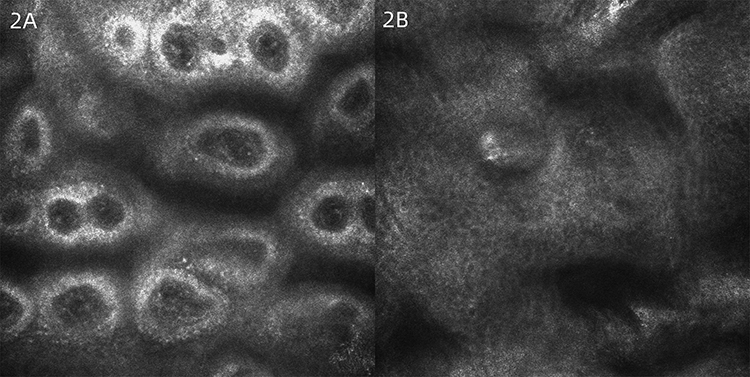 |
Figure 2 (A and B) Skin CT: Compared to the surrounding normal skin, the pigment ring in the basal layer of the leukoplakic area was completely absent, and no inflammatory cell infiltration was seen in the dermal papillae, with clear boundaries to the surrounding normal skin. |
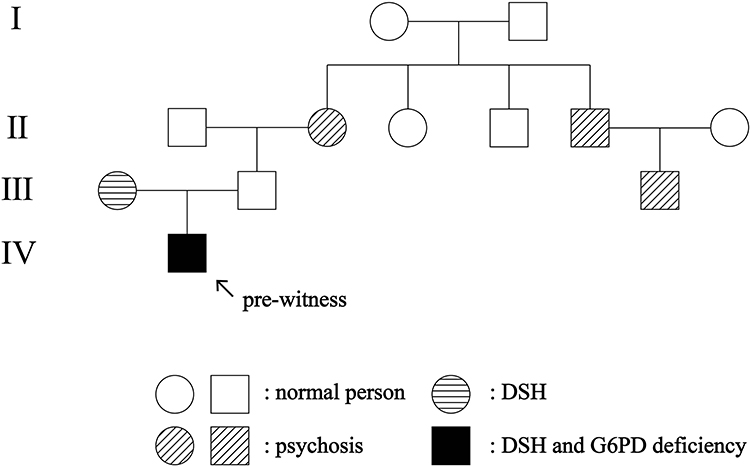 |
Figure 3 Family tree of dyschromatosis symmetrica hereditaria, glucose-6-phosphate dehydrogenase deficiency, psychosis in four generations. |
Discussion
Dyschromatosis symmetrica hereditaria (DSH) is a rare autosomal dominant disorder, and in 2003, the team of Japanese scholar Professor Miyamura1 localized the causative gene of DSH to the 1q21.3 adenosine deaminase acting on RNA1 (ADAR1) gene. The latest research indicates that the E3 ubiquitin ligase SMURF2 can stabilize ADAR1p110 and promote its adenosine-to-inosine (A-to-I) editing function.8 In addition to genetic mutations, the disease may be associated with UV light, infection, and frostbite.9 Histopathological changes in DSH are non-specific,10 and Oiso et al11 showed that dermoscopic examination of DSH revealed different features of each pigmented spot, such as the degree of hyperpigmentation and epidermal-dermal structure and hypothesized that the pigmented spots have different melanocyte dysfunction, abnormal melanocyte-keratin-forming cell interactions, and impaired reticular crest structure.
Currently, DSH has been reported in combination with Aicardi-Goutie`res syndrome 6 (AGS6),2 psoriasis,3 acromegaly,4 eyelid hemangioma,5 intracranial hemangioma,6 and dystonia,7 while our case is unique in that G6PD deficiency is a mutation in the G6PD gene, an X-chromosome linked genetic defect,12 and this comorbidity has not been reported. There have been many reports of DSH in combination with neurological or psychiatric disorders, and patients may present with movement disorder, mental deterioration, tissue calcification, psychic alteration such as irritability and excitation. CT of the brain is a necessary test and often shows tissue calcification. In this case, the DSH and G6PD deficiencies were inherited from the mother, and the family history of psychiatric disorders was paternal. Considering that the patient had no psychiatric changes and was only a 7-year-old child, we did not perform cerebrospinal fluid, brain computed tomography (CT) and electroencephalogram (EEG) examinations, which will need to be improved later. We also recommend that the patient undergo genetic testing and analysis to further investigate the pathogenesis of DSH.
There is no effective treatment for DSH, Xu et al13 applied ultra-pulsed CO2 fractional laser to treat DSH lesions on the back of the hand with some effectiveness, Taki’s team14 proposed surgical treatment with flap grafting, the efficacy of which needs to be further verified, Kawakami’s team15 combined microdermal flap grafting and 308 nm excimer laser to treat skin pigmentation Kono et al16 suggested that daily sunscreen with sun protection factor (SPF) 50+ and UVA (PA) ++++ protection could control the disease progression. Considering the young age of the patient, a safer treatment was chosen, and the patient was instructed to apply a high level of sunscreen and wear sun-protective clothing to prevent sun exposure.
Conclusion
This case describes a rare case of DSH with G6PD deficiency and a family history of psychosis. Whether the two disorders have an impact on the development of DSH, or whether there is a correlation between the three, still needs further follow-up and discussion. We also need to conduct further studies on the pathogenesis of DSH.
Consent Statements
Written informed consent was provided by the patient and his parents to have the case details and any accompanying images published. Institutional approval was not required for this case study.
Acknowledgments
We thank the patient and his parents for their permission to publish this information.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 2003;73(3):693–699. doi:10.1086/378209
2. Jones HF, Stoll M, Ho G, et al. Autosomal dominant ADAR c.3019G>A (p. (G1007R)) variant is an important mimic of hereditary spastic paraplegia and cerebral palsy. Brain Dev. 2022;44(2):153–160. doi:10.1016/j.braindev.2021.10.001
3. Weng HY, Wu RW, Chen YT, et al. Novel ADAR1 mutations in three cases of psoriasis coexisting with dyschromatosis symmetrica hereditaria. J Eur Acad Dermatol Venereol. 2022;36(1):e54–e57. doi:10.1111/jdv.17620
4. Murata T, Yagi Y, Tanioka M, et al. Dyschromatosis symmetrica hereditaria with acral hypertrophy. Eur J Dermatol. 2011;21(4):649–650. doi:10.1684/ejd.2011.1486
5. Alshomar KM, Alkatan HM, Alrikabi AC, Al-Faky YH. A case of dyschromatosis symmetrica hereditaria with an associated eyelid hemangioma. Int J Surg Case Rep. 2021;79:73–75. doi:10.1016/j.ijscr.2021.01.012
6. Yanagishita S, Fukai K, Tsuruta D, et al. Dyschromatosis symmetrica hereditaria complicated by intracranial hemangiomas and Parry-Romberg syndrome. J Dermatol. 2016;43(9):1106–1108. doi:10.1111/1346-8138.13353
7. Tojo K, Sekijima Y, Suzuki T, et al. Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family with ADAR1 mutation. Mov Disord. 2006;21(9):1510–1513. doi:10.1002/mds.21011
8. Koganti P, Kadali VN, Manikoth Ayyathan D, et al. The E3 ubiquitin ligase SMURF2 stabilizes RNA editase ADAR1p110 and promotes its adenosine-to-inosine (A-to-I) editing function. Cell Mol Life Sci. 2022;79(5):237. doi:10.1007/s00018-022-04272-8
9. Zhang G, Shao M, Li Z, et al. Genetic spectrum of dyschromatosis symmetrica hereditaria in Chinese patients including a novel nonstop mutation in ADAR1 gene. BMC Med Genet. 2016;17:14. doi:10.1186/s12881-015-0255-1
10. Sheu HM, Yu HS. Dyschromatosis symmetrica hereditaria-a histochemical and ultrastructural study. Taiwan Yi Xue Hui Za Zhi. 1985;84(2):238–249.
11. Oiso N, Murata I, Hayashi M, et al. Dermoscopic features in a case of dyschromatosis symmetrica hereditaria. J Dermatol. 2011;38(1):91–93. doi:10.1111/j.1346-8138.2010.01110.x
12. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371(9606):64–74. doi:10.1016/S0140-6736(08)60073-2
13. Xu XG, Lv Y, Zhai JL, Li YH, Gao XH, Chen HD. Two novel mutations of the ADAR1 gene in Chinese patients with dyschromatosis symmetrica hereditaria successfully treated with fractional CO2 laser. J Eur Acad Dermatol Venereol. 2016;30(6):1035–1038. doi:10.1111/jdv.13090
14. Taki T, Kozuka S, Izawa Y, et al. Surgical treatment of speckled skin caused by dyschromatosis symmetrica hereditaria--case report. J Dermatol. 1986;13(6):471–473. doi:10.1111/j.1346-8138.1986.tb02978.x
15. Kawakami T, Otaguchi R, Kyoya M, Soma Y, Suzuki T. Patient with dyschromatosis symmetrica hereditaria treated with miniature punch grafting, followed by excimer light therapy. J Dermatol. 2013;40(9):771–772. doi:10.1111/1346-8138.12205
16. Kono M, Okamoto T, Takeichi T, Muro Y, Akiyama M. Dyschromatosis symmetrica hereditaria may be successfully controlled by topical sunscreen. Eur J Dermatol. 2018;28(6):840–841. doi:10.1684/ejd.2018.3415
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.