")
Back to Journals » International Medical Case Reports Journal » Volume 8
A case of choroidal osteoma in a 10-year-old child
Received 19 July 2015
Accepted for publication 29 September 2015
Published 2 November 2015 Volume 2015:8 Pages 273—275
DOI https://doi.org/10.2147/IMCRJ.S92693
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Madhusmita Behera,1 Manmath Kumar Das2
1Rotary Narayana Nethralaya, Kolkata, India; 2Vitreo-Retina Services, CL Gupta Eye Institute, Moradabad, India
Abstract: Choroidal osteoma is a rare, benign tumor, usually diagnosed in healthy adult women in their second or third decade of life. Though its etiology and pathogenesis are unclear, it is usually diagnosed due to its typical clinical features of yellowish-orange colored subretinal lesion at posterior pole and a dense echogenic plaque persisting even in lower gains on B-scan ultrasonography. Mostly unilateral (79%), the median age of diagnosis is 26 years. It is relatively rare in children. We report a case of choroidal osteoma in a 10-year-old boy.
Keywords: choroidal osteoma, choroidal osseous choristoma, choroidal tumor
Introduction
Choroidal osteoma (choroidal osseous choristoma) is a benign tumor of unknown etiology first described by Gass et al 1978.1 It is typically found in healthy young women, mostly unilateral, in the posterior pole of the eye, near the optic disc and becomes clinically apparent when it involves the macula. Though present from birth, it is usually diagnosed during the second or third decade of life. The diagnosis of choroidal osteoma is based on its very characteristic clinical picture, ultrasonography, and fundus fluorescein angiography findings.
Case report
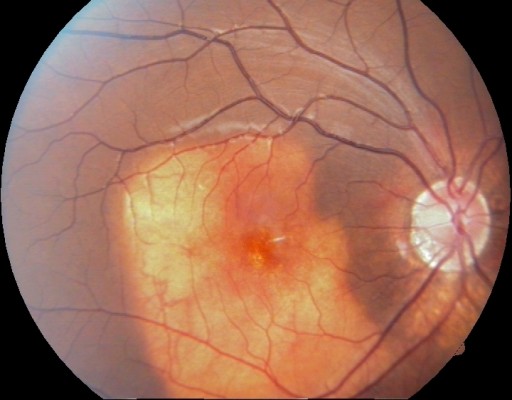
A 10-year-old boy presented with a decreased vision in the right eye. His best corrected visual acuity was 6/60, N6 in the right eye and 6/6, N6 in the left eye. Anterior segment was unremarkable in both eyes. Funduscopy of right eye revealed a large subretinal, orange colored lesion of approximately three disc diameters in size in the macula whereas that of left eye was normal (Figure 1). B-scan ultrasonography of the right eye demonstrated dense echogenic plaque persisting even at 50 db gain (Figure 2). On fundus fluorescein angiography, the lesion showed hyperfluorescence which was progressively increasing in mid-phase with intense staining in late phases. Clinically, there was no evidence of any subretinal neovascularization and optical coherence tomography was also normal. The patient was asked to come for regular follow-up. Over the next year on periodic follow-up, no variations of the mass were noted and vision also remained stable. Appropriate informed consent was taken from father of the child. Institutional Review Board approval was deemed not necessary for the following case study. All principles outlined in the Declaration of Helsinki were followed.
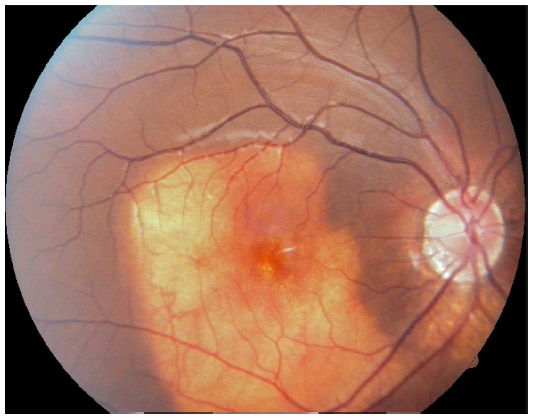 | Figure 1 Fundus picture showing a typical orange-yellowish lesion. |
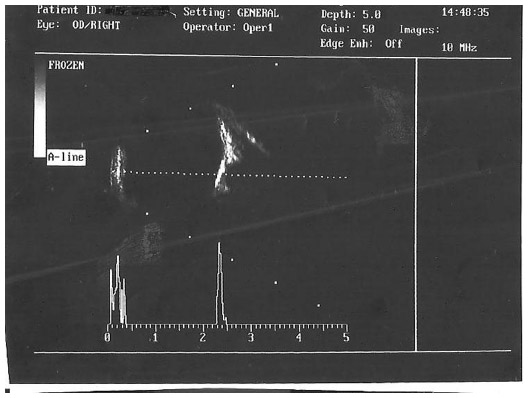 | Figure 2 B-scan ultrasonogram showing hyper-reflectivity of the lesion persisting even at 60 dB gain. |
Discussion
The most important complications of the tumor are subretinal neovascularization, subretinal and intraretinal hemorrhages, and serous and hemorrhagic retinal detachments.
Choroidal osteoma has traditionally been managed by observation because no specific treatment was believed to be effective unless it is associated with subretinal neovascularization. But recently, a few reports have described a successful treatment of subretinal neovascularization with intravitreal injections of anti-VEGF agents.2–4
Histopathologically, choroidal osteoma is composed of mature bone at the level of choroid. The overlying retinal pigment epithelium is usually intact, a finding quite different from osseous metaplasia in which there is no normal retinal pigment epithelium in the affected area.
Choroidal osteoma must be differentiated mainly from amelanotic choroidal melanoma, choroidal nevus, choroidal hemangioma, choroidal metastasis, granuloma, organized subretinal hemorrhage, sclerochoroidal calcification, posterior scleritis, and age-related macular degeneration. In our case, melanoma was ruled out as such lesions are usually elevated and associated with overlying drusen and retinal pigment epithelial proliferations and sometimes with neurosensory retinal detachment. Moreover, melanomas typically show high surface reflectivity and low internal reflectivity on ultrasonography. Choroidal hemangioma owing to its vascular structures typically shows high surface reflectivity with moderate to high internal reflectivity on ultrasound. Choroidal metastases are typically yellow lesions and should be considered in those with a known source of primary malignancy elsewhere. Organized subretinal hemorrhage can also mimic osteoma but is more common following blunt trauma. Posterior scleritis is usually a painful condition associated with a sudden drop in vision and is more common in women with a history of rheumatoid arthritis.
Though seen in adults, choroidal osteoma can present in children as well5 where it can pose a serious diagnostic challenge as it can mimic other non-pigmented lesions at posterior pole like retinoblastoma, congenital retinochoroidal coloboma, and congenital toxoplasmic retinochoroiditis. Retinoblastomas are typically white lesions whereas colobomas are non-pigmented, deeply excavated lesions associated with coloboma of iris and microcornea and nystagmus and so on. Congenital toxoplasma scars are pigmented lesions typically present in fovea and are usually bilateral.
To conclude, choroidal osteomas are benign tumors of uveal tract, of unknown cause, typically seen in young adults, unilateral and orange red in color. Fundus fluorescein angiography shows typical patchy hyperfluorescence with intense late staining whereas ultrasonography shows a highly reflective echo that persists at lower sensitivity. Magnetic resonance imaging shows the tumor to be hyperintense to vitreous in T1 images and hypointense to vitreous in T2 images.
Treatment is only by observation.
Disclosure
The authors report no conflicts of interest in this work.
References
Gass JDM, Guerry RK, Jack RL, Harris G. Choroidal osteoma. Arch Ophthalmol. 1978;96:428–435. | |
Yoshikawa T, Takahashi K. Long term outcomes of intravitreal injection of bevacizumab for choroidal neovascularization-associated with choroidal osteoma. Clin Ophthalmol. 2015;9:429–437. | |
Rao S, Gentile RC. Successful treatment of choroidal neovascularisation complicating a choroidal osteoma with intravitreal bevacizumab. Retin Cases Brief Rep. 2010;4(4):303–305. | |
Shields CL, Salazar PF, Demirci H, Benson WE, Shields JA. Intravitreal bevacizumab (avastin) and ranibizumab (lucentis) for choroidal neovascularization overlying choroidal osteoma. Retin Cases Brief Rep. 2008;2(1):18–20. | |
Fava GE, Brown GC, Shields JA, Broocker G. Choroidal osteoma in a 6-year–old Child. J Pediatr Ophthalmol Strabismus 1980;17(3):203–205. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.