Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
A Case-Control Study of the Relationship Between Genetic Polymorphism and Cretinism in Xinjiang
Authors Huang J, Wu H, Zhao G, Ma Y, An Y, Sun L, Li F, Wang S
Received 23 May 2023
Accepted for publication 11 August 2023
Published 23 August 2023 Volume 2023:16 Pages 785—794
DOI https://doi.org/10.2147/PGPM.S418722
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Jia Huang,1,2,* Haiyan Wu,2,* Guiqiang Zhao,3 Yan Ma,1 Yunping An,4 Li Sun,5 Fuye Li,1 Shengling Wang2
1Department of Public Health, Xinjiang Medical University, Urumqi, Xinjiang, 830054, People’s Republic of China; 2Division of Endemic Disease Prevention, Xinjiang Uygur Autonomous Region Center for Disease Control and Prevention, Urumqi, Xinjiang, 830002, People’s Republic of China; 3Infection Management Department, Affiliated Hospital of Traditional Chinese Medicine of Xinjiang Medical University, Urumqi, Xinjiang, 830000, People’s Republic of China; 4Kashgar District Center for Disease Control and Prevention, Kashgar, Xinjiang, 844000, People’s Republic of China; 5Ingisha County Center for Disease Control and Prevention, Kashgar, Xinjiang, 844500, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shengling Wang, Department of Division of Endemic Disease Prevention, Center for Disease Control and Prevention of Xinjiang Uighur Autonomous Region, No. 380 of Jianquan Road, Urumqi, Xinjiang, 830002, People’s Republic of China, Tel +86-18599067010, Fax +86-991-2614020, Email [email protected] Fuye Li, Department of Public Health, Xinjiang Medical University, No. 393 Xinyi Road, Urumqi, Xinjiang, 830054, People’s Republic of China, Tel +86-18599067010, Fax +86-991-2614020, Email [email protected]
Background: Cretinism is a subtype of congenital hypothyroidism, an endocrine disorder resulting from inadequate thyroid hormone production or receptor deficiency. Genetic abnormalities play a major role in the development of thyroid dysfunction.
Methods: We recruited 183 participants with cretinism and 119 healthy participants from the Xinjiang Uyghur Autonomous Region and randomly selected 29 tag single nucleotide polymorphisms (tSNPs) in TSHB, PAX8, TPO, NKX2-5, and TSHR in all participants. We compared genotype and allele frequencies between cases and controls utilizing the chi-squared test, logistic regression analysis, and haplotype analysis.
Results: Using the chi-squared test, a single SNP was found to be associated with cretinism (recessive model: rs3754363, OR = 0.46, 95% CI = 0.27– 0.80, P = 0.00519; genotype model: P = 0.01677). We stratified neurological, myxedematous, and mixed type and determined that another SNP was associated with a higher risk when comparing myxedematous type to the neurological type (rs2277923).
Conclusion: rs3754363 has a statistically significant protective effect on people with cretinism, while rs2277923 may play a greater role in promoting the development of neurocretinism.
Keywords: chi-squared test, cretinism, NKX2-5, SNPs
Introduction
Congenital hypothyroidism is an endocrine disorder caused by insufficient thyroid hormone production or receptor deficiency, which results in severe physical and mental retardation as well as low metabolic levels in children. Children with congenital hypothyroidism typically show no clinical symptoms or very mild ones during the neonatal period. Lack of thyroid hormones during fetal brain development can lead to severe brain dysfunction, which can manifest as severe sensory and motor disorders and mental retardation. Neonatal screening is the primary method for patient detection and diagnosis. Early diagnosis and treatment with thyroid hormone replacement does not interfere with the normal physical and nervous system development of the patient.1 In China, the incidence of congenital hypothyroidism is approximately 1/2000,2 with a higher incidence among females than males. Anterior neck enlargement due to goiter is a rare symptom of congenital hypothyroidism.3 Thyroid dysplasia, thyroxine synthesis and secretion disorder, thyroxine transport disorder, thyroxine metabolism disorder, thyrotropin/thyroxine resistance, and central hypothyroidism are typical causes of cretinism.4 The vast majority of thyroid hypoplasia cases are isolated incidents. Gene mutations have been linked to some cases, but the underlying pathogenic mechanisms remain unknown. NK2 homeobox 1 (NKX2.1),5 paired box 8 (PAX8),6 NK2 homeobox 5 (NKX2-5), forkhead box E1 (FOXEl), and thyroid stimulating hormone receptor (TSHR) have been identified as pathogenic genes in related research on hypothyroidism. Studies on gene abnormalities associated with thyroid hormone synthesis have focused primarily on thyroid peroxidase (TPO), dual oxidase 2 (DUOX2),7 dual oxidase maturation factor 2 (DUOXA2),8 TG, solute carrier family 5 member 5 (SLC5A5), solute carrier family 26 member 4 (SLC26A4),9 and others.10
Congenital hypothyroidism is also referred to as cretinism, and is a clinical term used to describe the wide clinical spectrum of physical and neurological sequelae in patients with severely delayed treatment and even untreated hypothyroidism, which may be congenital (ie, permanent and primary congenital hypothyroidism) or acquired (ie, due to environmental factors, fetal iodine deficiency or endemic cretinism). There is widespread iodine deficiency in the Xinjiang population and the region’s economic development is quite low. It is one of China’s historically endemic disease hotspots. The rate at which new cases of cretinism are being diagnosed in Xinjiang has dropped precipitously since China’s universal salt iodization policy went into effect. New cases of cretinism were confirmed again in southern Xinjiang in 2006, likely as a result of the unique eating habits of the region’s ethnic minorities. More than 200 cases of endemic cretinism persist today in southern Xinjiang.11 Since this study is a census of endemic cretinism patients in Xinjiang, it may shed light on the pathogenesis of this condition elsewhere in the world.
In 1908, McCarrison proposed the classification of cretinism into two clinical types—neurological type and myxedematous type.12 As the disease has progressed, the presence of mixed cretinism in patients has also been documented. Patients with neurotic cretinism, according to studies, have normal thyroid function despite suffering primarily from damage to the central nervous system, whereas patients with myxomatous cretinism have obvious hypothyroidism.13 Myxedematous cretinism, which causes stunted growth, skeletal retardation, and early sexual development, may be distinguished from other forms of cretinism by a persistent deficiency in postpartum thyroid hormones.
Recent advances in genetics have sparked widespread interest in the hereditary causes of cretinism. Numerous studies have linked polymorphisms in certain genes to the pathogenesis of cretinism. Here, we analyzed the genetic contributions to disease in the Uyghur people by looking into the single nucleotide polymorphisms (SNPs) in their genes. We set out to find a reliable biomarker for genetic testing during pregnancy to prevent cretinism, or a biomarker that can be combined with the correct endocrinologic evaluation of the patient at birth or during the early neonatal period to diagnose early cretinism in Xinjiang.
Methods
Study Population
From December 2016 to April 2019, 183 participants were enrolled in a molecular biology study at the Disease Control and Prevention Centre of Xinjiang Uyghur Autonomous Region. The participants were diagnosed with cretinism based on the People’s Republic of China’s unified diagnostic standard, “Diagnosis of endemic cretinism and endemic sub-cretinism”, with 140 neurotic, 19 myxedematous, and 24 mixed cases. During the same period, 119 healthy, unrelated controls were randomly selected from the medical examination center. The principle used to select the healthy control group is that all of the participants come from the same villages and had the same ethnicity, the gender distribution and age range were similar to that of the participants diagnosed with cretinism, and all participants were long-term residents of the area for at least a year. All participants were Uyghurs from Urumqi and the surrounding areas.
Data Collection
A professional nurse interviewed each participant, and a standard questionnaire was used to collect detailed information about demographic characteristics, radiation exposure, habits, and other factors. Each participant provided peripheral blood samples (3–5 mL). Before sample collection, all participants or their guardians signed a written consent form acknowledging the purpose and experimental procedures of the study. The research protocol was created in accordance with the Declaration of Helsinki and was approved by the Human Research Committee of the Center for Disease Control and Prevention of Xinjiang Uighur Autonomous Region (approval number: 20130113-001).
SNP Selection and Genotyping
We selected several genes associated with cretinism, including thyroid stimulating hormone subunit beta (TSHB), PAX8, TPO, NKX2-5, and TSHR. In the HapMap Chinese Han Beijing population, 29 SNPs were selected and successfully genotyped in all participants; their minor allele frequencies (MAF) were > 5% and the blood DNA was extracted using the GoldMag® nanoparticles method (GoldMag Ltd., Xi’an, China) while the DNA concentration was determined using a NanoDrop 2000C (Thermo Scientific, Waltham, Massachusetts, USA). Multiplexed SNP MassEXTEND assays were designed with the Sequenom MassARRAY Assay Design 3.0 Software (San Diego, California, USA).14 SNPs were genotyped utilizing Sequenom MassARRAY RS1000 (San Diego, California, USA). The Sequenom Typer 4.0 software (San Diego, California, USA) was utilized for data management and analysis.14,15
Data Analysis
Statistical calculations were performed utilizing Microsoft Excel (Redmond, WA, USA) and SPSS 19.0. (SPSS, Chicago, IL, USA). All statistical tests were two-sided, and P ≤ 0.05 was considered statistically significant. Each SNP frequency was evaluated for deviation from Hardy-Weinberg equilibrium (HWE) using an exact test among controls (P > 0.05). Using the chi-squared test, we compared genotype and allele frequencies between cases and controls. To estimate the association between genetic polymorphisms and cretinism risk, odds ratios (ORs) and 95% confidence intervals (CIs) were calculated using unconditional logistic regression with age and sex as adjustment factors.16 The most common genotype in the controls served as the reference group. In addition, we compared the three subtypes of cases and observed the variations between case types.
Multiple inheritance models (codominant, dominant, recessive, over dominant, and additive) were utilized to assess the associations between particular SNPs and cretinism risk using Plink.17 Finally, we evaluated pairwise linkage disequilibrium (LD), haplotype construction, and genetic association at polymorphism loci using Haploview (version 4.2) and Shesis.18,19
Results
Study Participants
We included a total of 302 participants; the mean age of cases was 15.05 ± 5.86 years, and the mean age of controls was 15.45 ± 5.60 years. The gender and age distributions did not differ significantly between the case and control groups (P > 0.05).
Hardy-Weinberg Equilibrium and Chi-Squared Test of SNPs
Based on comparisons with the control participants, all tested SNPs were in accordance with the Hardy-Weinberg equilibrium (HWE) (P > 0.05). Table 1 lists the basic characteristics of these SNPs. Using the chi-squared test, only one SNP, rs3754363, was associated with cretinism (recessive model: OR = 0.46, 95% CI = 0.27 to 0.80, P = 0.00519; genotype model: P = 0.01677) (Table 2). Considering the number of participants with cretinism, we stratified the above classification methods (neurological, myxedematous, and mixed type) and identified another SNP, rs2277923, which was associated with an increased risk when myxedematous type was compared with the neurological type (recessive model: OR = 3.95, 95% CI = 1.46—1.69, P = 0.00447; genotype model: P = 0.00677). The aforementioned SNP risk association remained significant after Fisher’s test (Table 3). There was no significant association between the other SNPs and cretinism.
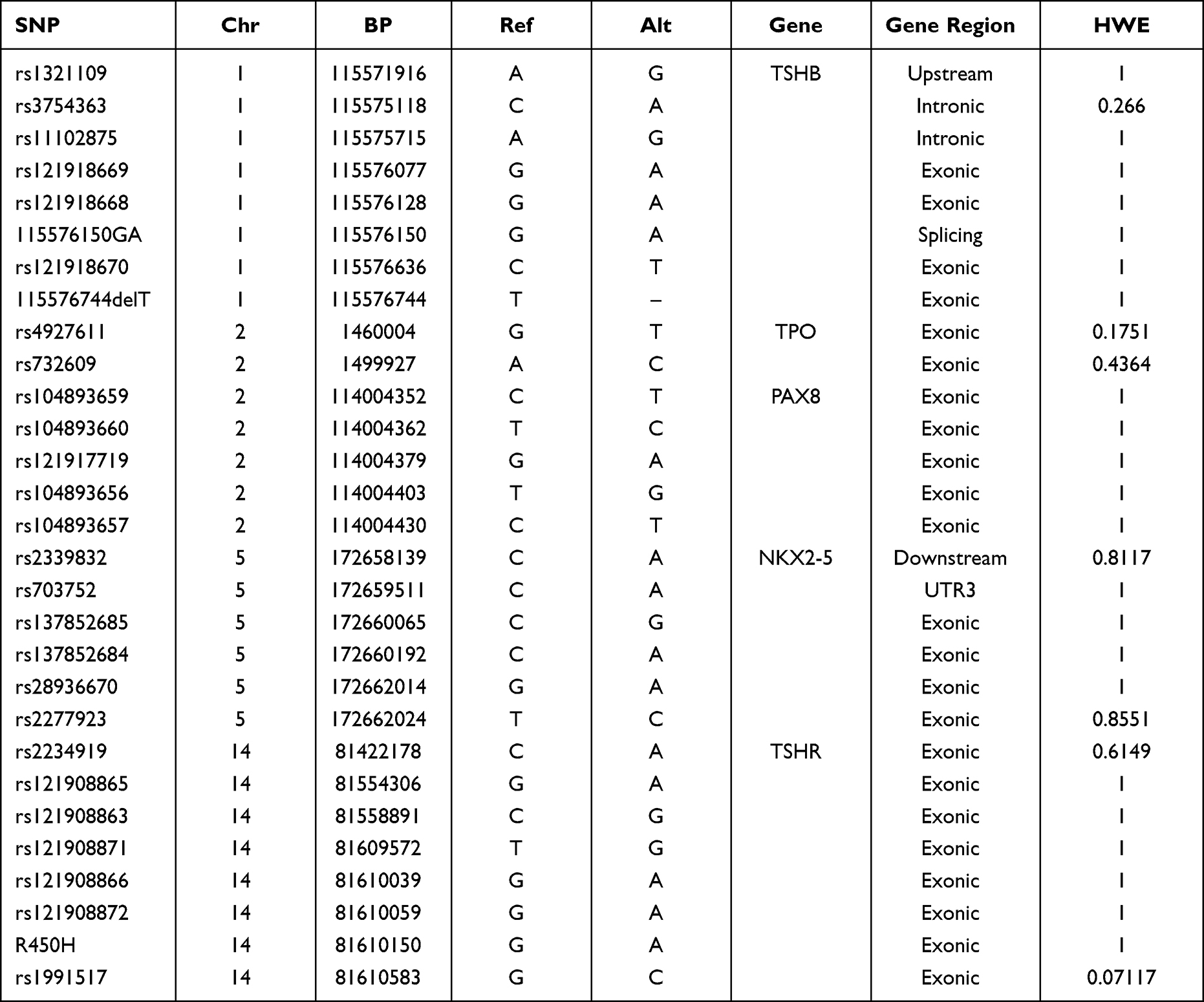 |
Table 1 Basic Information of SNPs Examined in the Research |
 |
Table 2 Chi-Square Test Results in Allele and Genotype Models of SNPs Between Cases and Controls |
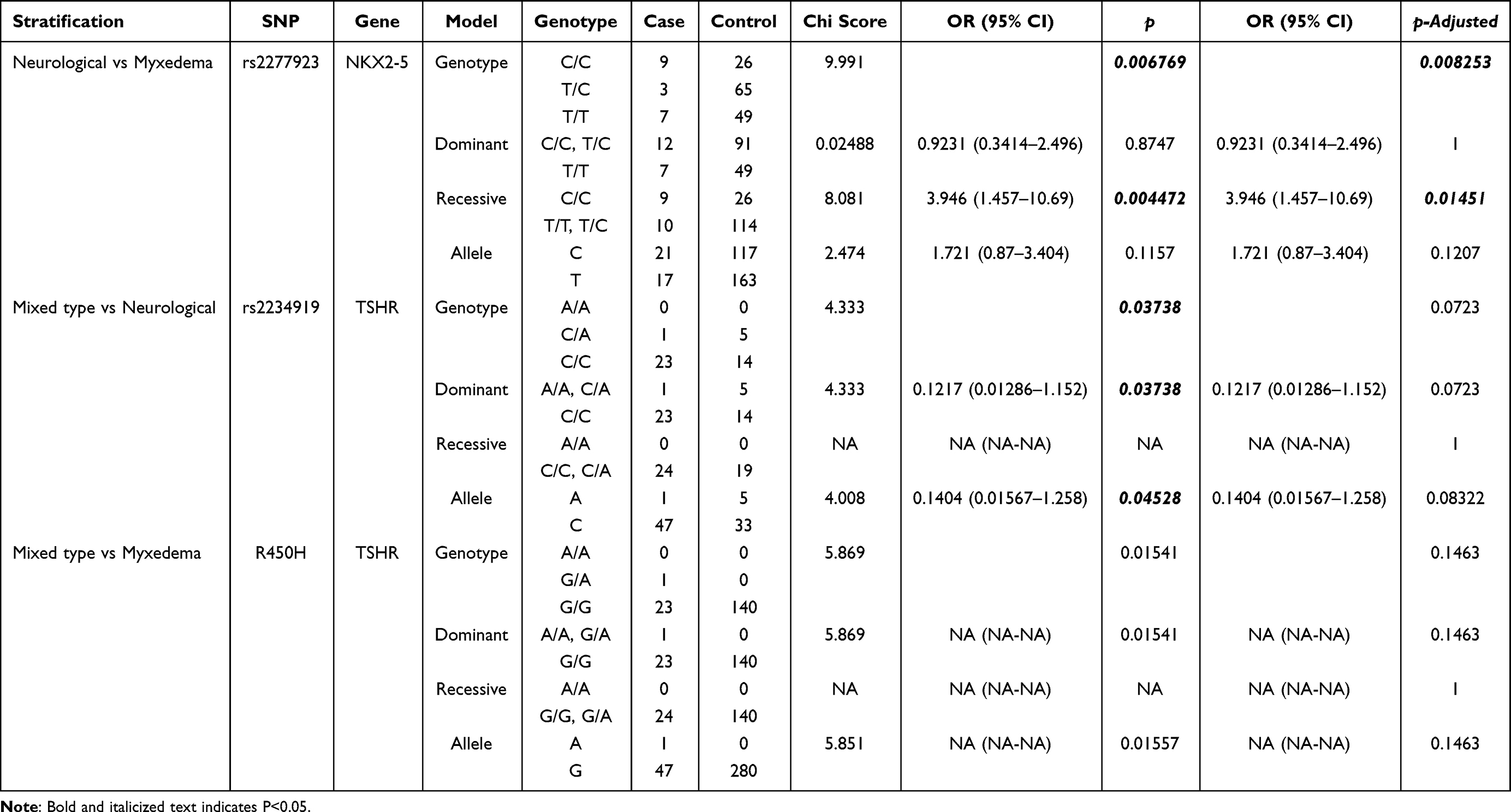 |
Table 3 Chi-Square Test Results in Allele and Genotype Model of SNPs in Different Stratification Analysis |
Logistic Regression Analysis Between SNPs and Cretinism Risk
We further investigated the relationship between 29 SNPs and cretinism risk using five logistic regression models (allele, dominant, recessive, genotype, and additive). As depicted in Table 4, the genotype AA of rs3754363 in the recessive model is associated with a decreased risk of cretinism (OR = 0.4639, 95% CI: 0.2691–0.7999, P = 0.00572; after correction, OR = 0.4716, 95% CI: 0.2725–0.816, P = 0.00722). When neurological and myxedematous cretinism were compared, genotype CC of rs2277923 is associated with increased cretinism risk in the recessive model (OR = 3.946, 95% CI: 1.457–10.69, P = 0.00692; after correction, OR = 3.572, 95% CI: 1.249–10.21, P = 0.01756). In addition, no models revealed an association between the other SNPs and the risk of neurological, myxedematous, or mixed-type cretinism (Table 5).
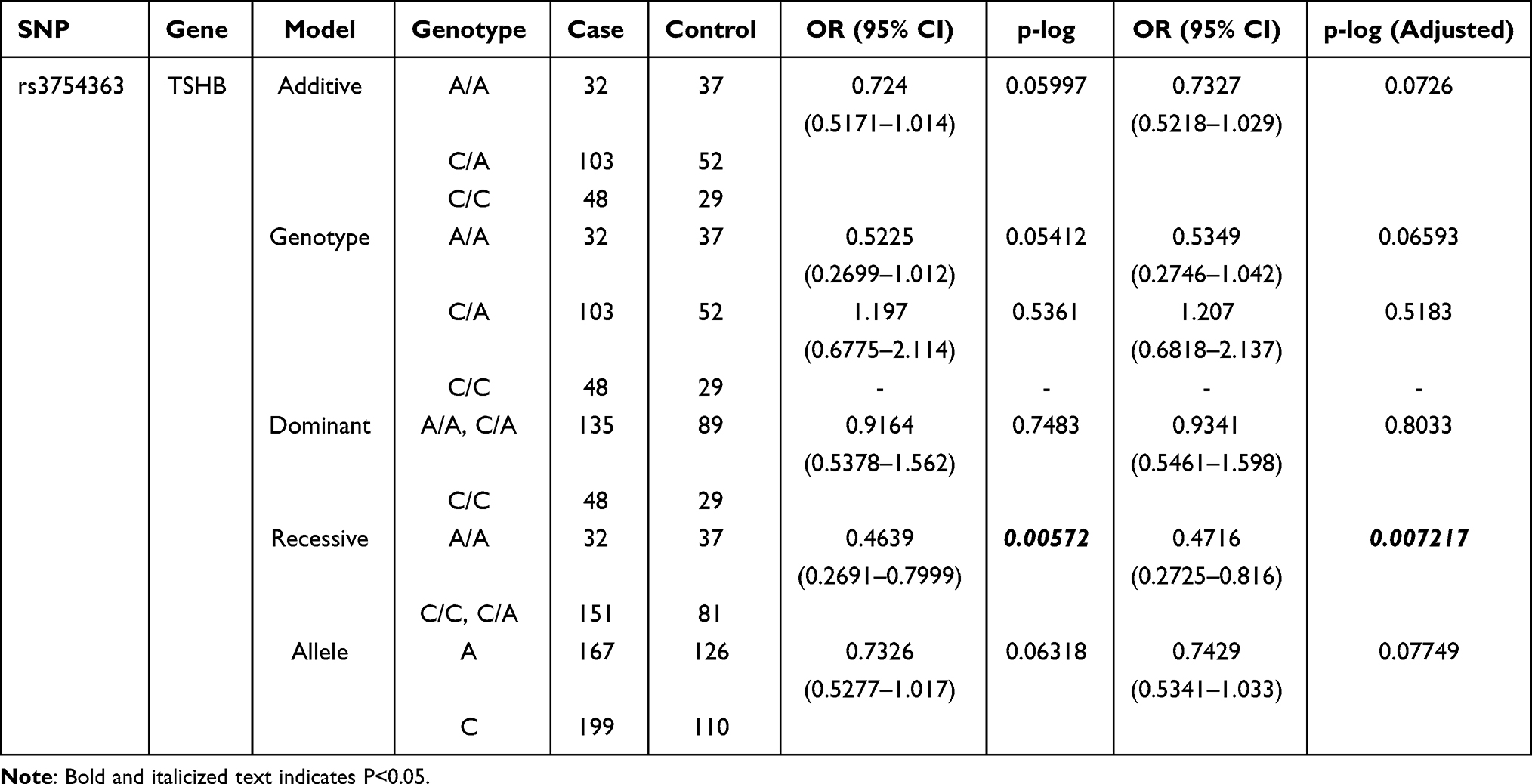 |
Table 4 Logistic Analysis Results of SNPs Associated with Cretinism in Case-Control Analysis |
 |
Table 5 Logistic Analysis Results of SNPs Associated with Cretinism in Neurological Myxedema Type Analysis |
Haplotype Analysis
The haplotype analysis of these SNPs revealed four distinct blocks. LD was composed of rs2339832 and rs703752, rs703752 and rs2277923, and rs3754363 and rs11102875 among rs2234919, R450H, and rs1991517, respectively. We did not identify any significant haplotypes that contribute to the pathogenesis of cretinism based on the results. No haplotypes affected the risk of cretinism significantly in the stratified analysis.
Discussion
While the iodine nutritional status of populations has improved thanks to salt iodization policies, iodine deficiency is still a major health issue in both developed and developing nations. To this day, endemic cretinism remains a major public health issue connected to the standard of living and socio-economic development of the population. Unfortunately, a lack of national-level data has hindered the study of endemic cretinism, despite its importance. Dementia, deaf-mutism, paralysis, and other permanent disabilities brought on by cretinism not only cause unimaginable pain for patients and their loved ones but also place an enormous strain on society as a whole. The etiology and pathogenesis of endemic cretinism are not completely understood. However, iodine deficiency is a concern in some regions of China. In this study, genetic polymorphism analysis was performed on biological samples from patients with cretinism to shed light on the disease’s etiology.
The results of our preliminary epidemiological investigation suggest that in addition to differences in social culture and lifestyle, the genetic background of the Uyghur people with endemic cretinism and subclinical cretinism in Xinjiang is different from that of other ethnic groups. There may be a correlation between the distribution of genes for iodine deficiency susceptibility and the emergence of endemic cretinism in different populations. Secondly, diagnostic criteria for endemic cretinism and actin have garnered significant attention. Our previous research found that after salt iodization policies were put into place in iodine-deficient areas, patients with cretinism exhibited the following patterns of thyroid function: patients with neurotype and mixed type exhibited normal thyroid function, while patients with myxedematous type exhibited hypothyroid function. When iodine nutrition is normal and stable after a period of iodine supplementation, the serum thyroid function index cannot be used as a diagnostic condition for Dike disease and Arc disease. Based on previous research and a review of the relevant literature, we propose the following hypothesis: genetic polymorphisms may provide a comprehensive and systematic approach for the pathogenesis of endemic cretinism.
Chromosomal abnormalities and single-gene mutations are the leading genetic causes of cretinism. Major symptoms include chronic systemic diseases and endocrine disorders (growth hormone deficiency and hypothyroidism). Despite this, many cases of dwarfism have unidentified pathological causes, and single-gene diseases account for a sizable percentage of these cases. Monogenic diseases associated with cretinism are generally rare and involve diverse aspects of individual growth and development, including intrauterine embryonic development, skeletal system, endocrine system, and metabolic system development. These diseases display complex and diverse clinical phenotypes (genetic diversity) as well as genetic heterogeneity.
NKX2.5 is a gene located on chromosome 5q34 that encodes a highly conserved homeobox protein with two exons and numerous essential transcription activation domains. It encodes 324 amino acids that can affect thyroid development during the embryonic stage. NKX2.5 belongs to the NKX2 family. The early stage is an essential component of the heart’s initial development. At this stage, NKX2.5 is expressed in the thyroid primordia. NKX2.5 plays a role in every stage of thyroid development, including formation, metastasis, differentiation, and proliferation.20–22 The DNA-binding proteins encoded by NK-2, called transcription activators, have a binding site in the [T (C/T) AAGTG] sequence found in both the promoter and enhancer of the target gene. NKx2-5, a member of the NK homeobox gene family NKx type 2, is located on human chromosome 5q35 and has two exons, exon 1, 799bp long, including 1a (46bp), 1b (93bp), and 1c (655bp). Exon 1c contains the start codon ATG, and exon 2 (983bp) contains the stop codon TAG and homology domain HD. In exon 1, an SNP (rs2277923, 63T>C) was identified as a synonymous mutation (Glu/Glu), and the allele frequency of this SNP was T = 0.4915. The SNP-related correlation analysis revealed a significant difference, indicating that rs2277923 and various types of cretinism may be associated. At present, there are few studies that have examined the correlation between rs2277923 and cretinism, whereas there are numerous studies that have examined the correlation between rs2277923 and congenital heart disease (CHD). Positive results were observed in subgroups of atrial septal defects.23 Rs2277923 is significantly more prevalent in the CHD population than in the control group in the Indian population.24 In Xinjiang, the Weiur ethnic group account for the vast majority of those diagnosed with endemic cretinism and subclinical cretinism. There are genetic differences between the people of this area and those of other regions, in addition to the obvious differences in culture and lifestyle. Variation in the prevalence and severity of endemic cretinism has been linked to differences in the distribution of genes that increase susceptibility to the effects of iodine deficiency. The current study has confirmed that the single nucleotide polymorphism rs2277923 leads to the transformation of codon 21 GAG into GAA, both of which are synonymous mutations, suggesting that the mutation site of NKX2.5 gene rs2277923 may play a role in the pathogenesis of endemic cretinism.
Thyrotropin is a glycoprotein heterodimer that is noncovalently bound and functions as a pituitary hormone. Its specificity is determined by the β subunit of thyroid stimulating hormone (TSH), which is encoded by the TSHB gene. TSHB is located at 1p22 and has three exons, the first of which is not encoded. Rs3754363 is an intron variant. Intron functions include many regulatory elements involved in gene transcription and RNA processing as well as numerous noncoding RNAs, including microRNAs and snoRNAs.25–27 Both introns and exons are essential eukaryotic genome components.28,29 Both introns and exons are genomic sequences that play an important role in maintaining the biological activity and optimal spatial structure of chromosomes prior to transcription; therefore, they need to share coevolutionary elements to perform their respective biological functions. It is only through the cooperation of introns and exons in the same gene that precise and efficient gene expression can occur at discrete times and locations after transcription.30,31 There is currently no relevant research on the function of rs3754363, and the function of introns is still in the preliminary stage of exploration. Therefore, more research into its mechanism and how it interacts with exons is necessary. Overall cretinism risk was reduced by rs3754363, according to our statistical analysis. Iodine deficiency was prevalent among the same ethnic group living in the same region, and the diet, life and cultural background were the same, however, some people were positively diagnosed with the disease, whereas others did not have the disease; this suggests that rs3754363 may have a protective effect, and the pathogenesis needs to be further studied.
Based on the above, we found that two SNPs were associated with the risk of cretinism: rs2277923 and rs3754363.
Limitations of this study: 1. There are few patients with cretinism, and the sample size is insufficient to be representative. If samples of patients with subclinical cretinism and healthy individuals can be included in future studies as the control group, the persuasiveness of this study can be enhanced. 2. The clinical data of congenital heart disease in patients with endemic cretinism were not collected in this study, and only the diagnostic criteria for endemic cretinism were used. In future studies, we will also add clinical data on diseases related to rs2277923.
Conclusion
In this study, we conducted a preliminary investigation into the relationship between cretinism and SNPs in the Xinjiang Uyghur population. We concluded that rs3754363 has a statistically protective effect in patients with cretinism. We found that rs2277923 is more strongly associated with neurocretinism than with myxedematous cretinism, based on our analysis of the two subtypes of cretinism. However, due to the low mutation rate and small sample size, we were unable to identify all mutants of the locus. Finally, the exploratory nature of this study calls for further research on specific clinical subtypes and a larger sample size.
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study.
Funding
This work was funded by the Key Discipline of the 14th 5-Year Plan in Xinjiang Uygur Autonomous Region-Public Health and Preventive Medicine.
Disclosure
The authors declare that they have no competing interests.
References
1. Delvecchio M, Vigone MC, Wasniewska M, et al. Final height in Italian patients with congenital hypothyroidism detected by neonatal screening: a 20-year observational study. Ital J Pediatr. 2015;41(1):82. doi:10.1186/s13052-015-0190-y
2. Cherella CE, Wassner AJ. Congenital hypothyroidism: insights into pathogenesis and treatment. Int J Pediatr Endocrinol. 2017;2017(1):11. doi:10.1186/s13633-017-0051-0
3. Gomes-Lima C, Wartofsky L, Burman K. Can reverse T3 assay be employed to guide T4 vs. T4/T3 therapy in hypothyroidism? Front Endocrinol. 2019;10:856. doi:10.3389/fendo.2019.00856
4. Nagasaki K, Asami T, Ogawa Y, Kikuchi T, Uchiyama MA. A study of the etiology of congenital hypothyroidism in the Niigata prefecture of Japan in patients born between 1989 and 2005 and evaluated at ages 5-19. Thyroid. 2011;21(4):361. doi:10.1089/thy.2010.0005
5. Teissier R, Guillot L, Carré A, et al. Multiplex ligation-dependent probe amplification improves the detection rate of NKX2. 1 mutations in patients affected by brain-lung-thyroid syndrome. Horm Res Paediatr. 2012;77(3):146–151. doi:10.1159/000337214
6. Carvalho A, Hermanns P, Rodrigues A-L, et al. A new PAX8 mutation causing congenital hypothyroidism in three generations of a family is associated with abnormalities in the urogenital tract. Thyroid. 2013;23(9):1074–1078. doi:10.1089/thy.2012.0649
7. De Marco G, Agretti P, Montanelli L, et al. Identification and functional analysis of novel dual oxidase 2 (DUOX2) mutations in children with congenital or subclinical hypothyroidism. J Clin Endocrinol Metab. 2011;96(8):E1335–E1339. doi:10.1210/jc.2010-2467
8. Hulur I, Hermanns P, Nestoris C, et al. A single copy of the recently identified dual oxidase maturation factor (DUOXA) 1 gene produces only mild transient hypothyroidism in a patient with a novel biallelic DUOXA2 mutation and monoallelic DUOXA1 deletion. J Clin Endocrinol Metab. 2011;96(5):E841–E845. doi:10.1210/jc.2010-2321
9. Banghova K, Al Taji E, Cinek O, et al. Pendred syndrome among patients with congenital hypothyroidism detected by neonatal screening: identification of two novel PDS/SLC26A4 mutations. Eur J Pediatr. 2008;167(7):777. doi:10.1007/s00431-007-0588-7
10. Szinnai G. Clinical genetics of congenital hypothyroidism. Endocr Dev. 2014;26:60–78.
11. Cui Y, Hunag J, Wang SL, et al. Analysis of a survey results on endemic cretinism in Southern Xinjiang. Chin J Endemiol. 2016;2016(8):593–596.
12. McCarrison R. Observations on endemic cretinism in the Chitral and Gilgit valleys. Ind Med Gaz. 1908;43(12):441–449.
13. Halpern JP, Boyages SC, Maberly GF, Collins JK, Eastman CJ, Morris JG. The neurology of endemic cretinism. A study of two endemias. Brain. 1991;114(2):825–841. doi:10.1093/brain/114.2.825
14. Gabriel S, Ziaugra L, Tabbaa D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet. 2009;60(1). doi:10.1002/0471142905.hg0212s60
15. Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39(3):347–351. doi:10.1038/ng1975
16. Bland JM, Altman DG. Statistics notes. The odds ratio. BMJ. 2000;320(7247):1468. doi:10.1136/bmj.320.7247.1468
17. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi:10.1086/519795
18. Shi YY, He L. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. 2005;15(2):97–98. doi:10.1038/sj.cr.7290272
19. Barrett JC. Haploview: visualization and analysis of SNP genotype data. Cold Spring Harb Protoc. 2009;2009(10):db ip71. doi:10.1101/pdb.ip71
20. Khatami M, Heidari MM, Tabesh F, Ordooei M, Salehifar Z. Mutation analysis of the NKX2.5 gene in Iranian pediatric patients with congenital hypothyroidism. J Pediatr Endocrinol Metab. 2017;30(1):e2653. doi:10.1515/jpem-2017-0084
21. Iwadate M, Takizawa Y, Kimura S. Abstract 357: NKX2-1 regulates SMAD and non-SMAD pathways in thyroid stem cells. Cancer Res. 2017;77(13 Supplement):357. doi:10.1158/1538-7445.AM2017-357
22. Monica D, Viviana C, Annamaria R, et al. Missense mutation in the transcription factor NKX2-5: a novel molecular event in the pathogenesis of thyroid dysgenesis. J Clin Endocrinol Metab. 2006;91(4):1428–1433. doi:10.1210/jc.2005-1350
23. Zhenling W, Li Z, Rong Z, et al. Associations between two genetic variants in NKX2-5 and risk of congenital heart disease in Chinese population: a meta-analysis. PLoS One. 2013;8(8):e70979. doi:10.1371/journal.pone.0070979
24. Ketharnathan S, Koshy T, Sethuratnam R, Paul S, Venkatesan V. Investigation of NKX2.5 gene mutations in congenital heart defects in an Indian population. Genet Test Mol Biomarkers. 2015;19(10):579–583. doi:10.1089/gtmb.2015.0112
25. Tom M, Robin R. An extensive network of coupling among gene expression machines. Nature. 2002;416(6880):499–506. doi:10.1038/416499a
26. Chen W, Feng PM, Lin H, Chou KC. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013;41(6):e68. doi:10.1093/nar/gks1450
27. Guil S, Soler M, Portela A, et al. Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nat Struct Mol Biol. 2012;19(7):664. doi:10.1038/nsmb.2315
28. Eddo K, Alon M, Gil A. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007;35(1):125–131. doi:10.1093/nar/gkl924
29. Meenakshi R, Namshin K, Yi X, Christopher L. The effect of intron length on exon creation ratios during the evolution of mammalian genomes. Rna-a Pub Rna Soc. 2008;14(11):2261. doi:10.1261/rna.1024908
30. Fawcett JA, Rouzé P, Peer YVD. Higher intron loss rate in Arabidopsis thaliana than A. lyrata is consistent with stronger selection for a smaller genome. Mol Biol Evol. 2012;29(2):849–859. doi:10.1093/molbev/msr254
31. Roy S, Hartl D. Very little intron loss/gain in plasmodium: intron loss/gain mutation rates and intron number. Genome Res. 2006;16(6):750–756. doi:10.1101/gr.4845406
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.