Back to Journals » International Journal of Nanomedicine » Volume 21
A Bubble-Driven Drug Delivery System Enhances Oral Absorption and Antipyretic Efficacy of Poorly Water-Soluble Andrographolide
Authors Zhou J ![]() , Luo W, Zeng L
, Luo W, Zeng L ![]() , Xu S, Wu R, Lu J, Li Y, Xiong W
, Xu S, Wu R, Lu J, Li Y, Xiong W
Received 30 October 2025
Accepted for publication 23 January 2026
Published 3 February 2026 Volume 2026:21 574265
DOI https://doi.org/10.2147/IJN.S574265
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. RDK Misra
Jieying Zhou,1,* Wei Luo,1,* Lingxin Zeng,1,* Shihao Xu,2 Ruohan Wu,3 Jiaqi Lu,4 Ying Li,1 Wei Xiong1
1School of Pharmacy, Shenzhen University Medical School, Shenzhen University, Shenzhen, 518055, People’s Republic of China; 2School of Chinese Pharmacy, Shenyang Pharmaceutical University, Shenyang, 110016, People’s Republic of China; 3Shenzhen College of International Education, Shenzhen, 518043, People’s Republic of China; 4Production Center, Raytor Instruments, Shenzhen, 518001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ying Li, School of Pharmacy, Shenzhen University Medical School, Shenzhen University, 1066 Xueyuan Avenue, Nanshan District, Shenzhen, 518055, People’s Republic of China, Email [email protected] Wei Xiong, School of Pharmacy, Shenzhen University Medical School, Shenzhen University, 1066 Xueyuan Avenue, Nanshan District, Shenzhen, 518055, People’s Republic of China, Tel +86075526913026, Email [email protected]
Purpose: While bubble-carrier systems hold great promise for enhancing the oral bioavailability of poorly water-soluble drugs, a deeper understanding of their underlying absorption mechanisms is still required. This study aimed to address this gap by developing a bubble-driven delivery system (BDDS) using the poorly water-soluble andrographolide (AG) as a model drug, and systematically evaluating its solubilization capacity and intestinal permeability.
Methods: The BDDS was optimized through the screening of effervescent formulations (citric acid: Na2CO3 = 6:5) and bile salts (sodium deoxycholate, SDC). Its performance was comprehensively assessed via in vitro dissolution tests, an everted intestinal sac assay, Caco-2 cell monolayer experiments, pharmacokinetic studies, and yeast-induced fever rat models.
Results: The BDDS achieved 75.0%– 88.7% dissolution in gastrointestinal pH media, which was markedly superior to that of commercial dripping pills (DP; 30.5– 50.2%). Its apparent permeability coefficient was 7.7-fold and 3.6-fold higher than that of free AG and DP, respectively. Transport studies indicated that SDC enhanced AG permeability primarily via the apical sodium-dependent bile acid transporter (ASBT)-mediated pathway, a mechanism further confirmed by inhibition with linerixibat. The relative oral bioavailability of BDDS versus DP reached 257.1%. BDDS demonstrated enhanced efficacy by significantly inhibiting key pro-inflammatory cytokines and thermoregulatory mediators in yeast-induced fever rats.
Discussion: The BDDS effectively overcomes AG’s low solubility (by SDC solubilization and CO2 bubbles) and permeability (via ASBT transport), outperforming conventional formulations in terms of preparation simplicity and storage stability.
Conclusion: BDDS is a promising strategy to improve oral absorption and therapeutic efficacy of BCS Class IV drugs like AG, with ASBT-mediated transport as a key mechanism.
Keywords: bubble, poorly water-soluble, intestinal permeability, oral bioavailability, andrographolide
Introduction
Oral dosing is a preferred and convenient drug administration route due to its high patient compliance and low manufacturing costs.1 However, approximately 70% of commercial drugs and 90% of new chemical entities suffer from poor aqueous solubility.2 This limitation is particularly critical for Biopharmaceutics Classification System (BCS) Class IV drugs,3 which exhibit both low solubility and low intestinal permeability, leading to inadequate oral bioavailability and compromised clinical efficacy. Addressing the oral absorption barrier of poorly water-soluble drugs, especially BCS Class IV agents, remains a major challenge in pharmaceutical research.
To improve the oral bioavailability of poorly water-soluble drugs, various formulation strategies have been developed and applied in commercial production, such as solid dispersions,4 cyclodextrin complexes,5 and nanocrystal formulations.6 Nevertheless, these technologies have inherent limitations that hinder their broader application: solid dispersions are prone to drug recrystallization during storage, reducing long-term stability;7 cyclodextrins require high doses to achieve effective solubilization, which may cause gastrointestinal irritation;8 and nanocrystal preparation often involves organic solvents, posing risks of residual toxicity and aggregation.9 Thus, there is an urgent need for a simple, safe, and effective delivery system that can simultaneously enhance both drug solubility and intestinal permeability.
Recently, Sung et al proposed the use of a bubble-carrier system to enhance the oral bioavailability of lipophilic curcumin and paclitaxel.10,11 The system contained a powdered mixture of an acid initiator citric acid (CA), a foaming agent (sodium bicarbonate, NaHCO3), a surfactant bile salt, and a poorly water-soluble drug. After an enteric-coated capsule containing the described powdered mixture was orally administered, the CA was deprotonated, which produced an acidic environment in which NaHCO3 underwent an effervescent reaction to infuse the intestinal lumen with CO2 bubbles. These CO2 bubbles were instantly stabilized by bile salts; they then burst and self-aggregated into micelles in the intestinal fluid, enhancing the water solubility and oral absorption of the drug.
Despite the attention, this bubble-carrier system has received for its facile preparation and without organic solvent residues, the underlying mechanisms by which it improves oral drug absorption remain unclear. Critical knowledge gaps include: (1) the specific role of bile salts in coordinating drug solubilization and intestinal permeability; (2) whether apical sodium-dependent bile acid transporter (ASBT)-mediated transport—an established pathway for bile acid absorption in the terminal ileum—contributes to the system’s permeability-enhancing effect;12,13 and (3) the system’s efficacy in improving the oral absorption of BCS Class IV drugs, which have dual defects of low solubility and permeability.
To address these gaps, the present study selected andrographolide (AG) as the model drug. AG is a clinically used anti-inflammatory and antipyretic agent,14 but it belongs to BCS Class IV with extremely low aqueous solubility (0.074 mg/mL) and poor intestinal permeability.15 Its poor oral absorption severely limits its therapeutic efficacy, making it an ideal candidate to evaluate novel delivery systems. Building on the design principle of the previous bubble-carrier system, we developed a bubble-driven delivery system (BDDS) by rationally screening effervescent formulations (acid initiators and foaming agents) and bile salt compositions, and optimizing drug-loading capacity (Figure 1). Consequently, this study had three primary objectives. First, we characterized the BDDS and its enhancement of AG solubility under gastrointestinal pH. Second, we assessed its effect on AG intestinal permeability and probed the role of ASBT-mediated transport. Third, we validated its ability to improve AG oral bioavailability and antipyretic efficacy in vivo.
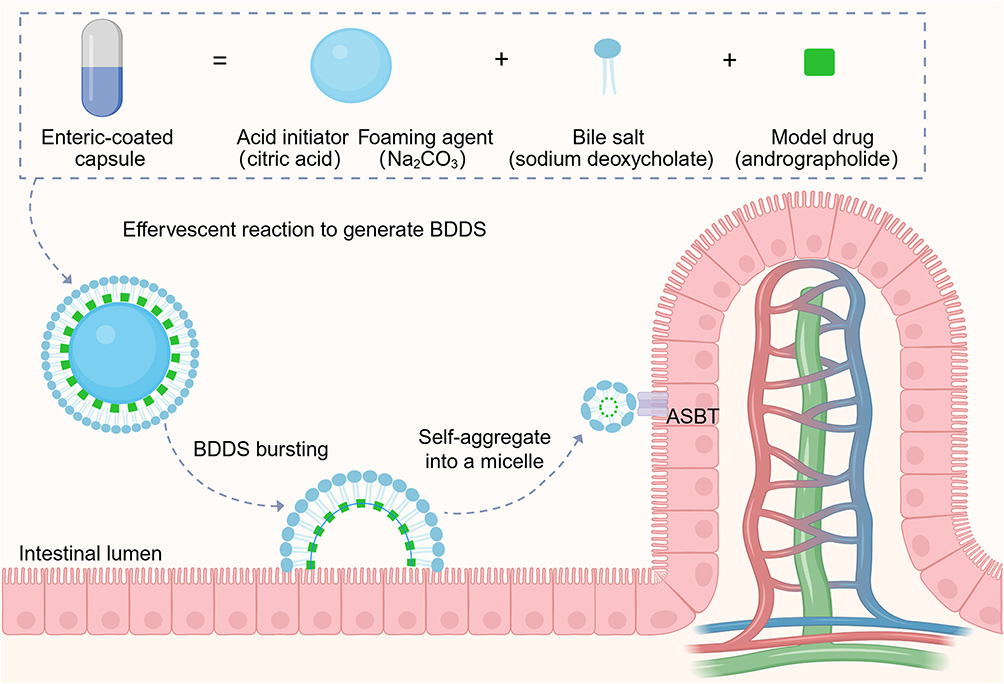 |
Figure 1 Formation of the bubble-driven delivery system (BDDS) and intestinal absorption of andrographolide after bubble bursting. |
Materials and Methods
Chemicals and Reagents
The following chemicals and reagents of analytical grade were used in this study: AG, coumarin 6 (C6), sodium deoxycholate (SDC), taurocholic acid (TCA), ursodeoxycholic acid (UDCA), NaHCO3, Na2CO3, CA, and tartaric acid (TA) (all purities ≥ 98%; sourced from Aladdin, Shanghai, China). Linerixibat (purity ≥ 98%) was obtained from Bide Pharmatech Ltd (Shanghai, China). Commercial dropping pills (DP, developed using solid dispersions with PEG6000 and PEG4000) were provided by Tasly Pharmaceutical Group Co., Ltd (Tianjin, China).
BDDS Formulation
Effervescent Formulation Composition and Ratios
Effervescent activity was estimated by observing the foam volume in a test tube after an effervescent reaction, as previously reported.10 Briefly, 2, 4, 6, 8, or 10 mg of acid initiator (CA or TA) and 5 mg of foaming agent (NaHCO3 or Na2CO3) were placed into a test tube, then 3 mL of PBS buffer (pH, 6.8) was added. After recording the maximum height of foam produced, the pH of the resulting solution was determined.
AG to Effervescent Formulation Ratio
After obtaining the optimal effervescent formulation composition and ratio (CA:Na2CO3 = 6:5, w/w), we investigated the weight ratio of AG to effervescent formulation with a dissolution test.11 A mixture of AG (5 mg) and the effervescent composite (CA 25 mg + Na2CO3 20.8 mg; CA 50 mg + Na2CO3 41.7 mg; CA 75 mg + Na2CO3 62.5 mg) was placed in a dissolution vessel (volume 250 mL) containing 250 mL of distilled water at 37°C. The dissolution was carried out using a dissolution tester (Raytor Instruments, Shenzhen, China) with a small paddle rotating at 100 rpm. At the indicated time intervals, 2 mL of each sample was withdrawn and replaced with distilled water to maintain a constant volume. The AG content of the dissolution test samples was determined using high-performance liquid chromatography (HPLC).16 Briefly, the analysis was performed on a Waters 2965 series HPLC system equipped with an Agilent ZORBAX SB-C18 column (4.6 × 250 mm, 5 μm). The mobile phase, consisting of water and methanol in a 50:50 (v/v) ratio, was delivered at a flow rate of 1.0 mL/min. The column temperature was maintained at 25°C, and the detection wavelength was set at 225 nm. The injection volume was 10 μL.
Addition of Surfactant
A mixture of AG (5 mg; 14.3 μmol), effervescent composite [CA, 50 mg (237.9 μmol); Na2CO3, 41.7 mg (393.4 μmol)], and surfactant (SDC, 2, 5, 10 or 25 mg; 4.8, 12.1, 24.1 or 60.3 μmol, respectively) was placed in a dissolution cup with 250 mL of distilled water. The dissolution of AG was determined as previously described in AG to Effervescent Formulation Ratio.
Screening of Surfactant
A mixture of AG (5 mg; 14.3 μmol), effervescent composite [CA, 50 mg (237.9 μmol); Na2CO3, 41.7 mg (393.4 μmol)], and surfactant (SDC, TCA, or UDCA, 5 mg; 12.1, 9.7, or 12.7 μmol, respectively) was placed in a dissolution cup with 250 mL of distilled water. The dissolution was determined as previously described in AG to Effervescent Formulation Ratio.
Characterization of the BDDS After Effervescent Reaction
The size and zeta potential of the micelles generated by SDC after effervescent reaction were characterized using dynamic light scattering (DLS).17 The morphology of the micelles generated by SDC after effervescent reaction was observed using transmission electron microscopy (TEM, Hitachi, Japan).18 To observe CO2 bubble diffusion behavior, AG was replaced with fluorescent C6. The C6-containing BDDS was placed in a confocal dish and observed by fluorescence microscopy (Nikon, Japan).10
Dissolution Test
The dissolution of free AG, DP and BDDS (each equivalent to 5 mg of AG) was assessed for 0.1 M HCl (pH 1.2) and PBS (pH 6.8 and 7.4). Briefly, each sample was placed in a 250 mL dissolution cup containing 250 mL of the respective medium at 37°C, with a stirring speed of 100 rpm in the dissolution tester. At the indicated time intervals, 2 mL of each sample was collected and replaced with an equal volume of fresh media. The concentration of AG in the samples was determined by HPLC.16
Intestinal Permeability
Measurement of intestinal permeability was performed via the everted intestinal sac method, as previously described.19 Briefly, a section of the ileum (12 cm) was isolated and collected from an overnight-fasted male Sprague-Dawley (SD) rat. The intestinal segment was carefully flushed with warm PBS and gently turned inside out over a glass rod. One end of the segment was tied with suture thread (4–0), and the section was filled with 1.5 mL of Krebs-Ringer (KR) solution (Pricella Biotechnology Co., Ltd., Wuhan, China). The other end of the segment was sealed and cannulated with a polyethylene catheter (polyethylene tubing: 1.8-mm internal diameter, 2.4-mm outside diameter). The intestine segment was submerged in a tube containing oxygenated KR solution (10 mL) maintained at 37°C in the presence free AG (0.4 mg/mL) or DP or BDDS (equal to AG 0.4 mg/mL). To investigate the role of ASBT in BDDS’s effect on the intestinal permeability of AG, the intestine segments were pre-treated with linerixibat (10 μM) for 30 min to inhibit the transport function of the ASBT. After that, at the indicated time intervals (15, 30, 45, 60, 90, and 120 min), 300 μL of sample was collected through the polyethylene catheter and replaced with fresh KR solution to maintain a constant volume. At the end of the experiment, the sealed intestine was carefully removed to measure its length and width. The AG content in the collected samples was determined via HPLC.16 The apparent permeability coefficient (Papp) was calculated using the following equation, as used previously:20
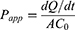
where dQ/dt is the rate of appearance of AG in the everted intestine (receiver compartment), C0 is the initial concentration of AG in the tube (donor compartment), and A is the total area of the everted intestine.
Parallel Artificial Membrane Permeability Assay (PAMPA)
The PAMPA was performed using a microdissolution penetration analysis system (NCE DP, Raytor Instruments, Shenzhen, China). Briefly, the donor chamber was filled with 20 mL of a PBS solution (pH 6.8) containing the test sample (Free AG, AG+SDC, AG+Na2CO3, or BDDS) at an AG concentration of 200 μg/mL, while the receiver chamber was filled with 20 mL of PBS (pH 6.8). Permeation was carried out at 37°C with agitation at 500 rpm. At predetermined time points (5, 15, 30, 45, 60, 90, and 120 min), 1 mL of sample was withdrawn from the receiver chamber and immediately replenished with an equal volume of fresh PBS (pH 6.8). The collected samples were analyzed by HPLC. The apparent permeability coefficient (Papp) was calculated using the following equation:
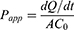
where dQ/dt is the rate of appearance of AG in the receiver chamber, C0 is the initial concentration of AG in the donor chamber, and A is the area of the artificial membrane.
Potential Mechanism of BDDS’s Impact on Intestinal Absorption
Caco-2 cells (passage numbers 10 to 15, Procell Life Science&Technology Co., Ltd., Wuhan, China) were seeded into a 6-well plate (2 × 105 cells/well) and allowed to adhere to the wells overnight. At 90% confluence, the culture medium was replaced with media containing AG (200 μM) in the presence and/or absence of SDC (169 μM), CA+Na2CO3, and the BDDS. After incubation for 2 h, the treated cells were washed with ice-cold PBS and lysed in 300 μL 80% methanol, and the AG content in the cell lysate was quantified by HPLC. To investigate the role of ASBT in BDDS’s effect on the oral absorption of AG, the Caco-2 cells were pre-treated with linerixibat (1 μM) for 30 min to inhibit the transport function of the ASBT.
Cellular Internalization Mechanism
To investigate the endocytosis pathways involved in BDDS uptake, HT-29 cells were pretreated for 0.5 h with specific endocytosis inhibitors, including chlorpromazine (50 μM), indomethacin (100 μM), colchicine (10 μM), and quercetin (10 μM). The effect of these inhibitors on cellular uptake was measured using the HPLC method.
Animal Studies
Male SD rats were obtained from the Guangdong Medical Laboratory Animal Center (Foshan, China) and maintained under standard conditions at 22°C, humidity 60% ± 10%, and a 12-h light/dark cycle, with free access to fresh water and rodent chow. All experimental protocols were performed in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals and were approved by the Animal Research Ethics Committee (Approval No. IACUC-202500157), Shenzhen University, Shenzhen, China.
Pharmacokinetic Study
Overnight-fasted rats (aged 6 weeks, 225 ± 25 g) were orally administered DP or BDDS (both equivalent to 40 mg/kg AG) (n = 6). Blood samples (0.5 mL) were collected in heparinized tubes at the indicated timepoints (0.25, 0.5, 0.75, 1, 2, 3, 4, 8, 12, and 24 h). All the obtained blood samples were centrifuged at 4000 rpm for 5 min, and the plasma samples were stored at −80 °C for analysis. After a pharmacokinetic study, the rats were sacrificed by CO2 inhalation. The plasma sample preparation was performed according to our previously reported methods.17 Briefly, 10 μL bilobalide (100 ng/mL, analytical standard, Aladdin, Shanghai, China) was added into 100 μL plasma samples. After vortex mixing, ethyl acetate (1 mL) was added to the mixture mentioned above and vortexed for 3 min. After that, these samples were centrifuged at 1,4000 rpm for 10 min. The supernatant was transferred into a plastic centrifuge tube and evaporated under a vacuum drying oven at 40°C. Finally, the residue was reconstituted with methanol (90 μL) and for LC-MS/MS (Xevo TQD, Waters, USA) analysis. The conditions of LC-MS/MS were added in supporting information (Supplementary Figure 1). The plasma concentrations versus time profiles were analyzed using DAS software (Shanghai, China).
Antipyretic Study
Rectal Temperature Measurements
Male SD rats (aged 4 weeks, 130 ± 10 g) were subjected to rectal temperature (Tr) measurements for 3 consecutive days. Specifically, a digital thermometer (temperature range: 32 − 44°C precision: ± 0.01°C; Shangnong Electronic Technology Co., Ltd., Heze, China) coated with Vaseline was inserted into the rectum of each rat (2–3 cm deep) for 1 min to measure Tr. Those rats with a Tr difference exceeding 0.5°C and higher than 38°C were excluded from the following study.
Yeast-Induced Fever Rat Model
An antipyretic study was performed using a yeast-induced fever rat model, as previously reported.21 Briefly, overnight-fasted rats were injected subcutaneously with yeast suspension (20%, w/w, Oxoid, UK) and randomly divided into three groups (n = 8): Model (saline, p.o).; DP (equal to 50 mg/kg of AG, p.o).; and BDDS (equal to 50 mg/kg of AG, p.o). groups.22 An equal volume of normal saline (0.9% w/v NaCl) was injected subcutaneously into the normal control group. These four groups of rats were given the respective therapeutics at 3 h post-injection. Tr was measured every hour for 3–8 h after modeling. At 8 h post-injection, all rats were sacrificed by CO2 inhalation and for the collection of serum and hypothalamic tissue samples.
Pyrogen Determination
Rat serum inflammatory cytokine (IL-1β and IL-6) concentrations were determined using commercial ELISA kits (Neobioscience, Shenzhen, China). Briefly, 100 μL of each sample or standard was added into the wells, and the plate was sealed and incubated at 37°C in the dark for 90 min. Thereafter, the plate underwent five wash cycles. Next, 100 μL of biotinylated detection antibody working solution was added to each well and incubated for 60 min. After another five washes, 100 μL of enzyme conjugate working solution was added and incubated for 30 min. Finally, following the addition of 100 μL TMB substrate and subsequent stop solution, the absorbance at 450 nm was measured within 3 min.
PGE2 and cAMP levels in hypothalamus samples were also determined with commercial ELISA kits (Meimian, Jiangsu, China). In brief, 50 µL of a mixture containing 10 µL of hypothalamic homogenate (0.1 g/mL) or standard and 40 µL of diluent was added into each well. The plate was sealed and incubated at 37°C in the dark for 60 min. Thereafter, it was washed five times. Next, 50 µL of HRP-labeled antibody working solution was added per well and incubated for 60 min. Following another five washes, color was developed by sequential addition of TMB substrate and stop solution. Absorbance at 450 nm was measured within 15 min.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Inc., CA). A value of p < 0.05 versus the control group was considered significant for all tests.
Results
Screening of Effervescent Formulation
The constitution of the effervescent formulation is a key factor in the development of a BDDS. Thus, we first screened various amounts of acid initiators (CA or TA) and foaming agents (Na2CO3 or NaHCO3) using the CO2 bubble volume as an indicator. Once the PBS (pH 6.8) was added into the test tube containing the described acid initiator and foaming agent, CO2 bubbles were immediately produced, as shown in Figure 2A and a high-resolution video included in the Supplementary Material. When the Na2CO3 or NaHCO3 was fixed at 5 mg, the foam volume increased with an increasing amount of CA or TA (Figure 2B and C). Considering the physiological pH of the small intestine ranges from 5 to 7, we selected the effervescent formulation with a CA to Na2CO3 weight ratio of 6:5 (molar ratio 3:5) for subsequent studies.
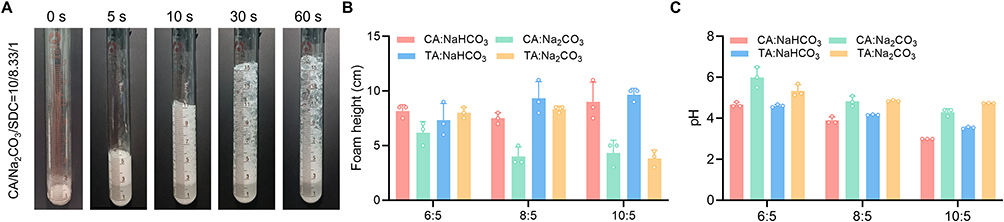 |
Figure 2 Screening of effervescent formulations of BDDS. (A) Foam volume after addition of PBS. Foam height (B) and pH (C) of different foaming agent reaction systems with the different acid initiator (CA and TA). |
Optimization of BDDS Formulation
To determine the optimum proportions for BDDS loading, we investigated drug solubilization via the AG dissolution. Due to the poor water solubility of AG, the dissolution of free AG (containing 5 mg AG) in water was 31.6% in 120 min (Figure 3A). Interestingly, the combined AG and effervescent compositions (namely AG, CA, and Na2CO3) enhanced AG dissolution, achieving approximately 60% (Figure 3A). We then tested three AG and effervescent compositions in the presence of SDC and found the formulation (AG:CA:Na2CO3:SDC = 5:50:41.7:5, w/w) had the best AG dissolution (Figure 3B), thereby the proportion of AG to effervescent formulation in subsequent studies was 5:50:41.7 (AG:CA:Na2CO3). We further confirmed 5 mg to be the optimal SDC dose for the BDDS formulation (Figure 3C). Due to the solubilization of bile salts, we screened different types of bile salts (SDC, TCA, and UDCA) for use in the BDDS. SDC exhibited the greatest potency in improving the AG dissolution (Figure 3D). Therefore, the optimal formulation of BDDS was determined to contain a powdered mixture of CA acid initiator (50 mg), Na2CO3 foaming agent (42 mg), SDC surfactant (5 mg), and AG (5 mg).
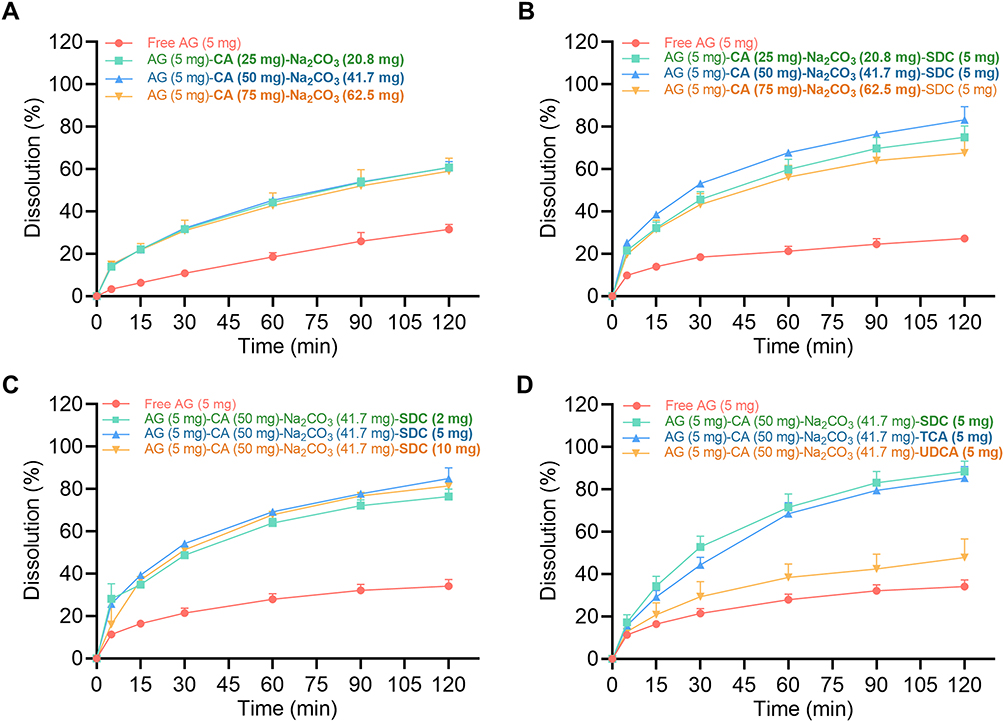 |
Figure 3 Screening of BDDS loading compositions. The effects of (A) effervescent composition, (B) effervescent composition in the presence of SDC, (C) SDC dose, and (D) bile salt type on AG dissolution. Data are expressed as mean ± SD (n = 3). |
Characterization of the BDDS After Effervescent Reaction
After identifying the optimal BDDS candidate, we characterized the micelles generated by SDC after effervescent reaction. Under TEM observation, the micelles generated by BDDS exhibited uniform spherical morphologies, with larger sizes compared to their counterparts (AG+CA+Na2CO3 and AG+SDC) (Figure 4A). DLS analysis revealed a mean particle size of 267.7 nm (Figure 4B) and a zeta potential of −32.4 mV (Figure 4C) for those micelles generated by BDDS after effervescent reaction. To better visualize the bubbles after the effervescent reaction, we replaced AG with fluorescent C6 and observed C6-BDDS under a fluorescence microscopy. We found that the green-fluorescent C6 stuck to the surfaces of the CO2 bubbles (Figure 4D). These results indicated that the SDC underwent self-assembly into micelles following the effervescent reaction, which might facilitate AG dissolution.
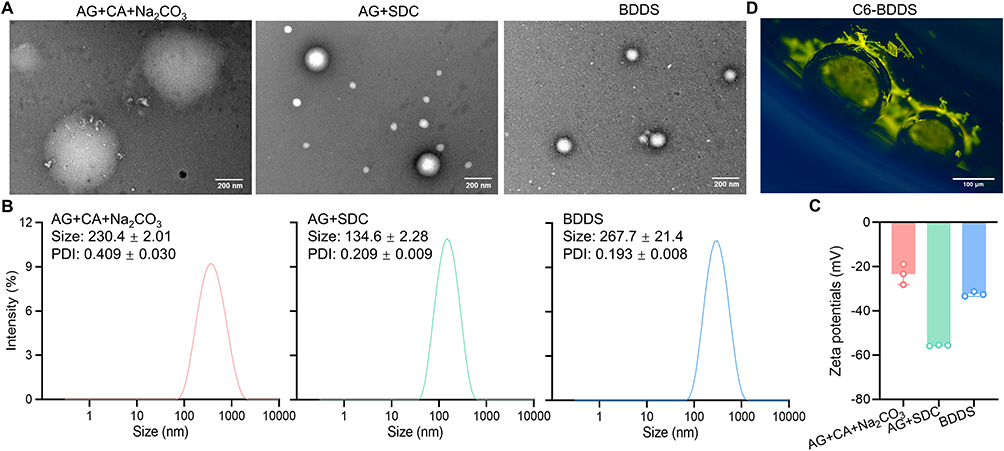 |
Figure 4 Characterization of the optimal BDDS candidate. (A) Representative TEM images of the self-emulsified BDDS, scale bar = 500 nm or 200 nm. (B) Particle size and (C) zeta potential of the self-emulsified BDDS. (D) Representative fluorescence microscopy of C6 on bubbles, scale bar = 100 μm. |
Enhanced Dissolution of AG by BDDS
The dissolution of AG was investigated in different pH environments. Only 28.8%, 25.6%, and 26.3% of free AG was dissolute within 120 min in media at pH 1.2, 6.8, and 7.4, respectively (Figure 5A–C). Commercial DP was able to increase the AG solubility to a certain extent (30.5%, 43.3%, and 50.2%, respectively). As expected, the non-enteric-coated BDDS markedly enhanced the dissolution of AG to 75.0%, 85.8%, and 88.7% in pH 1.2, 6.8, and 7.4 respectively, indicating the BDDS significantly improved AG dissolution.
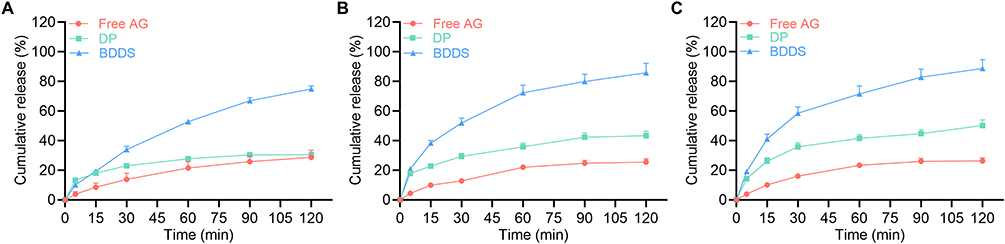 |
Figure 5 Dissolution of AG from BDDS in different pH environments. Dissolution of AG in (A) 0.1 M HCl (pH 1.2), (B) PBS (pH 6.8), and (C) PBS (pH 7.4) from free AG, AG dripping pill (DP), and the BDDS. Data are expressed as mean ± SD (n = 3). |
Potential Mechanism Behind BDDS’s Enhancement of Intestinal Permeability
After confirming the AG solubilization of the BDDS, we investigated the intestinal AG permeability via the everted intestinal sac method. As illustrated in Figure 6A, the permeation of AG from the BDDS (0.4 mg/mL) across the everted rat intestine was measured. The Papp of BDDS (6.37 ± 0.37×10−6 cm/s) was determined to be 7.7-fold and 3.6-fold that of free AG (0.83 ± 0.61×10−6 cm/s) and DP (1.79 ± 0.61×10−6 cm/s) (Figure 6B), respectively, suggesting the BDDS significantly enhanced the intestinal permeability of AG.
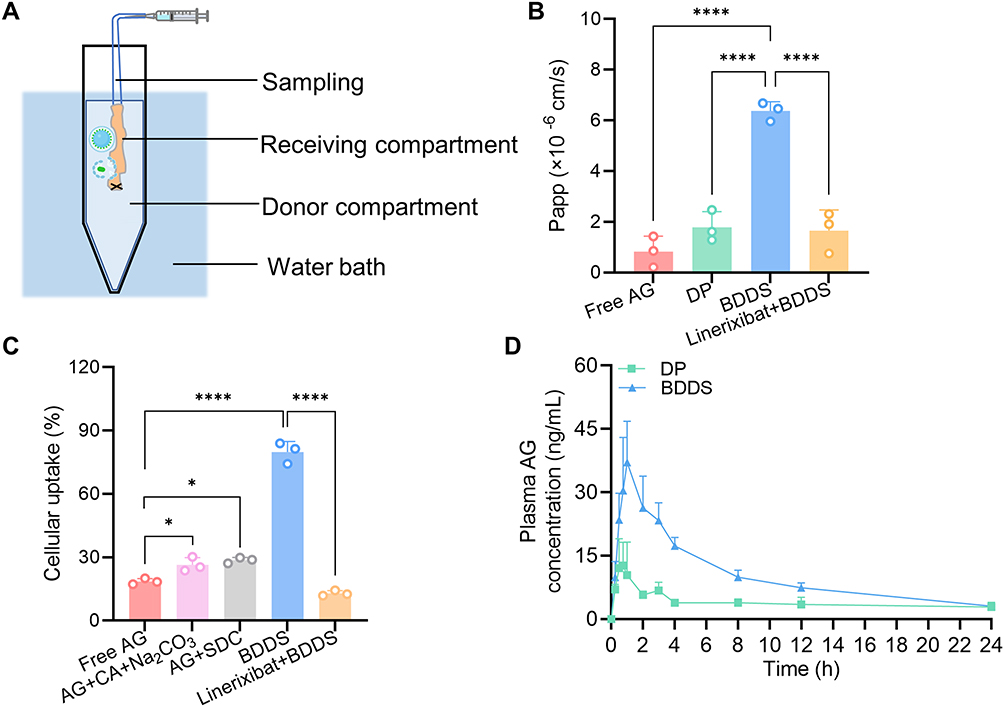 |
Figure 6 Oral absorption of AG from BDDS and the potential mechanism enhancing intestinal permeability. (A) Schematic of everted intestinal sac method. (B) Apparent permeability coefficient (Papp). (C) Cellular uptake in Caco-2 cells. (D) Plasma concentration-time curves of BDDS after oral administration in rats. * p < 0.05; **** p < 0.0001 versus the free AG or BDDS group. |
To explore the potential mechanism by which the BDDS enhances the intestinal permeability of AG, we determined its uptake by Caco-2 cells. Although effervescent composition (CA and Na2CO3) and SDC enhanced cellular uptake of AG to some extent, BDDS increased cellular AG uptake by 4.3-fold compared with free AG (Figure 6C). Given the bile acids undergo ASBT-dependent transport, we hypothesized the ASBT-mediated transport plays a key role in enhancing AG uptake. As expected, when cells were pre-treated with the specific ASBT inhibitor linerixibat (1 μM) for 30 min, the AG uptake dropped dramatically (Figure 6C). Consistent with the cellular uptake results, pre-treated with linerixibat (10 μM) for 30 min also inhibited BDDS intestinal permeability (Figure 6B). These findings indicated that the BDDS significantly increased AG intestinal permeability through ASBT-mediated transport.
Enhanced Oral Bioavailability
The plasma concentration-time profile of AG following oral administration of DP and BDDS is presented in Figure 6D, with the corresponding pharmacokinetic parameters summarized in Table 1. In line with the dissolution and intestinal permeability results, the BDDS demonstrated a marked enhancement in systemic exposure, as evidenced by a significantly higher Cmax (42.3 vs 18.6 ng/L) and extended tmax (1.2 vs 0.8 h) compared to DP. The relative oral bioavailability AG from BDDS in comparison to DP was 257.1%. These results demonstrate that the BDDS significantly enhanced the oral bioavailability of AG by simultaneously improving its water solubility and intestinal permeability.
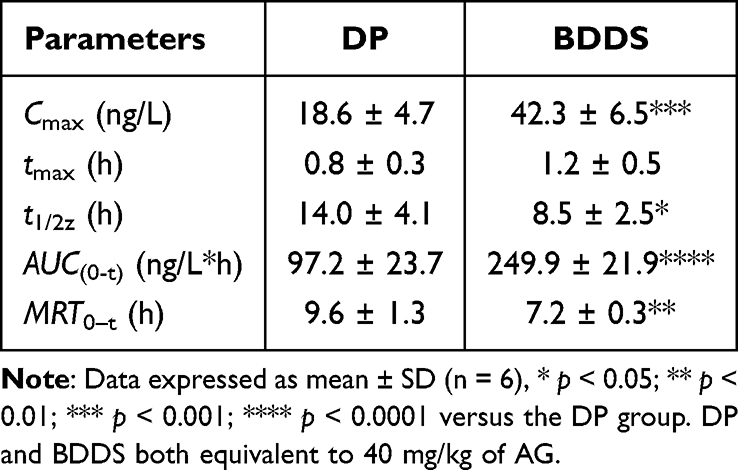 |
Table 1 Pharmacokinetic Parameters of Rats After Oral Administration of DP and BDDS |
Antipyretic Effect in Yeast-Induced Fever Rat Model
The antipyretic effect of the BDDS was evaluated in a yeast-induced fever rat model (Figure 7A). After subcutaneous injection of yeast suspension (20%, w/w, 10 mL/kg), the Tr of the model rats showed a marked increase (> 0.5°C) at 3 h post-injection (Figure 7B), and the upward trend lasted until 8 h post-injection. Notably, the BDDS effectively inhibited the Tr of the yeast-induced fever rats over the 3-h to 8-h observation times and exhibited a better antipyretic effect than DP.
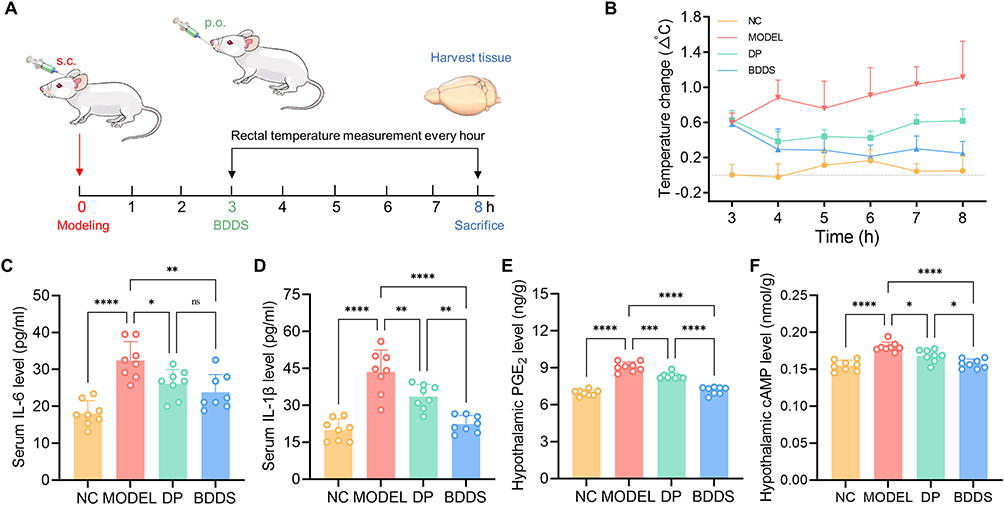 |
Figure 7 Antipyretic effect of BDDS in yeast-induced fever rat model. (A) Schematic illustration of treatment regimens. (B) Changes in rectal temperature after injection of yeast suspension. Levels of endothermic factors (C) IL-6 and (D) IL-1β in the sera of rats. Levels of thermoregulatory indicators (E) PGE2 and (F) cAMP in the hypothalamus of rats. Data are expressed as mean ± SD (n = 8). ns No significance; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001 versus the MODEL group. |
To further elucidate the potential antipyretic effects of the BDDS, the serum and hypothalamus of rats at 8 h post-injection were collected for biochemical analysis. Endogenous and exogenous pyrogens, including IL-1β and IL-6, can directly result in febrile illness.23 We found the levels of serum pro-inflammatory cytokines IL-1β and IL-6 were significantly inhibited in the BDDS-treated rats (Figure 7C and D), demonstrating the BDDS weakened peripheral inflammation to achieve antipyretic effects. Additionally, these pyrogens trigger the hypothalamus to release thermoregulatory indicators (PGE2 and cAMP) that also result in febrile illness.24 Compared to DP, BDDS exhibited significantly stronger inhibition of PGE2 and cAMP levels in the hypothalamus of yeast-induced febrile rats (Figure 7E and F), underscoring its enhanced antifebrile efficacy in this model. Taken together, these results demonstrate that the BDDS significantly enhances the oral delivery of AG, thereby contributing to its improved antifebrile efficacy in the yeast-induced fever rat model.
Discussion
In this study, we successfully developed BDDS to simultaneously address the poor water solubility and low intestinal permeability of AG. In contrast to previously reported bubble carriers,25,26 our key innovation lies in the rational selection of the bile salt component. Our results demonstrated that BDDS promoted cell uptake via ASBT-mediated transport more effectively than both the bubble carrier group (AG+CA+Na2CO3) and the bile salt group (AG+SDC) (Figure 6C). While BDDS showed a 3.1-fold higher apparent permeability than free AG in the parallel artificial membrane permeability assay (PAMPA) model (Supplementary Figure 2), its intestinal permeability measured by the everted sac method was increased by 7.7-fold (Figure 6B). This greater enhancement in intestinal permeability compared to the PAMPA result further supports ASBT-mediated transport as a key mechanism for BDDS uptake. These findings collectively underscore that the combination of the bubble carrier and SDC is essential for the performance of BDDS.
Additionally, the performance of BDDS was comprehensively evaluated against four reported AG formulations: solid dispersions (SD),27 mesoporous silica nanoparticles (MSNs),28,29 nanoemulsions (NEs) and self-microemulsifying drug delivery systems (SMEDDS).30,31 The comparison focused on four critical parameters: dissolution profile, intestinal absorption mechanism, pharmacokinetic performance, and industrial applicability.
First, the low aqueous solubility of AG is the primary barrier to oral absorption. Free AG showed low dissolution (25.6–28.8% at 120 min) (Figure 5A–C). BDDS achieved 75.0–88.7% dissolution at 120 min via CO2 bubbles (disrupting AG aggregates) and SDC micelles (preventing precipitation). By contrast, the AG-SD achieved 80% dissolution (120 min, pH 1.2) by converting AG to amorphous state but required a high AG: excipient ratio (1:8) and risked recrystallization. AG-MSNs only achieved 45% dissolution due to the slow nanocrystal diffusion from mesopores. AG-NEs and AG-SMEDDS reached 90.0–97.7% dissolution (at 15 min) via nano/microemulsions but relied on pH-sensitive emulsification kinetics. Therefore, the advantage of the developed BDDS was the stable dissolution across GI pH 1.2–7.4 (superior to pH-dependent AG-NEs/SMEDDS) and used only GRAS excipients, avoiding GI irritation from high surfactants in AG-NEs (Cremophor EL) and AG-SMEDDS (Labrasol).
Second, compared to free AG, BDDS showed a 7.7-fold increase in intestinal permeability and a 4.3-fold increase in cellular uptake. These enhancements were attributed to ASBT-mediated transport (Figure 6B and C) and clathrin-mediated endocytosis (Supplementary Figure 3), respectively. This mechanism effectively overcomes the permeability limitations typical of BCS IV drugs, outperforming those formulations reliant solely on passive diffusion. For instance, AG-SD enhanced permeability by 2.7-fold through the amorphization of AG, though they lacked active transport components. In contrast, AG-NEs achieved an 8.2-fold improvement in permeability, primarily via P-glycoprotein inhibition. Intestinal permeability was not evaluated for the other AG formulations.
Third, the BDDS achieved a relative oral bioavailability of 257.1% compared to the commercial DP. This value was lower than those reported for AG-SD (297.7%), AG-NEs (594.3%), and AG-SMEDDS (a 15-fold increase) relative to their respective controls, but higher than that of AG-MSNs (207.7% vs free AG). However, it is important to note that these pharmacokinetic parameters were derived from different research groups and did not use the same control group, which limits direct comparability.
Fourth, the preparation of the BDDS via simple powder mixing offers distinct advantages in simplicity and industrial applicability over other AG formulations. Specifically, AG-SD required solvent evaporation, AG-MSNs involved high-temperature calcination at 550°C for template removal, AG-NEs relied on high-pressure homogenization, and AG-SMEDDS faced challenges with phase separation during storage.
We acknowledge that our study had some limitations. Firstly, the mechanism by which CO2 bubbles enhance membrane permeability was not fully explored. Although recent evidence indicates CO2 may expand and loosen membrane structure,32 future studies should directly investigate its contribution to BDDS efficacy. Secondly, while the BDDS demonstrated favorable oral bioavailability and antipyretic efficacy, its clinical translation may face several challenges. These include the difficulty in quantitatively monitoring the persistence, transformation, and collapse dynamics of CO2 bubbles within the human gastrointestinal tract, as well as the need for more comprehensive safety and biocompatibility data on the GRAS components.
Conclusion
We successfully developed a BDDS to enhance the oral absorption and antipyretic efficacy of AG, a BCS class IV drug. The BDDS significantly improved AG dissolution in gastrointestinal pH environments, which attributed to CO2 bubbles disrupting AG aggregates and sodium deoxycholate forming solubilizing micelles. Furthermore, it enhanced intestinal permeability via ASBT-mediated transport, as confirmed by the significant reduction in AG uptake and permeability upon pre-treated with the ASBT inhibitor linerixibat. Moreover, BDDS achieved a relative oral bioavailability of 257.1% compared to DP and demonstrated superior antipyretic effects in a yeast-induced febrile rat model. Overall, this study provides evidence that the BDDS is a promising pharmaceutical approach to promoting the oral absorption of BCS class IV drugs.
Data Sharing Statement
Data are available upon reasonable request from the corresponding author (Dr. Wei Xiong).
Acknowledgments
This study was supported by the Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515010481), and the Shenzhen Science and Technology Program (No. RCBS20221008093120049, 860000002111304, 827-00074220). The authors would like to thank the Instrumental Analysis Center of Shenzhen University for their technical support.
Disclosure
The authors declare that they have no competing interests in this work. This paper has been uploaded to SSRN as a preprint: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=5402815
References
1. Nardin I, Köllner S. Successful development of oral SEDDS: screening of excipients from the industrial point of view. Adv Drug Deliv Rev. 2019;142:128–13. doi:10.1016/j.addr.2018.10.014
2. Vinarov Z, Abrahamsson B, Artursson P, et al. Current challenges and future perspectives in oral absorption research: an opinion of the UNGAP network. Adv Drug Deliv Rev. 2021;171:289–331. doi:10.1016/j.addr.2021.02.001
3. Roy P, Das S, Auddy RG, et al. Engineered andrographolide nanoparticles mitigate paracetamol hepatotoxicity in mice. Pharm Res. 2013;30:1252–1262. doi:10.1007/s11095-012-0964-5
4. Tran PHL, Tran TTD. Nano-sized solid dispersions for improving the bioavailability of poorly water-soluble drugs. Curr Pharm Des. 2020;6(38):4917–4924. doi:10.2174/1381612826666200701134135
5. Topuz F, Uyar T. Recent advances in cyclodextrin‐based nanoscale drug delivery systems. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2024;
6. McGuckin MB, Wang J, Ghanma R, et al. Nanocrystals as a master key to deliver hydrophobic drugs via multiple administration routes. J Control Release. 2022;345:334–353. doi:10.1016/j.jconrel.2022.03.012
7. Mamidi H, Palekar S, Patel H, et al. Formulation strategies for the development of high drug-loaded amorphous solid dispersions. Drug Discov Today. 2023;28:103806. doi:10.1016/j.drudis.2023.103806
8. Kurkov SV, Loftsson T. Cyclodextrins. Int J Pharm. 2012;453:167–180. doi:10.1016/j.ijpharm.2012.06.055
9. Agrahari V, Agrahari V. Facilitating the translation of nanomedicines to a clinical product: challenges and opportunities. Drug Discov Today. 2018;23(5):974–991. doi:10.1016/j.drudis.2018.01.047
10. Chuang E-Y, Lin K-J, Huang T-Y, et al. An intestinal “transformers”-like nanocarrier system for enhancing the oral bioavailability of poorly water-soluble drugs. ACS Nano. 2018;12(7):6389–6397. doi:10.1021/acsnano.8b00470
11. Chen K-H, Miao Y-B, Shang C-Y, et al. A bubble bursting-mediated oral drug delivery system that enables concurrent delivery of lipophilic and hydrophilic chemotherapeutics for treating pancreatic tumors in rats. Biomaterials. 2020;255:120157. doi:10.1016/j.biomaterials.2020.120157
12. Li M, Wang Q, Li Y, et al. Apical sodium-dependent bile acid transporter, drug target for bile acid related diseases and delivery target for prodrugs: current and future challenges. Pharmacol Therapeut. 2020;212:107539. doi:10.1016/j.pharmthera.2020.107539
13. Deng F, Bae YH. Bile acid transporter-mediated oral drug delivery. J Control Release. 2020;327:100–116. doi:10.1016/j.jconrel.2020.07.034
14. Zhang Y, Hu X, Liu X, et al. Dry state microcrystals stabilized by an HPMC film to improve the bioavailability of andrographolide. Int J Pharm. 2015;493(1–2):214–223. doi:10.1016/j.ijpharm.2015.07.057
15. Gao H, Su Y, Wang W, et al. Integrated computer-aided formulation design: a case study of andrographolide/ cyclodextrin ternary formulation. Asian J Pharm Sci. 2021;16(4):494–507. doi:10.1016/j.ajps.2021.03.006
16. Suo X, Zhang H, Wang Y. HPLC determination of andrographolide in rat whole blood: study on the pharmacokinetics of andrographolide incorporated in liposomes and tablets. Biomed Chromatogr. 2007;21(7):730–734. doi:10.1002/bmc.812
17. Ding B, Zheng Z, Su J, et al. Unraveling the impact of stabilizers on nanocrystal preparation and oral absorption: a case study of poorly soluble andrographolide. Nano Lett. 2025;25(2):820–827. doi:10.1021/acs.nanolett.4c05230
18. Zhang Q, Li Y, Wang S, et al. Chitosan-based oral nanoparticles as an efficient platform for kidney-targeted drug delivery in the treatment of renal fibrosis. Int J Biol Macromol. 2024;256:128315. doi:10.1016/j.ijbiomac.2023.128315
19. Murthy A, Ravi PR, Kathuria H, et al. Self-assembled lecithin-chitosan nanoparticles improve the oral bioavailability and alter the pharmacokinetics of raloxifene. Int J Pharm. 2020;588:119731. doi:10.1016/j.ijpharm.2020.119731
20. Fredlund L, Winiwarter S, Hilgendorf C. In vitro intrinsic permeability: a transporter-independent measure of Caco-2 cell permeability in drug design and development. Mol Pharm. 2017;14(5):1601–1609. doi:10.1021/acs.molpharmaceut.6b01059
21. Su F, Zhu E, Bai C, et al. A comparative study on the antipyretic effect and underlying mechanisms of different bile-fermented Arisaemas. J Ethnopharmacol. 2025;337:118951. doi:10.1016/j.jep.2024.118951
22. Chen H-W, Huang C-S, Li -C-C, et al. Bioavailability of andrographolide and protection against carbon tetrachloride-induced oxidative damage in rats. Toxicol Appl Pharmacol. 2014;280(1):1–9. doi:10.1016/j.taap.2014.07.024
23. Dangarembizi R, Erlwanger KH, Rummel C, et al. Brewer’s yeast is a potent inducer of fever, sickness behavior and inflammation within the brain. Brain Behav Immun. 2018;68:211–223. doi:10.1016/j.bbi.2017.10.019
24. Xie F, Xu L, Zhu H, et al. The potential antipyretic mechanism of ellagic acid with brain metabolomics using rats with yeast-induced fever. Molecules. 2022;27(8):2465. doi:10.3390/molecules27082465
25. Pisay M, Bhaskar KV, Mehta CH, et al. Drug-carrier miscibility in solid dispersions of glibenclamide and a novel approach to enhance its solubility using an effervescent agent. AAPS PharmSciTech. 2022;23(8):284. doi:10.1208/s12249-022-02437-z
26. Darweesh RS, Al-Qawasmi FS, Khanfar MS. Ezetimibe oral solid lipid nanoparticle by effervescent dispersion method: in vitro characterization and in vivo pharmacokinetic study in rats. Pharm Dev Technol. 2025;30(3):268–279. doi:10.1080/10837450.2025.2471461
27. Yen -C-C, Liang Y-K, Cheng C-P, et al. Oral bioavailability enhancement and anti-fatigue assessment of the andrographolide loaded solid dispersion. Int J Mol Sci. 2020;21(7):2506. doi:10.3390/ijms21072506
28. Zhou W, Li B, Min R, et al. Mucus-penetrating dendritic mesoporous silica nanoparticle loading drug nanocrystal clusters to enhance permeation and intestinal absorption. Biomater Sci. 2023;11(3):1013–1030. doi:10.1039/d2bm01404a
29. Abu-Dief AM, Alsehli M, Al-Enizi A, et al. Recent advances in mesoporous silica nanoparticles for targeted drug delivery applications. Curr Drug Deliv. 2022;19(4):436–450. doi:10.2174/1567201818666210708123007
30. Yen CC, Chen YC, Wu MT, et al. Nanoemulsion as a strategy for improving the oral bioavailability and anti-inflammatory activity of andrographolide. Int J Nanomedicine. 2018;13:669–680. doi:10.2147/IJN.S154824
31. Sermkaew N, Ketjinda W, Boonme P, et al. Liquid and solid self-microemulsifying drug delivery systems for improving the oral bioavailability of andrographolide from a crude extract of Andrographis paniculata. Eur J Pharm Sci. 2013;50:459–466. doi:10.1016/j.ejps.2013.08.006
32. Zhang H, Shao X, Dehez F, et al. Modulation of membrane permeability by carbon dioxide. J Comput Chem. 2020;41:421–426. doi:10.1002/jcc.26063
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.