Back to Journals » International Journal of Nanomedicine » Volume 12
A bone-resorption surface-targeting nanoparticle to deliver anti-miR214 for osteoporosis therapy
Authors Cai M, Yang L, Zhang S, Liu J, Sun Y, Wang X
Received 16 April 2017
Accepted for publication 25 July 2017
Published 13 October 2017 Volume 2017:12 Pages 7469—7482
DOI https://doi.org/10.2147/IJN.S139775
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Mingxiang Cai,1,* Li Yang,2,* Shufan Zhang,1 Jiafan Liu,2 Yao Sun,1 Xiaogang Wang2
1Engineering Research Center of Tooth Restoration and Regeneration, Department of Oral Implantology, School of Stomatology, Tongji University, Shanghai, 2Department of Cell Biology, Institute of Biomedicine, College of Life Science and Technology, Jinan University, Guangzhou, China
*These authors contributed equally to this work
Abstract: With increasing fracture risks due to fragility, osteoporosis is a global health problem threatening postmenopausal women. In these patients, osteoclasts play leading roles in bone loss and fracture. How to inhibit osteoclast activity is the key issue for osteoporosis treatment. In recent years, miRNA-based gene therapy through gene regulation has been considered a potential therapeutic method. However, in light of the side effects, the use of therapeutic miRNAs in osteoporosis treatment is still limited by the lack of tissue/cell-specific delivery systems. Here, we developed polyurethane (PU) nanomicelles modified by the acidic peptide Asp8. Our data showed that without overt toxicity or eliciting an immune response, this delivery system encapsulated and selectively deliver miRNAs to OSCAR+ osteoclasts at bone-resorption surface in vivo. With the Asp8-PU delivery system, anti-miR214 was delivered to osteoclasts, and bone microarchitecture and bone mass were improved in ovariectomized osteoporosis mice. Therefore, Asp8-PU could be a useful bone-resorption surface-targeting delivery system for treatment of osteoclast-induced bone diseases and aging-related osteoporosis.
Keywords: osteoporosis, microRNA, bone resorption, targeting delivery, nanoparticle
Introduction
Due to increased numbers of aged people globally, osteoporosis (OP) has become a worldwide health issue.1,2 OP is a systemic bone metabolism disorder with increased bone loss and fracture risk.3,4 OP occurs when the equilibrium of bone resorption and bone formation is disturbed.5 As such, there are two main strategies in OP therapy: to enhance new bone formation and inhibit bone resorption.6 In aged bone, the number and activities of osteoblasts decreases, and the activity of osteoclasts is much higher than osteoblasts.7 Therefore, to correct the imbalance in bone remodeling, inhibiting the formation and activity of osteoclasts should be a preferred method.8 To enhance bone-formation ability, drugs are delivered to the bone-formation area or osteoblasts.9–11 To inhibit the function of osteoclasts efficiently, the delivery system should preferentially deliver drug to the bone-resorption area or osteoclasts.12,13 Common medications used for osteoclast targeting OP treatments are bisphosphonates or modified bisphosphonates.14,15 However, these have severe side effects, such as heart attacks and osteonecrosis of jaw bone.16 These limitations have provided an incentive to search for efficient and safe therapy methods.
Gene therapy is a novel strategy that regulates gene expression to treat disease by delivering exogenous small nucleic acids, such as siRNA or miRNA.17,18 However, the clinical applications of gene therapy are seriously limited by nucleic acid degradation and lack of tissue- or cell-specific.19–21 Therefore, investigation of a bone-resorption surface/osteoclast-specific delivery system is urgent for future OP treatment. To generate a targeted delivery system, three factors should be considered: carrier, targeting director, and drugs.22 Several delivery strategies have been studied to deliver miRNAs, such as silica nanoparticles,23 ultrasmall magnetic nanoparticles,24 liposomes,25 and exosomes.26 Among various delivery systems, polyurethane (PU) is a polymer composed of organic units joined by carbamate (urethane) links. It has been confirmed that PU has excellent biocompatibility, low cytotoxicity, and good mechanical flexibility, which is good for drug encapsulation and increased stability.27–29 Peptides are a good choice for guiding specific targeting. Bone-resorption surfaces are characterized by highly crystallized hydroxyapatite.30 The peptide Asp8, which is highly negatively charged, has been reported as a bone-resorption area-targeting tool.31 In this study, Asp8 was used as the targeting part of the delivery system. Mounting evidence has shown that miRNA drugs are good candidates for OP treatment through regulation of the activity of functional cells. For instance, miR31-5p, a positive regulator of osteoclast formation and bone resorption through targeting rhodamine A, has been considered an effective therapeutic target for OP.32 Also, miR20a has been found to be overexpressed in osteoblasts derived from dexamethasone induction by downregulating RANKL expression and subsequently suppressing osteoclastogenesis and bone resorption.33
In this study, to regulate the gene expression of osteoclasts in OP, miR214 was chosen as an effective therapeutic miRNA strategy. miR214 has been considered an important metabolism-regulation miRNA, and plays a crucial role in gluconeogenesis34 and especially bone remodeling.35 In vivo, elevated miR214 levels are correlated with a lower degree of bone formation in aged patients.36 miR214 targets ATF4 directly to inhibit osteoblast activity, and plays a critical role in osteoclast differentiation by targeting the PTEN–PI3k–Akt pathway, which shows great potential as an OP-treatment target.37–39 Here we report the assembly and function of a new targeted delivery system for delivery of miR214 drugs to bone-resorption areas for OP-targeted therapy.
Materials and methods
Animals
Sixty C57 BL/6 mice (weight 15–25 g) were obtained from the laboratory animal center at Tongji University. All animal work was carried out following the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, 1985). All experimental protocols were approved by the animal care and use committee of Tongji University (TJ-2016-0432).
Synthesis of PU
PU was synthesized in two steps: preparation of hard PU segments, and polymerization of hard segments and soft segments. The hard segments were obtained through a reaction between L-lysine diisocyanate and N-methyldiethanolamine in acetone solution. The reaction system was stirred constantly at 60°C for 24 hours to form an isocyanate terminated prepolymer in a closed reaction vessel. The isocyanate-terminated PU prepolymer solutions were cooled to room temperature, and poly(ε-caprolactone) diol polyethylene glycol (PEG) and acetone were added to initiate the polymerization reaction. The reactions continued at 60°C with constant stirring for 72 hours in a closed reaction vessel. Then, ethanol was added and the reaction kept at 60°C for 48 hours. The reaction mixture was precipitated in petroleum to obtain pure PU, and the final products were dried in a vacuum oven at 25°C. PU micelles were prepared by high-speed centrifugation and dialysis.
Synthesis of Asp8-PU
D8-PEG–COOH (1 mg) was dissolved in deionized water and reacted with 1 mg 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride and 1 mg N-hydroxysuccinimide at 4°C for 10 minutes. PU micelles were subsequently added to D8 peptide solutions and the mixture reacted overnight at 4°C. Then, the reaction solution was purified by dialysis (molecular weight cutoff 3,500 Da) three times, washing with PBS (500 μL containing 10 μL of 0.5 M EDTA solution, pH 7.4). Pure Asp8-PU was freeze-dried and stored at −20°C.40
Peptide-binding assays
Peptide-binding assays were performed in accordance with a previous report.41 Conjugated Cy3 was incubated with cells at a final concentration of 20 μg/mL for 6 hours at 37°C. Then, unbound peptides were removed from the well by washing and the nuclei were stained with 4′,6-diamidino-2-phenylindole or dihydrochloride (DAPI). The binding ability of peptides was analyzed using microscopy and flow cytometry.
Preparation of miRNA formulations
The preparation of different miRNA formulations was performed in accordance with a previous study.42 PU or Asp8-PU was diluted with double-distilled H2O and sonicated for 10 minutes. Then, miRNA solution was added to PU or Asp8-PU solution and immediately vortexed at 4°C. The prepared miRNA formulations were placed on vortexed movement before using in all experiments.
Characterization of Asp8-PU
Average size and ζ-potential of Asp8-PU were detected by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).43 The morphology of Asp8-PU was assessed with transmission electron microscopy (Tecnai 10; Philips, Amsterdam, the Netherlands).44 For serum-stability assays, free miRNA or Asp8-PU-miRNA (N:P 15:1 wt%) complexes were incubated with 50% fetal bovine serum (FBS) for up to 24 hours. Based on incubation time, aliquots from each sample were electrophoresed on a 2% agarose gel in tris-acetic acid-EDTA (TAE) buffer at 130 V for 30 minutes. Afterward, RNA was analyzed on an ultraviolet illuminator. The loading capacity of Asp8-PU-miRNA was assessed by agarose-gel electrophoresis. Firstly, Asp8-PU and miRNA was mixed with different mass ratio. And Asp8-PU:miRNA (w:w) ratios ranging from 0:1 to 25:1 were prepared. Asp8-PU-miRNA complexes were mixed with 1 μL 6× DNA gel-loading buffer, and the mixture was loaded onto 2% agarose gel (0.5 mg/mL ethidium bromide). Electrophoresis was run with TAE buffer at 130 V for 30 minutes. At last, RNA was analyzed on an ultraviolet illuminator.
Cell culture
Bone-marrow cells were collected from femora and tibiae of ovariectomized (Ovx) mice and cultured in α-minimum essential medium containing 10% FBS and 1% penicillin and streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). All cells were maintained in 5% CO2 at 37°C.
Toxicity assays in vivo
Mice were injected with 200 μL saline, PU, and Asp8-PU via tail vein (PU and Asp8-PU were dissolved in 200 μL RNase/DNase-free saline at a dose of 10 mg/kg). After administration for 24 hours, hearts, livers, spleens, and kidneys were collected for hematoxylin-eosin-staining analysis. Biochemical parameters, ie, ALT, AST, creatine kinase isoenzyme (CK-MB), and blood urea nitrogen (BUN), were analyzed using a clinical chemistry analyzer (Cruinn Diagnostics, Dublin, Ireland). Levels of serum IL6, IL2, TNFα, and IFNγ were determined with enzyme-linked immunosorbent assay (Thermo Fisher Scientific).45
Tissue distribution of miRNA in vivo
Mice were divided into two groups (PU and Asp8-PU). Mice in the PU-miRNA group were injected with fluorescein amidite-labeled miRNA with PU via tail vein, while the Asp8-PU group received the fluorescein amidite-labeled miRNA delivered by Asp8-PU, both at a dose of 10 mg/kg. Twelve hours later, all the mice were killed and the major organs (long bones, kidneys, lungs, and liver) collected from each mouse for detection of the fluorescence signal using molecular imaging software (Bruker, Billerica, MA, USA). The concentration of Asp8-PU was 1 mg/mL, administered at 0.2 mL/mouse/injection.
Cell-selective delivery in vivo
Femora from the PU–anti-miR214 or Asp8-PU–anti-miR214 group were sectioned with a diamond sand saw and microgrinding system for 15 μm (Exakt, Norderstedt, Germany). Calcein staining was employed to label bone-formation areas. Femora were decalcified with 10% ethylene diamine tetraacetic acid (EDTA). After dehydration, femur tissue was embedded in paraffin, and 4 μm sections were prepared for staining. Cell-selective delivery efficiency was evaluated by histological analysis. Green fluorescence signals indicating Asp8-PU localizations were observed by florescence microscopy (Nikon, Tokyo, Japan). Anti-CTSK (ab19027) was used for labeling osteoclasts. Goat antimouse short heavy and light chain secondary antibody was used at 1:1,000 dilution (Abcam, Cambridge, UK). DAPI (5%; Sigma-Aldrich, St Louis, MO, USA) was used for localization of cell nuclei.
FACS isolation of osteoclasts from mice
Bone-marrow stromal cells were collected from the femora and tibiae of Ovx mice. PerCP-labeled mouse osteoclast associated receptor (OSCAR) antibodies (AAST0108101, 1:50; R&D Systems, Minneapolis, MN, USA) was used for fluorescence-activated cell sorting (FACS) to isolate osteoclasts. After being washed with PBS and 1% bovine albumin (BSA), cells were directly stained with the PerCP-labeled OSCAR antibodies. Then, both stained and unstained cell populations were washed three times for FACS analysis. Selected OSCAR+ and OSCAR− cells were used for real-time polymerase chain reaction (PCR) analysis.
Cell-specific knockdown efficiency of miR214 in vivo
A total of 36 3-month-old female C57BL/6 mice were ovariectomized and administered with saline, PU–anti-miR214 or Asp8-PU–anti-miR214 at 16 mg/mL at day 2 after ovariectomy. The mice in each group were killed at 24 and 48 hours after administration (n=6 per time point in each treatment group). Bone-marrow cells were isolated for sorting OSCAR+ and OSCAR− by FACS. miR214 expression and mRNA expression of TRAP and CTSK in the OSCAR+ and OSCAR− cells were measured by real-time quantitative PCR. Furthermore, another 12 3-month-old Ovx mice were intravenously pretreated with Asp8 or PBS (n=6 for each treatment) at day 1 after ovariectomy. Then, half of the mice were administered with PU–anti-miR214 and the other half treated with Asp8-PU–anti-miR214 16 mg/mL 24 hours after pretreatment. Then, mice in each group were killed 24 and 48 hours later (n=6 for per time point in each treatment group). Bone-marrow cells were isolated for sorting OSCAR+ and OSCAR− cells by FACS. miR214 expression and mRNA expression of TRAP and CTSK in the OSCAR+ and OSCAR− cells were measured by real-time PCR.
Dose–response pattern and persistence of miR214 knockdown in vivo
To determine the dose–response pattern during in vivo administration, 120 3-month-old female C57BL/6 mice were ovariectomized, divided into three groups, and subjected to saline, PU–anti-miR214, or Asp8-PU–anti-miR214 at 2, 4, 8, 16, or 32 mg/kg (n=6 for each dose) via tail-vein injection at day 2 after ovariectomy: 2 days after administration, all mice were killed, and bone-marrow cells were collected from bilateral femora. FACS using OSCAR antibody sorted osteoclasts from bone-marrow cells. miR214 expression in OSCAR+ cells was quantified by real-time PCR analysis. To determine the knockdown persistence of Asp8-PU–anti-miR214 in vivo, 120 3-month-old female C57BL/6 mice were ovariectomized, divided into two groups, and injected with PU–anti-miR214 or Asp8-PU–anti-miR214 16 mg/mL (n=6 for each group). At 0, 2, 4, 6, 8, 10, 12, 14, and 16 days after treatment, the mice were killed (n=6 for each time point), and bone-marrow cells were collected from bilateral femora. Osteoclasts were sorted from bone-marrow cells by FACS using OSCAR antibodies. miR214 expression in OSCAR+ cells at each time point was quantified by real-time PCR analysis. Osteoclast-knockdown efficiency of 2 weeks’ Asp8-PU–anti-miR214 treatment was tested by TRAP staining with undecalcified femur sections.
Therapeutic evaluation of anti-miR214 delivered by Asp8-PU to Ovx mice
For anti-OP analysis, Ovx mice were divided into three groups. The Ovx control group was untreated, the Ovx + antiscramble group was treated with 200 μL Asp8-PU–antiscramble and the Ovx + anti-miR214 group was treated with 200 μL Asp8-PU–anti-miR214, with a miRNA dose of 16 mg/kg at an interval of 1 week. After drug administration for 1 month, all mice were killed. miR214 levels in osteoclasts sorted from all mice were detected by real-time PCR. Proximal tibiae were collected to show three-dimensional trabecular architecture by microcomputed tomography (micro-CT). Bone-mineral density of all mice was calculated by micro-CT.
Micro-CT analysis
For distal femora, the whole secondary spongiosa of the left distal femur from each mouse was scanned ex vivo using a micro-CT system (μCT50; Scanco Medical, Brüttisellen, Switzerland). Briefly, 100 slices with a voxel size of 10 μm were scanned in the region of the distal femur, beginning at the growth plate and extending proximally along the femur diaphysis. Eighty continuous slices beginning at 0.1 mm from the most proximal aspect of the growth plate in which both condyles were no longer visible were selected for analysis. All trabecular bone from each selected slice was segmented for three-dimensional reconstruction (σ=1.2, supports two, threshold 180 hounsfield unit [HU]) to calculate bone-mineral density (BMD).46
Statistical analyses
All numerical data are expressed as means ± SE. Significant differences among groups were analyzed by one-way analysis of variance with a post hoc test to determine group differences in the study parameters. All statistical analyses were performed with SPSS version 16.0. P<0.05 was considered statistically significant.
Results
Characterization of Asp8-PU
In brief, a series of monomers were synthesized and PU was prepared via a three-step polymerization reaction (Figure 1A). With transmission electron microscopy, we observed that most nanoparticles were uniform in size and morphology (Figure 1B). Asp8-PU nanoparticles were ~80 nm, with narrow size distribution and average ζ-potential 13.6 mV (Figure 1C). The nucleic acid-binding capacity of Asp8-PU nanomicelles was assessed by gel electrophoresis, and data indicated that nucleic acid mobility was retarded by Asp8-PU nanomicelles at Asp8-PU:nucleic acid (w:w) weight ratios greater than 10:1 (Figure 1D). After incubation with serum for 6 hours, electrophoretic assays indicated that a band representing free nucleic acid was degraded, but the Asp8-PU–nucleic acid band was still visible after incubation with serum for 24 hours (Figure 1E).
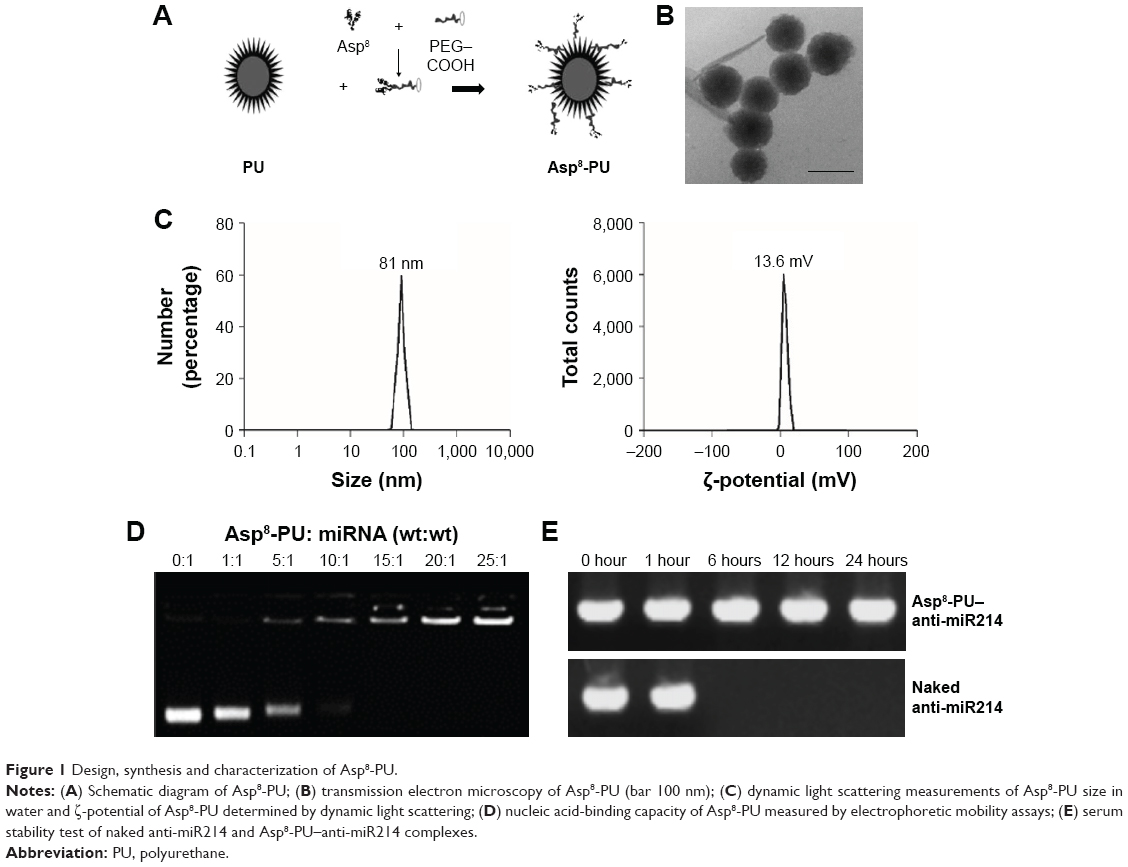 | Figure 1 Design, synthesis and characterization of Asp8-PU. |
Safety evolution of Asp8-PU in vivo
Hemagglutination assays confirmed that no agglutination occurred when erythrocytes were incubated with PU or Asp8-PU, indicating that both were compatible (Figure 2A). There were no statistically significant differences in CK-MB, ALT, or blood urea nitrogen in mice after Asp8-PU injection (Figure 2B). Toxicity assays in vivo demonstrated that both Asp8-PU and PU had no significant side effects on hearts, livers, spleens, or kidneys (Figure 2C). Moreover, administration of Asp8-PU or PU did not elicit inflammatory response, as reflected in unaltered levels of inflammatory cytokines IL2, IL6, TNFα, and IFNγ (Figure 2D).
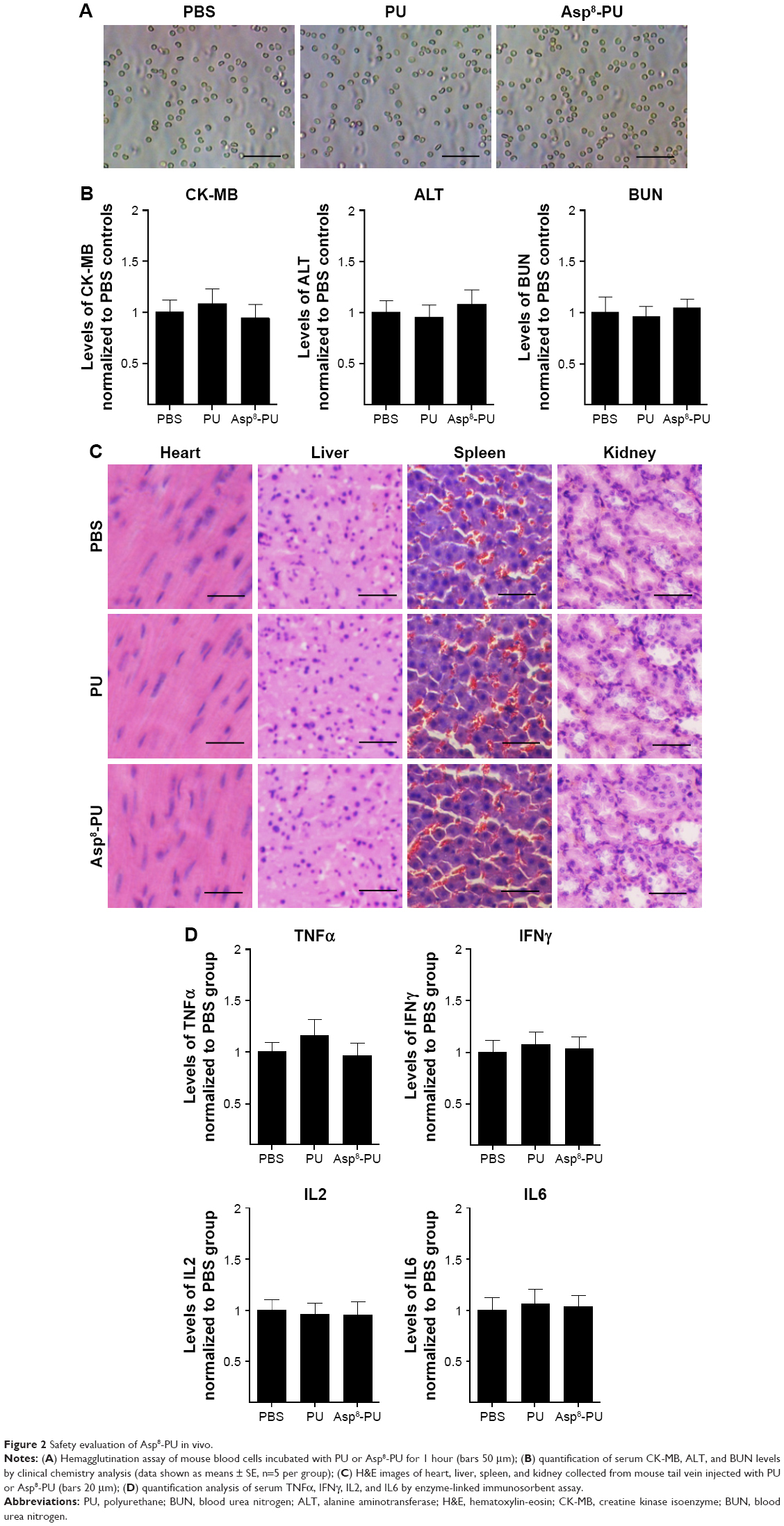 | Figure 2 Safety evaluation of Asp8-PU in vivo. |
Tissue-selective distribution and cell-selective delivery of miRNA by Asp8-PU in vivo
To detect the bioavailability of anti-miR214 with or without Asp8-PU, fluorescence imaging was used to show the distribution of PU and Asp8-PU to organs (Figure 3A). Fluorescence intensity of bone in Asp8-PU group was higher than that in the PU group, while less miRNA was delivered to lungs, liver, and kidneys. To investigate the local distribution of Asp8-PU delivery, localization of miRNA delivered by PU or Asp8-PU was assessed. Bone-formation surfaces (labeled with calcein, a green fluorescent calcium-binding dye that labels new bone deposition at bone-formation surfaces) were barely colabeled with miRNA delivered by Asp8-PU (red fluorescence, Figure 3B) in vivo. We next determined whether Asp8-PU delivered miRNA to bone-resorption surfaces/osteoclasts in vivo by analyzing colocalization of Cy3-miR 214 with CTSK (an osteoclast marker). Numerous instances of colocalization of Cy3-miR 214 with CTSK-positive cells of mice treated with Asp8-PU were observed, whereas few instances of such overlapping staining were observed in the PU groups (Figure 3C).
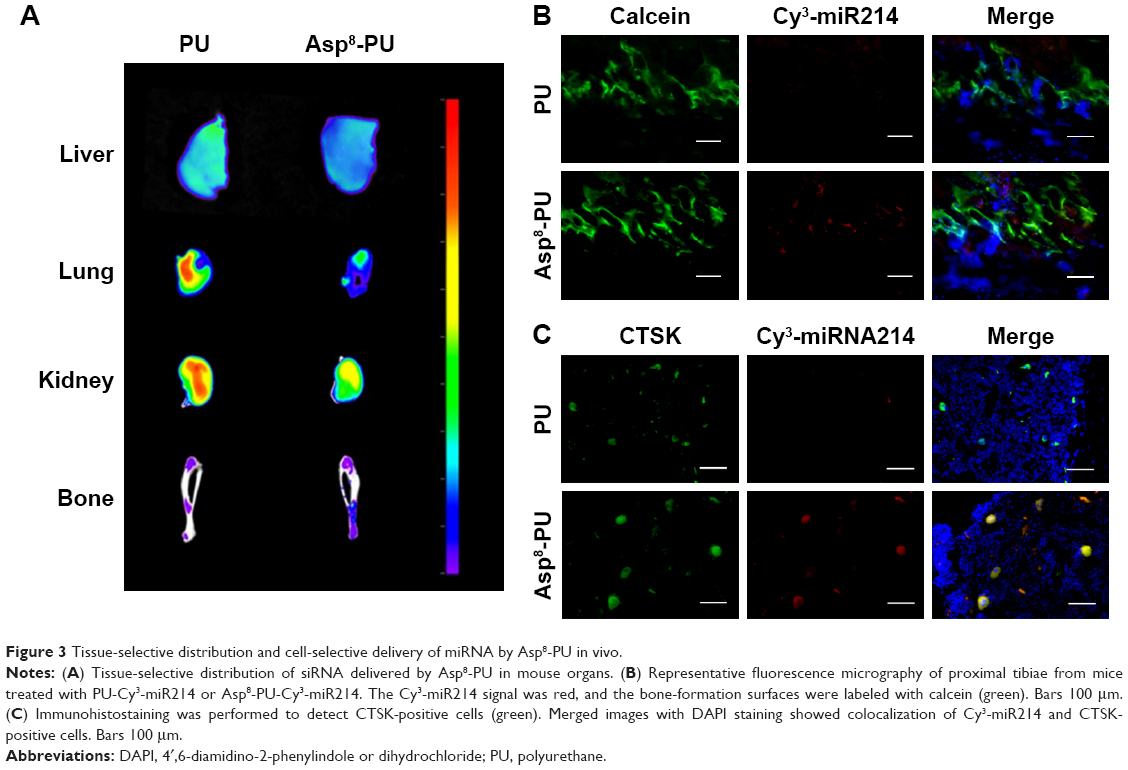 | Figure 3 Tissue-selective distribution and cell-selective delivery of miRNA by Asp8-PU in vivo. |
Dose–response pattern and persistence of miR214 knockdown in vivo
FACS determined the dose–response pattern and persistence of miR214 knockdown in OSCAR+ cells by PU–anti-miR214 or Asp8-PU–anti-miR214 in combination with real-time quantitative PCR analysis. The knockdown efficiency of miR214 increased in a dose-dependent manner at 2–24 mg/kg, and almost 70% knockdown efficiency was achieved 16 mg/kg in the Asp8-PU–anti-miR214 group. However, no obvious miR214 knockdown (over 50%) was achieved at the same dose in the PU–anti-miR214 group (Figure 4A). After a single injection of Asp8-PU–anti-miR214 at a dose of 16 mg/kg, almost 80% miR214 knockdown was found at 48 hours, and over 50% miR214-knockdown efficiency was maintained for 6 days. However, no obvious miR214 knockdown (over 50%) was observed at any time point after the same dose in the PU–anti-miR214 group (Figure 4B). In addition, by TRAP staining, fewer osteoclasts labeled TRAP-positive were observed with Asp8-PU–anti-miR214 treatment group than PU–anti-miR214 (Figure 4C and D).
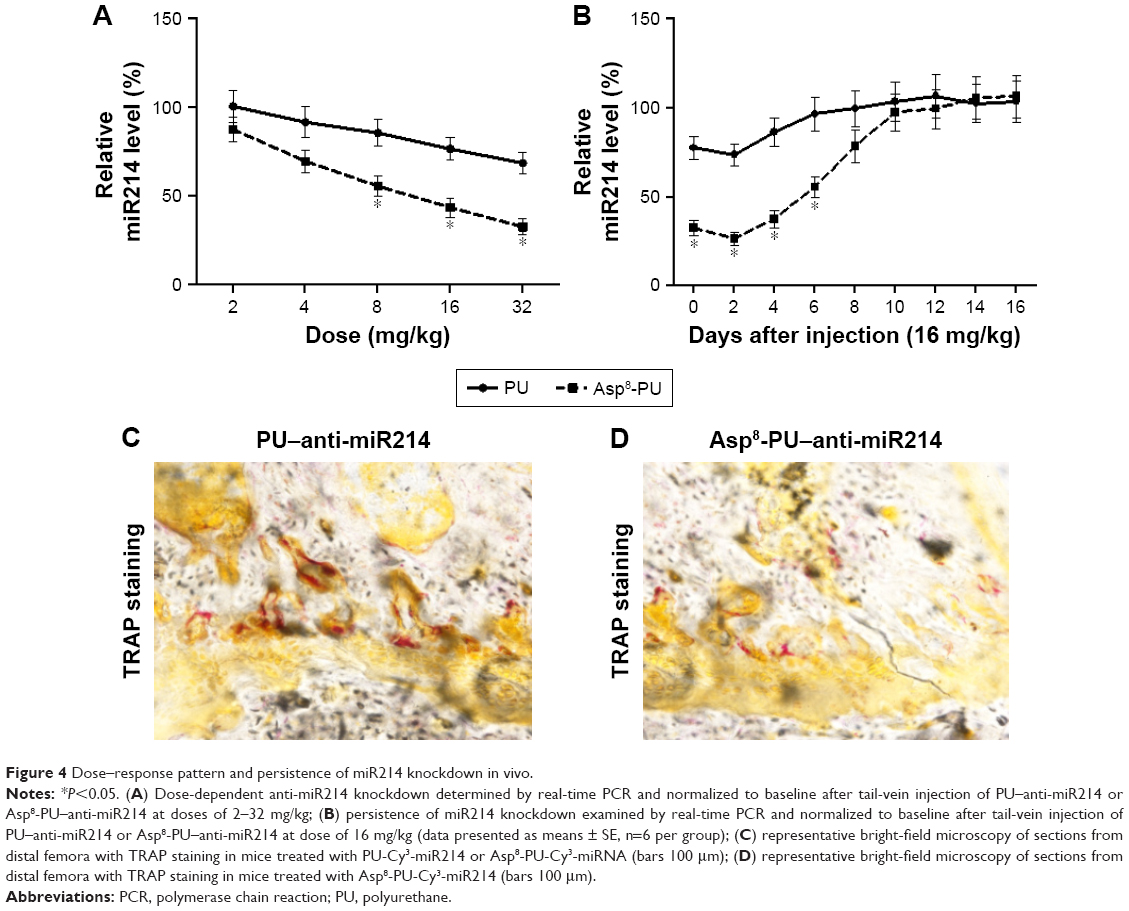 | Figure 4 Dose–response pattern and persistence of miR214 knockdown in vivo. |
Cell-specific knockdown efficiency of miR214 in vivo
FACS and real-time PCR analysis were performed to examine the knockdown efficiency of miR214 in bone-marrow-derived OSCAR+ cells from the 3-month-old Ovx mice treated with PU–anti-miR214 or Asp8-PU–anti-miR214, respectively. Data showed that miR214-knockdown efficiency in OSCAR+ cells was significantly higher than that in OSCAR− cells at both 24 and 48 hours after treatment with Asp8-PU–anti-miR214 (Figure 5A). Therefore, TRAP and CTSK expression were significantly downregulated in OSCAR+, while no distinct difference was found in OSCAR− cells after Asp8–anti-miR214 treatment (Figure 5B). To confirm further the delivery efficiency of Asp8-PU, both OSCAR+ and OSCAR− cells were pretreated with Asp8. No significant differences in knockdown efficiency of miR214 or TRAP- and CTSK-expression levels were found between OSCAR+ and OSCAR− cells after pretreatment with PU–anti-miR214 Asp8, respectively (Figure 5C and D).
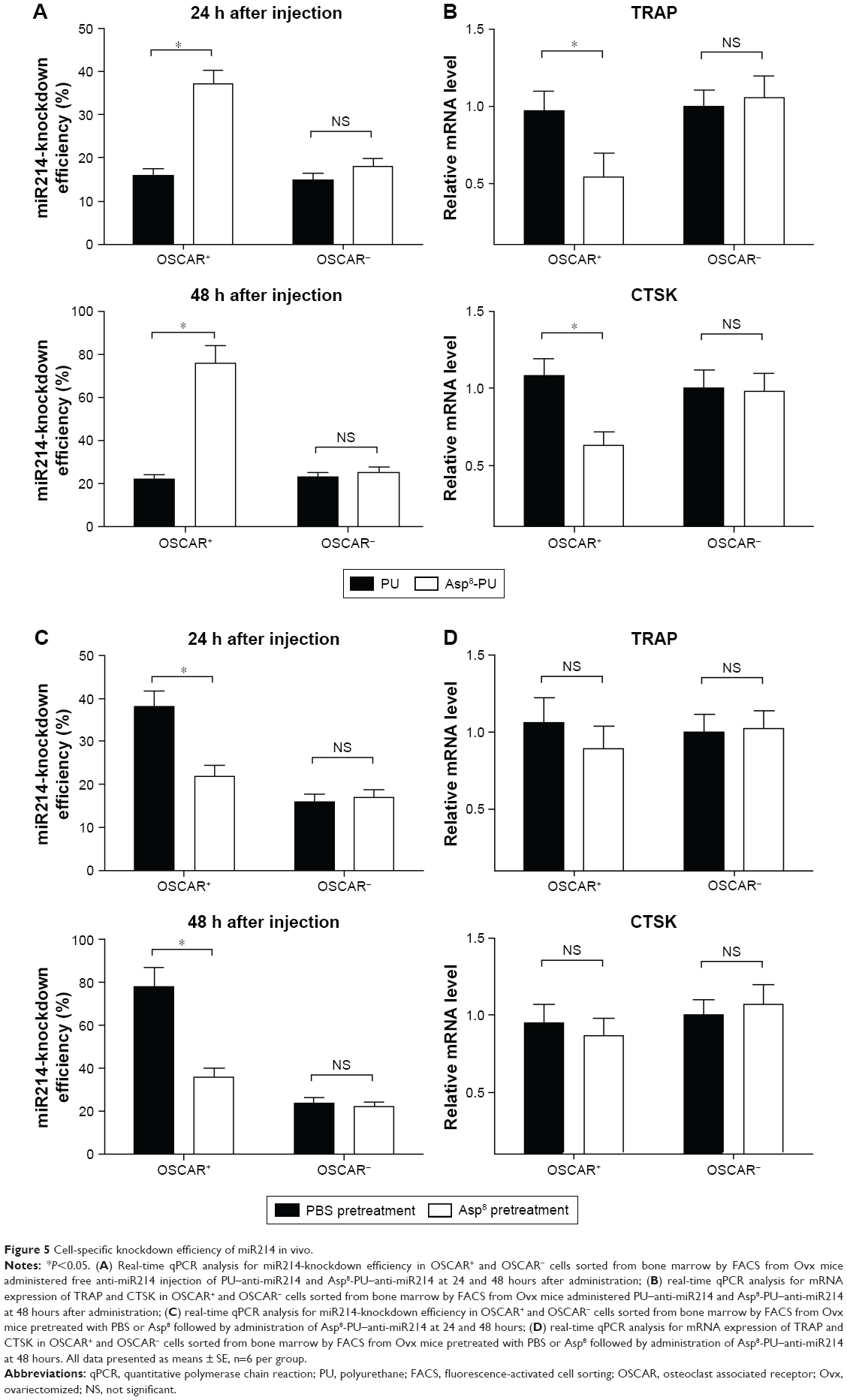 | Figure 5 Cell-specific knockdown efficiency of miR214 in vivo. |
Antiosteoporotic efficacy of Asp8-PU–anti-miR214
We studied the therapeutic effects of anti-miR214 in an Ovx mouse model of OP. First, we noted that in osteoclasts, miR214 efficiency decreased 80% after Asp8-PU–anti-miR214 treatment (Figure 6A). Bone microarchitecture and BMD were measured with micro-CT. Improved bone microarchitecture and greater bone mass were found in mice treated with Asp8-PU–anti-miR214 (Figure 6B). Micro-CT measurements showed increased trabecular thickness, trabecular number, and structure model index in distal femora and decreased trabecular spacing after Asp8-PU–anti-miR214 therapy (Figure 6C). In addition, micro-CT quantification data and histomorphometric analysis confirmed that the Ovx + Asp8-PU–anti-miR214 group had significantly increased BMD and bone volume:total volume ratio (Figure 6D). Moreover, osteoclast numbers were lower in the Ovx + Asp8-PU–anti-miR214 group than control groups (Figure 6E).
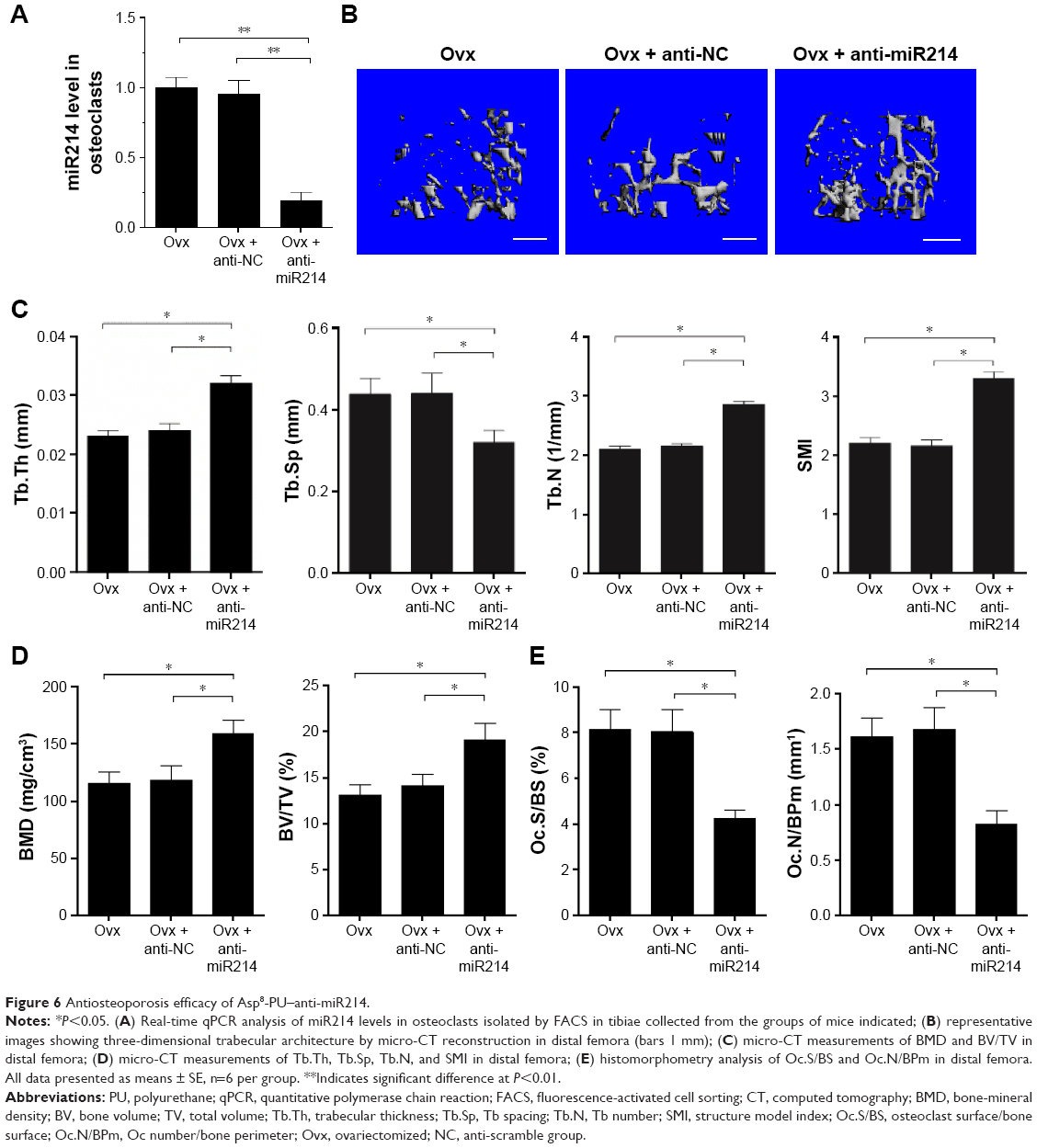 | Figure 6 Antiosteoporosis efficacy of Asp8-PU–anti-miR214. |
Discussion
For OP treatment in postmenopausal women, emphasis has always been on how to regulate osteoblasts to promote bone formation or inhibit bone loss caused by osteoclasts.47,48 Estrogen or parathyroid hormone is widely used in promoting bone formation. Estrogen-replacement therapy is used in the prevention of postmenopausal OP with significant risk of OP.49,50 However, estrogen has never been approved for the treatment of OP. Teriparatide (recombinant human parathyroid hormone 1–34) is used in the treatment of postmenopausal OP with a high risk of fracture.51 Teriparatide has a serious side-effect warning, because high doses of teriparatide can upregulate the occurrence of osteosarcomas in rats.52 On the other hand, osteoclasts could be set as an essential target for OP treatment. Agents that can inhibit OP include bisphosphonates and monoclonal antibodies against RANKL.53 Unfortunately, persistence with these drugs results in poor long-term OP prognosis.54 Bisphosphonates are the most widely used drugs for the treatment of OP. Efficacy and safety beyond 10 years have not yet been established. Denosumab (human monoclonal antibody against RANKL) is used in the treatment of postmenopausal women with high fracture risk, but infections, dermatitis, rashes, and eczema may occur. Therefore, in future, both effective drugs and efficient drug-delivery systems are needed for preventing OP before fractures occur.55,56
To facilitate OP treatment, a bone-resorption area-targeting delivery system could be a good tool. For targeted delivery, selecting a good molecule to guide the drug carrier toward bone-resorption areas is a key issue. As novel therapeutic reagents, peptides have numerous characteristics: high absorbability and water-solubility, imitation of local structural features of proteins, and versatile capabilities.57,58 To select a good delivery engine for bone, Asp-rich peptides show good potential. Asp is a negatively charge amino acid, and can bind to positive molecules, such as calcium in bone. Several Asp-rich peptides show bone-tissue affinity, such as Asp8, (Asp–Ser–Ser)6, and SDSSD (Ser-Asp-Ser-Ser-Asp).9 Peptides of six repeating sequence (Asp–Ser–Ser)6 have been found to bind favorably to lowly crystallized hydroxyapatite in vivo.28 The short peptide SDSSD could specifically bind to osteoblasts and deliver small-nucleus drugs to inhibit Ovx-induced OP. Peptides of eight repeating sequences of aspartate Asp8 have been reported to preferentially bind to highly crystallized hydroxyapatite, which is assumed to be an active bone-resorption area. Therefore, we employed Asp8 as the guide for delivering the miRNA drugs.
Employing a proper drug carrier is also a key issue in developing a bone-resorption-targeting delivery system: PU nanomicelles, which were able to encapsulate small nucleic acids via electrostatic interactions. Previous clinical results have confirmed that PU has high drug-loading efficiency, excellent biocompatibility, low cytotoxicity, and good mechanical flexibility.59,60 Serum-stability data suggested that PU protected miRNAs from degradation by endogenous enzymes, which confirmed it as a good carrier for small nucleic acid drugs. As such, Asp8-PU was developed as a bone-resorption-targeting system to deliver and release miRNAs. With this delivery system, Cy3-miR214 was successfully delivered to TRAP-positive bone-resorption surfaces. In contrast, Asp8 was used to pretreat cells and delivery efficiency was significantly downregulated by half. Moreover, cytotoxicity tests showed prominent biocompatibility of both the Asp8 peptide and PU carrier.
It is known that in bone-marrow monocytes, expression of miR214 is upregulated during osteoclastogenesis. In addition, overexpression of miR214 in bone-marrow monocytes promotes osteoclastogenesis. Here, we delivered anti-miR214 to bone-resorption areas to inhibit the formation of osteoclasts. Osteoclasts are derived from monocyte–macrophage precursors that arise from multipotent hematopoietic stem cells. Many inflammatory cytokines have been reported to be involved in osteoclastogenesis through transcription factors that positively or negatively modulate osteoclasts. such as IL2,61 IL6,62 TNFα,63 and IFNγ.64 Based on our quantification analysis by enzyme-linked immunosorbent assay, serum TNFα, IFNγ, IL2, and IL6 levels were not affected (Figure 2D). Therefore, it can be inferred that the downregulation of osteoclast activity was a result of the selective delivery of miR214 by Asp8-PU, rather than an inflammatory response. The data showed that targeted delivery of anti-miR214 inhibit osteoclast formation on bone-resorption surfaces. It is known that OSCAR+ cells gather and fuse to form osteoclasts in bone marrow. As such, OSCAR is always used as a marker of premature osteoclasts.65 In this study, we found that OSCAR+ cells significantly decreased in number compared to the control group after injection of anti-miR214 with Asp8-PU. In addition, osteoclast-related genes were also downregulated, such as TRAP and CTSK. Accordingly, the bone mass of Ovx mice increased markedly after Asp8-PU-miR214 treatment. As for clinical use of this delivery system, more experiments on large mammals still need to be employed to confirm the effects and to test the proper dosage and biosafety. Also, the molecular mechanism of how Asp8 binds to OSCAR+ cells and the method of clinical conservation of Asp8-PU still need further investigation. In sum, we believe that Asp-8-PU could be a potential tool for specific delivery of gene drugs to bone-resorption areas to inhibit bone resorption and reverse estrogen-deficiency-related OP.
Acknowledgments
This study was supported by the Key Project of Chinese National Programs for Research and Development (2016YFC1102705), National Science Technology Support Plan Projects (2014BAI04B07), National Natural Science Foundation Projects (81370971, 81722031, 81770873 to WXG, 81470715, 81771043 to SY), Shanghai Health System Academic Leader Program (2017BR009); Guangdong Natural Science Funds (2014A030313358, 2015A030313333), Major Science Project in Guangdong Province (2014KZDXM011), Science and Technology Planning Project of Guangdong Province (2013B090500105, 2014A020210015), Guangdong Natural Science Funds for Distinguished Young Scholars (S2013050013880), and the 2012 Recruitment Program of Global Experts 1000 Plan (YS).
Disclosure
The authors report no conflicts of interest in this work.
References
Lippuner K. [Osteoporosis: a global challenge?]. Ther Umsch. 2012;69(3):135–136. German. | ||
Melton LJ 3rd, Johnell O, Lau E, Mautalen CA, Seeman E. Osteoporosis and the global competition for health care resources. J Bone Miner Res. 2004;19(7):1055–1058. | ||
Nik J, Lai PS, Ng CJ, Emmerton L. A qualitative study of community pharmacists’ opinions on the provision of osteoporosis disease state management services in Malaysia. BMC Health Serv Res. 2016;16:448. | ||
Høiberg MP, Rubin KH, Hermann AP, Brixen K, Abrahamsen B. Diagnostic devices for osteoporosis in the general population: a systematic review. Bone. 2016;92:58–69. | ||
Sipos W, Pietschmann P, Rauner M. Strategies for novel therapeutic approaches targeting cytokines and signaling pathways of osteoclasto- and osteoblastogenesis in the fight against immune-mediated bone and joint diseases. Curr Med Chem. 2008;15(2):127–136. | ||
Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289(5484):1508–1514. | ||
Yu B, Wang CY. Osteoporosis: the result of an ‘aged’ bone microenvironment. Trends Mol Med. 2016;22(8):641–644. | ||
Yang YM, Li P, Cui DC, et al. Effect of aged bone marrow microenvironment on mesenchymal stem cell migration. Age (Dordr). 2015;37(2):16. | ||
Sun Y, Ye X, Cai M, et al. Osteoblast-targeting-peptide modified nanoparticle for siRNA/microRNA delivery. ACS Nano. 2016;10(6):5759–5768. | ||
Guo HH, Yu CC, Sun SX, et al. Adenovirus-mediated siRNA targeting TNF-α and overexpression of bone morphogenetic protein-2 promotes early osteoblast differentiation on a cell model of Ti particle-induced inflammatory response in vitro. Braz J Med Biol Res. 2013;46(10):831–838. | ||
Katsuyama T, Otsuka F, Terasaka T, et al. Regulatory effects of fibroblast growth factor-8 and tumor necrosis factor-α on osteoblast marker expression induced by bone morphogenetic protein-2. Peptides. 2015;73:88–94. | ||
Newa M, Lam M, Bhandari KH, Xu B, Doschak MR. Expression, characterization, and evaluation of a RANK-binding single chain fraction variable: an osteoclast targeting drug delivery strategy. Mol Pharm. 2014;11(1):81–89. | ||
Liu J, Dang L, Li D, et al. A delivery system specifically approaching bone resorption surfaces to facilitate therapeutic modulation of microRNAs in osteoclasts. Biomaterials. 2015;52:148–160. | ||
Reid IR, Heap SW, King AR, Ibbertson HK. Two-year follow-up of biphosphonate (APD) treatment in steroid osteoporosis. Lancet. 1988;2(8620):1144. | ||
Khajuria DK, Vasireddi R, Trebbin M, Karasik D, Razdan R. Novel therapeutic intervention for osteoporosis prepared with strontium hydroxyapatite and zoledronic acid: in vitro and pharmacodynamic evaluation. Mater Sci Eng C Mater Biol Appl. 2017;71:698–708. | ||
Mozzati M, Arata V, Gallesio G. Tooth extraction in osteoporotic patients taking oral bisphosphonates. Osteoporos Int. 2013;24(5):1707–1712. | ||
Adair JE, Weitzman MD. Applying the speed-dating model and other approaches to foster future leaders for the American Society of Gene and Cell Therapy. Mol Ther. 2014;22(8):1397–1398. | ||
Chen H, Chen L, Sun L, Zhen H, Li X, Zhang Q. A small interfering RNA targeting the KLF6 splice variant, KLF6-SV1, as gene therapy for gastric cancer. Gastric Cancer. 2011;14(4):339–352. | ||
Hydbring P, Badalian-Very G. Clinical applications of microRNAs. F1000Res. 2013;2:136. | ||
Giza DE, Fuentes-Mattei E, Bullock MD, et al. Cellular and viral microRNAs in sepsis: mechanisms of action and clinical applications. Cell Death Differ. 2016;23(12):1906–1918. | ||
Yanokura M, Banno K, Iida M, et al. MicroRNAS in endometrial cancer: recent advances and potential clinical applications. EXCLI J. 2015;14:190–198. | ||
Cheng KW, Hsu SH. A facile method to prepare superparamagnetic iron oxide and hydrophobic drug-encapsulated biodegradable polyurethane nanoparticles. Int J Nanomedicine. 2017;12:1775–1789. | ||
Tivnan A, Orr WS, Gubala V, et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PLoS One. 2012;7(5):e38129. | ||
Yigit MV, Ghosh SK, Kumar M, et al. Context-dependent differences in miR-10b breast oncogenesis can be targeted for the prevention and arrest of lymph node metastasis. Oncogene. 2017;36(18):2628. | ||
Rai K, Takigawa N, Ito S, et al. Liposomal delivery of microRNA-7-expressing plasmid overcomes epidermal growth factor receptor tyrosine kinase inhibitor-resistance in lung cancer cells. Mol Cancer Ther. 2011;10(9):1720–1727. | ||
Ohno SI, Takanashi M, Sudo K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21(1):185–191. | ||
Yu S, Ding J, He C, Cao Y, Xu W, Chen X. Disulfide cross-linked polyurethane micelles as a reduction-triggered drug delivery system for cancer therapy. Adv Healthc Mater. 2014;3(5):752–760. | ||
Gencturk A, Kahraman E, Güngör S, Ozhan G, Ozsoy Y, Sarac AS. Polyurethane/hydroxypropyl cellulose electrospun nanofiber mats as potential transdermal drug delivery system: characterization studies and in vitro assays. Artif Cells Nanomed Biotechnol. 2017;45(3):655–664. | ||
Akduman C, Ozgüney I, Kumbasar EP. Preparation and characterization of naproxen-loaded electrospun thermoplastic polyurethane nanofibers as a drug delivery system. Mater Sci Eng C Mater Biol Appl. 2016;64:383–390. | ||
Wang D, Miller SC, Shlyakhtenko LS, et al. Osteotropic peptide that differentiates functional domains of the skeleton. Bioconjug Chem. 2007;18(5):1375–1378. | ||
Carinci F. Restoration of incisor area using one-piece implants: evaluation of crestal bone resorption. Dent Res J (Isfahan). 2012;9 Suppl 2:S151–S154. | ||
Mizoguchi F, Murakami Y, Saito T, Miyasaka N, Kohsaka H. miR-31 controls osteoclast formation and bone resorption by targeting RhoA. Arthritis Res Ther. 2013;15(5):R102. | ||
Ell B, Kang Y. MicroRNAs as regulators of bone homeostasis and bone metastasis. Bonekey Rep. 2014;3:549. | ||
Li K, Zhang J, Yu J, et al. MicroRNA-214 suppresses gluconeogenesis by targeting activating transcriptional factor 4. J Biol Chem. 2015;290(13):8185–8195. | ||
Wu Y, Li Z, Yang M, et al. MicroRNA-214 regulates smooth muscle cell differentiation from stem cells by targeting RNA-binding protein QKI. Oncotarget. 2017;8(12):19866–19878. | ||
Wang X, Guo B, Li Q, et al. miR-214 targets ATF4 to inhibit bone formation. Nat Med. 2013;19(1):93–100. | ||
Zhao C, Sun W, Zhang P, et al. miR-214 promotes osteoclastogenesis by targeting Pten/PI3k/Akt pathway. RNA Biol. 2015;12(3):343–353. | ||
Wang X, Chen J, Li F, et al. MiR-214 inhibits cell growth in hepatocellular carcinoma through suppression of β-catenin. Biochem Biophys Res Commun. 2012;428(4):525–531. | ||
Wang X, Shen E, Wang Y, et al. Cross talk between miR-214 and PTEN attenuates glomerular hypertrophy under diabetic conditions. Sci Rep. 2016;6:31506. | ||
Wu C, Gong F, Pang P, et al. An RGD-modified MRI-visible polymeric vector for targeted siRNA delivery to hepatocellular carcinoma in nude mice. PLoS One. 2013;8(6):e66416. | ||
Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–712. | ||
Choi KY, Silvestre OF, Huang X, et al. A nanoparticle formula for delivering siRNA or miRNAs to tumor cells in cell culture and in vivo. Nat Protoc. 2014;9(8):1900–1915. | ||
Ding M, Song N, He X, et al. Toward the next-generation nanomedicines: design of multifunctional multiblock polyurethanes for effective cancer treatment. ACS Nano. 2013;7(3):1918–1928. | ||
Khatri N, Baradia D, Vhora I, Rathi M, Misra A. cRGD grafted liposomes containing inorganic nano-precipitate complexed siRNA for intracellular delivery in cancer cells. J Control Release. 2014;182:45–57. | ||
Zhou HF, Yan H, Pan H, et al. Peptide-siRNA nanocomplexes targeting NF-κB subunit p65 suppress nascent experimental arthritis. J Clin Invest. 2014;124(10):4363–4374. | ||
Iwaniec UT, Wronski TJ, Liu J, et al. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22(3):394–402. | ||
Tanaka S. [Osteoporosis in rheumatoid arthritis: role of osteoclast differentiation and cytokines]. Clin Calcium. 2001;11(5):598–601. Japanese. | ||
Binder NB, Niederreiter B, Hoffmann O, et al. Estrogen-dependent and C-C chemokine receptor-2-dependent pathways determine osteoclast behavior in osteoporosis. Nat Med. 2009;15(4):417–424. | ||
Sato Y, Tando T, Morita M, et al. Selective estrogen receptor modulators and the vitamin D analogue eldecalcitol block bone loss in male osteoporosis. Biochem Biophys Res Commun. 2017;482(4):1430–1436. | ||
Gjoksi B, Ghayor C, Siegenthaler B, Ruangsawasdi N, Zenobi-Wong M, Weber FE. The epigenetically active small chemical N-methyl pyrrolidone (NMP) prevents estrogen depletion induced osteoporosis. Bone. 2015;78:114–121. | ||
Mazziotti G, Formenti AM, Adler RA, et al. Glucocorticoid-induced osteoporosis: pathophysiological role of GH/IGF-I and PTH/vitamin D axes, treatment options and guidelines. Endocrine. 2016;54(3):603–611. | ||
Barbehenn EK, Lurie P, Wolfe SM. Osteosarcoma risk in rats using PTH 1-34. Trends Endocrinol Metab. 2001;12(9):383. | ||
Heckt T, Keller J, Peters S, et al. Parathyroid hormone induces expression and proteolytic processing of Rankl in primary murine osteoblasts. Bone. 2016;92:85–93. | ||
Schwarz EM, Ritchlin CT. Clinical development of anti-RANKL therapy. Arthritis Res Ther. 2007;9 Suppl 1:S7. | ||
Aho J, Edinger M, Botker J, Baldursdottir S, Rantanen J. Oscillatory shear rheology in examining the drug-polymer interactions relevant in hot melt extrusion. J Pharm Sci. 2016;105(1):160–167. | ||
Khajuria DK, Disha C, Vasireddi R, Razdan R, Mahapatra DR. Risedronate/zinc-hydroxyapatite based nanomedicine for osteoporosis. Mater Sci Eng C Mater Biol Appl. 2016;63:78–87. | ||
Cao J, Wang R, Gao N, et al. A7RC peptide modified paclitaxel liposomes dually target breast cancer. Biomater Sci. 2015;3(12):1545–1554. | ||
Xiao Y, Reis LA, Feric N, et al. Diabetic wound regeneration using peptide-modified hydrogels to target re-epithelialization. Proc Natl Acad Sci U S A. 2016;113(40):E5792–E5801. | ||
Camacho KM, Menegatti S, Mitragotri S. Low-molecular-weight polymer-drug conjugates for synergistic anticancer activity of camptothecin and doxorubicin combinations. Nanomedicine (Lond). 2016;11(9):1139–1151. | ||
Sun W, Zhao C, Li Y, et al. Osteoclast-derived microRNA-containing exosomes selectively inhibit osteoblast activity. Cell Discov. 2016;2:16015. | ||
Wang Y, Grainger DW. RNA therapeutics targeting osteoclast-mediated excessive bone resorption. Adv Drug Deliv Rev. 2012;64(12):1341–1357. | ||
Suzuki M, Hashizume M, Yoshida H, Shiina M, Mihara M. Intercellular adhesion molecule-1 on synovial cells attenuated interleukin-6-induced inhibition of osteoclastogenesis induced by receptor activator for nuclear factor κB ligand. Clin Exp Immunol. 2011;163(1):88–95. | ||
Kitaura H, Kimura K, Ishida M, Kohara H, Yoshimatsu M, Takano-Yamamoto T. Immunological reaction in TNF-α-mediated osteoclast formation and bone resorption in vitro and in vivo. Clin Dev Immunol. 2013;2013:181849. | ||
Wang L, Liu S, Zhao Y, et al. Osteoblast-induced osteoclast apoptosis by Fas ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ. 2015;22(10):1654–1664. | ||
Kim JH, Kim K, Youn BU, Jin HM, Kim N. MHC class II transactivator negatively regulates RANKL-mediated osteoclast differentiation by downregulating NFATc1 and OSCAR. Cell Signal. 2010;22(9):1341–1349. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.