Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
(2R,6R)-Hydroxynorketamine Alleviates Electroconvulsive Shock-Induced Learning Impairment by Inhibiting Autophagy
Authors Zhong X, Ouyang C, Liang W, Dai C, Zhang W
Received 25 August 2020
Accepted for publication 11 January 2021
Published 3 February 2021 Volume 2021:17 Pages 297—304
DOI https://doi.org/10.2147/NDT.S278422
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yu-Ping Ning
Xiaomei Zhong,1 Cong Ouyang,2 Wanyuan Liang,2 Cunying Dai,2 Weiru Zhang2
1Department of Geriatric Psychiatry, The Affiliated Brain Hospital of Guangzhou Medical University, Guangzhou, Guangdong, 510370, People’s Republic of China; 2Institute of Neuroscience, The Affiliated Brain Hospital of Guangzhou Medical University, Guangzhou, Guangdong, 510370, People’s Republic of China
Correspondence: Xiaomei Zhong
Department of Geriatric Psychiatry, The Affiliated Brain Hospital of Guangzhou Medical University, Guangzhou, Guangdong, 510370, People’s Republic of China
Tel +8620-8126-8084
Fax +8620-8189-1391
Email [email protected]
Purpose: Learning impairment after electroconvulsive therapy (ECT) is common. Ketamine, an anesthetic used for ECT, has been demonstrated to attenuate cognitive impairment after ECT. However, the mechanism by which ketamine occurs in this case is still unknown. We aimed to explore the role of ketamine metabolite (2R,6R)-hydroxynorketamine [(2R,6R)-HNK] in the protection against learning impairment and investigate whether autophagy is involved in the protective effect.
Materials and Methods: A rat depression model received electroconvulsive shock (ECS; simulated ECT in animal models) daily for 3 days. The Morris water maze was used to assess the spatial learning function of the rats. Western blotting was used to detect the expression of Beclin-1, light chain (LC)3-II/LC3-I, p62, mammalian target of rapamycin (mTOR), and p-mTOR in the hippocampus.
Results: The escape latency for the maze in the ECS group was significantly longer than that in the sham ECS group (P=0.042). Meanwhile, the escape latency in the (2R,6R)-HNK+ECS group was significantly shorter than that in the ECS group (P=0.005). The LC3-II/LC3-I ratio and Beclin-1 expression level significantly increased, and the p62 expression level significantly decreased in the ECS group, compared with those in the sham ECS group (all P< 0.001). The (2R,6R)-HNK+ECS group showed lower LC3-II/LC3-I ratio (P< 0.001) and Beclin-1 expression level (P< 0.001) and higher p62 (P< 0.001) and p-mTOR expression levels (P=0.048) than did the ECS group. After small-molecule enhancer of rapamycin 28 (SMER28) administration, the role of (2R,6R)-HNK in protecting against learning impairment and inhibiting autophagy was abrogated, showing no difference in the escape latency; the difference in the LC3-II/LC3-I ratio and p62 expression level between the SMER28+(2R,6R)-HNK+ECS and ECS groups was not as significant as that between the (2R,6R)-HNK+ECS and ECS groups (P< 0.05– 0.01 vs P< 0.001).
Conclusion: (2R,6R)-HNK yields cognitive protection by suppressing autophagy through the mTOR signaling pathway in the ECS-treated rat hippocampus.
Keywords: electroconvulsive shock, learning impairment, (2R 6R)-hydroxynorketamine, autophagy
Introduction
Major depressive disorder (MDD) is a globally prevalent psychiatric disorder that develops into a severe socio-economic challenge. Over a third of patients with MDD do not have an adequate response to first-line antidepressant treatments; that is, they have treatment-resistant depression (TRD). These patients are at an increased risk of suicide and tend to have a more severe course of illness. Several effective pharmaco-therapeutic strategies that are currently available for patients with TRD include combining more than one antidepressant, switching to a different antidepressant, or augmenting an antidepressant with non-antidepressant medication. It is still unclear which of these treatment approaches should be applied to a given patient and in what order.1 Owing to this ambiguity, electroconvulsive therapy (ECT) is one of the preferred treatments for TRD. ECT is a standard psychiatric treatment for certain psychiatric illnesses and has been proven to be effective for TRD.2 However, this treatment may lead to learning impairment.2 Further, there are still critics postulating that ECT may be associated with neurotoxic injury.
Ketamine has emerged as a novel and rapid-acting antidepressant. Intravenous injection of low-dose ketamine rapidly reduces depressive symptoms and suicidal ideation in patients with affective disorders.3 There is an unmet clinical need for efficient drugs with a rapid onset of antidepressant effects. Esketamine has recently been licensed by the US Food and Drug Administration and European Drug Agency for use in TRD. Ketamine is a noteworthy anesthetic agent used mainly for the initiation and maintenance of anesthesia. Because of its anesthetic and antidepressant effects, ketamine has become progressively popular in ECT anesthesia.4–7
The common side effect after ECT is cognitive impairment. Patients experience disorientation and anterograde amnesia after each ECT treatment.8 The use of ECT is limited owing to its adverse effects on cognitive function. A previous study has shown that ketamine as an anesthetic for ECT exhibits cognitive protection for patients with MDD.7 However, the postulated molecular mechanism of ketamine for ECT against cognitive impairment remains unclear. Neurotoxic injury, such as excitotoxic damage related to excessive glutamatergic transmission through the N-methyl-D-aspartate receptor during ECT, may be one of the postulated molecular mechanisms for cognitive impairment. In addition, there is evidence that electroconvulsive seizures enhance autophagy signaling in the rat hippocampus.9 The excitotoxicity of glutamate can induce autophagy. Autophagy imbalance is involved in the pathophysiological process of many diseases. Once autophagy is overactivated, it will lead to programmed cell death.10 Studies have shown that the decline in cognitive function caused by traumatic damage is closely related to the activation of autophagy.11,12 There is evidence that ketamine can mitigate excitotoxic neuronal damage mediated by the effect of rapidly leading to increased synaptic signaling proteins,13 increasing the number and function of new spine synapses,13 and attenuating autophagy.11 The metabolism of ketamine to (2R,6R)-hydroxynorketamine [(2R,6R)-HNK] is essential for the antidepressant effects, which involve early and sustained α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor activation, as evidenced by an increase in GluA1 and GluA2 production in hippocampal synapses.14 There is evidence that mammalian target of rapamycin (mTOR) signaling increases in the presence of (2R,6R)-HNK.15 Autophagy is inhibited by mTOR activation.16 Recent findings support the potential use of (2R,6R)-HNK for the treatment of MDD.14,15
The side effects of cognitive impairment after ECT are related to neurotoxic injury and enhancement of autophagy signaling.9 Considering the effect of (2R,6R)-HNK on mTOR activation,15 we speculate that administration of (2R,6R)-HNK in ECT may yield potential cognitive protection by suppressing the excessive activation of autophagy. In this study, the learning ability of depressed rats after electroconvulsive shock (ECS; simulated ECT in animal models) was assessed, and the effects of (2R,6R)-HNK on the expression of autophagy markers and mTOR in the ECS-treated rat hippocampus were explored.
Materials and Methods
Animals
This study was approved by the Guangzhou Medical University Animal Committee (No. GY2020-103) and complied with the Chinese Guide for the Care and Use of Laboratory Animals (Laboratory Animal Guideline for the ethical review of animal welfare). Clean healthy adult male Sprague-Dawley (SD) rats (240–260 g; 2–3 months old) were used in this experiment. The SD rats were kept under standard laboratory conditions with ad libitum access to food and water for 1 week. The experiment was conducted in accordance with the standard ethical guidelines for the care and use of laboratory animals.
After the model of depression was established, 30 rats were randomly divided into five groups: sham ECS, ECS, (2R,6R)-HNK+ECS, autophagy enhancer small-molecule enhancer of rapamycin 28 (SMER28)+(2R,6R)-HNK+ECS, and autophagy inhibitor 3-methyladenine (3-MA)+(2R,6R)-HNK+ECS groups. A detailed flowchart is shown in Figure 1.
 |
Figure 1 Flowchart for the behavior test. Abbreviations: CUMS, chronic unpredicted mild stress; SPT, sucrose preference test; MWM, Morris water maze; ECS, electroconvulsive shock. |
Chronic Unpredictable Mild Stress (CUMS)
The CUMS method was used to establish a rat model of depression. The rats were subjected randomly to the following mild stressors: (1) feeding of foreign matter, (2) swimming in ice water at 4°C for 5 min, (3) cage tilting at 45° for 24 h, (4) wet caging for 24 h, (5) food deprivation for 24 h, (6) water deprivation for 24 h, and (7) tail pinching for 1 min. All rats were exposed randomly to two stressors every day for 21 days.
Sucrose Preference Test
The sucrose preference test was performed to assess depressive behavior symptoms in all rats before and after conducting the CUMS method. The percentage of sucrose solution consumed relative to the total liquid consumed during the test was calculated and used as a measure of sucrose preference.
Drugs
(2R,6R)-HNK (Sigma, USA) was dissolved in saline (final concentration: 10 mg/mL). SMER28 (MedChemExpress, USA) was dissolved in 10% DMSO (final concentration: 13.21 mg/mL). 3-MA (Sigma, USA) was dissolved in saline (final concentration: 100 nmol/mL). In the (2R,6R)-HNK+ECS group, 3-MA+(2R,6R)-HNK+ECS group, and SMER28+(2R,6R)-HNK+ECS group, (2R,6R)-HNK (1 mL/kg) and etomidate (5 mg/kg) were administered simultaneously via intraperitoneal injection, while 3-MA (5 µL) or SMER28 (20 µL) was administered via intraperitoneal injection 30 min before anesthesia.
ECS
ECS was performed after the CUMS method and the first Morris water maze. Electrical stimulation (55–60 mA, duration: 0.5 s, frequency: 120–125 Hz, amplitude: 0.8 A, and wave width: 1.5 ms)17,18 was performed to induce tonic-clonic convulsion using lightweight alligator clips attached to the ears wetted with saline. The rats in the sham ECS group received sham ECS (clips on the ears, no shock).
Morris Water Maze
The Morris water maze, a gold standard test used to study spatial memory and learning developed by Richard G. (1984), was used in this study. Six rats in each group were examined in this experiment. The Morris water maze equipment is a circular swimming pool (150 cm in diameter, with four quadrants). The escape latency experiment was performed after the ECS for four times once a day for 5 days. Each rat was placed into four different quadrants of the pool. The video analysis system automatically recorded the time it took the rats to find the hidden platform, which is regarded as the escape latency. When the rats failed to reach the platform within 60 s, they were guided to the platform and stayed there for 10 s. After 10 s of being kept on the platform, the rats were removed therein. The final score was the mean escape latency from day 1 to day 5 of the Morris water maze experiment.
Tissue Preparation and Western Blot Analysis
The hippocampal tissue was cut and sheared. Thereafter, it was added to the lysate containing RIPA, PMSF, and phosphatase inhibitor at 100 mg/mL. The resulting lysates were centrifuged at 12,000 rpm for 15 minutes at 4°C, and the supernatants were collected. Protein concentrations were measured using a BCA Protein Assay Kit (TaKaRa, China). Loading buffer containing mercaptoethanol was added to the sample, which was then boiled for 5 minutes. Equal amounts of proteins were loaded into wells, analyzed via electrophoresis on sodium dodecyl sulfate polyacrylamide gels, and transferred to polyvinylidene difluoride membranes (GE Healthcare, Germany). After blocking with nonfat milk, the membranes were further probed with specific primary antibodies against mTOR, p-mTOR, microtubule-associated protein light chain (LC)3-I, LC3-II, p62, Beclin-1, and β-actin (Abcam, Britain). The membrane was kept overnight at 4°C. After washing, the membranes were incubated with secondary antibodies (Beyotime, China) conjugated with horseradish peroxidase. Immunoreactions were visualized using an ECL detection system, and the intensities of the protein bands were analyzed.
Statistical Analysis
Statistical analysis was performed using the Statistical Package for Social Sciences (IBM SPSS) version 22.0 (Chicago, IL, USA). All data were expressed as means ± SEMs. Statistical significance was determined using one-way ANOVA, followed by the Bonferroni post hoc test, where appropriate. P-values of <0.05 were considered statistically significant.
Results
Morris Water Maze
There were no significant differences found in the escape latency and space exploration times among the groups before ECS treatment. One-way ANOVA revealed that there were group differences in the average escape latency (F=4.868, P=0.005) after ECS treatment. The Bonferroni post hoc analyses indicated that the ECS group showed a significantly longer escape latency than did the sham group (P=0.042), (2R,6R)-HNK+ECS group (P=0.005), and 3-MA+(2R,6R)-HNK+ECS group (P=0.031) (Figure 2). No difference in the escape latency was observed between the (2R,6R)-HNK+ECS, 3-MA+(2R,6R)-HNK+ECS, and SMER28+(2R,6R)-HNK+ECS groups and the sham group (Figure 2).
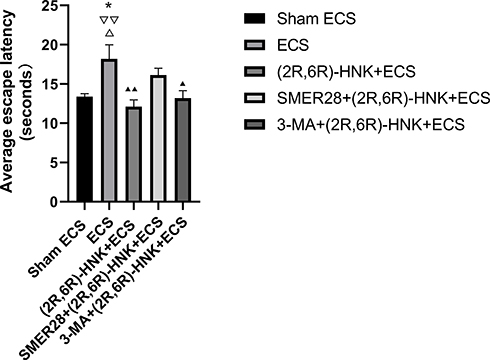 |
Figure 2 Effects of ECS and (2R,6R)-HNK on the learning of animals based on the Morris water maze results. This figure presents the results of the Morris water maze experiment after ECS treatment. A longer escape latency indicates worse learning performance. The final score was the mean escape latency from day 1 to day 5 of the Morris water maze experiment. *P<0.05 compared with the sham ECS group. ▲P<0.05, ▲▲P<0.01, compared with the ECS group. ▽▽P<0.01 compared with the (2R,6R)-HNK+ECS group. ΔP<0.05 compared with the 3-MA+(2R,6R)-HNK+ECS group. Data were analyzed using one-way analysis of variance, with the Bonferroni post hoc test. Bars represent the means ± SEMs; n=6 per group. Abbreviations: ECS, electroconvulsive shock; (2R,6R)-HNK, (2R,6R)-hydroxynorketamine; 3-MA, 3-methyladenine; SMER28, small-molecule enhancer of rapamycin 28. |
Western Blotting
One-way ANOVA revealed that there were group differences in the Beclin-1 expression level (F=73.369, P<0.001), p62 expression level (F=145.925, P<0.001), and LC3-II/LC3-I ratio (F=43.485, P<0.001) (Figure 3A–D).
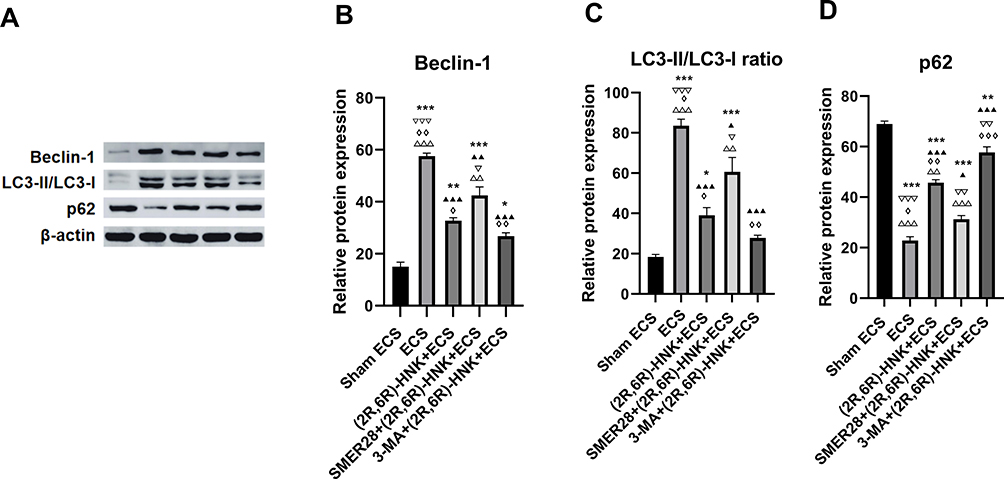 |
Figure 3 Western blot analysis of Beclin-1, LC3-II/LC3-I ratio and p62 expression in the hippocampus. (A) Western blot analysis; (B) expression of Beclin-1; (C) ratio of LC3-II/LC3-I; (D) expression of p62. *P<0.05, **P<0.01, ***P<0.001 compared with the sham ECS group. ▲P<0.05, ▲▲P<0.01, ▲▲▲P<0.001 compared with the ECS group. ▽P<0.05, ▽▽P<0.01, ▽▽▽P<0.001 compared with the (2R,6R)-HNK+ECS group. ◊P<0.05, ◊◊P<0.01, ◊◊◊P<0.001 compared with the SMER28+(2R,6R)-HNK+ECS group. ΔΔP<0.01, ΔΔΔP<0.001 compared with the 3-MA+(2R,6R)-HNK+ECS group. Data were analyzed using one-way analysis of variance, with the Bonferroni post hoc test. Bars represent the means ± SEMs; n=3 per group. Abbreviations: ECS, electroconvulsive shock; (2R,6R)-HNK, (2R,6R)-hydroxynorketamine; 3-MA, 3-methyladenine; SMER28, small-molecule enhancer of rapamycin 28; LC, light chain. |
No group differences were found in the mTOR expression level (F=0.113, P=0.975) (Figure 4A and B). One-way ANOVA revealed that there was a group difference in the p-mTOR expression level (F=9.976, P=0.002) among the five groups (Figure 4A and C).
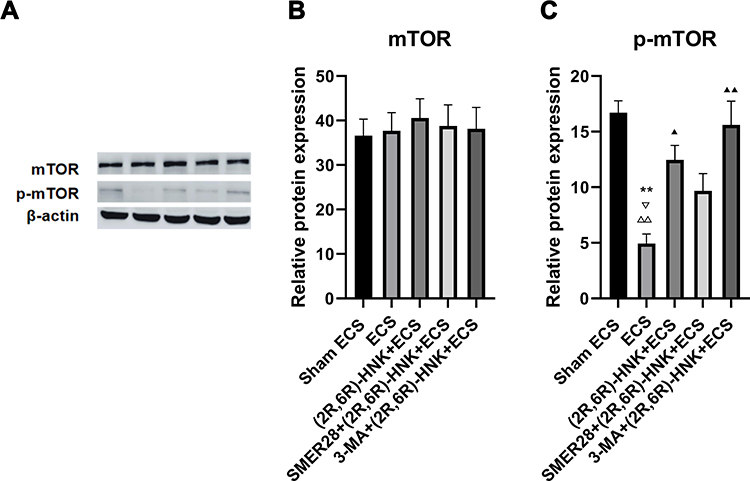 |
Figure 4 Western blot analysis of mTOR and p-mTOR expression in the hippocampus. (A) Western blot analysis; (B) expression of mTOR; (C) expression of p-mTOR. **P<0.01 compared with the sham ECS group. ▲P<0.05, ▲▲P<0.01, compared with the ECS group. ▽P<0.05 compared with the (2R,6R)-HNK+ECS group. ΔΔP<0.01 compared with the 3-MA+(2R,6R)-HNK+ECS group. Data were analyzed using one-way analysis of variance, with the Bonferroni post hoc test. Bars represent the means ± SEMs; n=3 per group. Abbreviations: ECS, electroconvulsive shock; (2R,6R)-HNK, (2R,6R)-hydroxynorketamine; 3-MA, 3-methyladenine; SMER28, small-molecule enhancer of rapamycin 28; mTOR, mammalian target of rapamycin. |
Role of ECS in Inducing Autophagy
The analysis of the critical proteins in the autophagy pathway showed that the p62 expression level significantly decreased (P<0.001), and the LC3-II/LC3-I ratio (P<0.001) and Beclin-1 expression level (P<0.001) significantly increased in the ECS group when compared with those in the sham ECS group (Figure 3B–D).
Role of (2R,6R)-HNK in ECS-Induced Autophagy
(2R,6R)-HNK significantly attenuated ECS-induced autophagy; the groups treated with (2R,6R)-HNK then showed a higher p62 expression level (P<0.001) and lower LC3-II/LC3- I ratio (P<0.001) and Beclin-1 expression level (P<0.001) than did the ECS group (Figure 3B–D).
Effect of SMER28 on (2R,6R)-HNK-Inhibited Autophagy
Infusion of SMER28 attenuated the role of (2R,6R)-HNK in inhibiting autophagy; a lower p62 expression level (P=0.001) and higher LC3-II/LC3-I ratio (P=0.033) and Beclin-1 expression levels (P=0.045) were then observed in the SMER28+(2R,6R)-HNK+ECS group than in the (2R,6R)-HNK+ECS group (Figure 3B–D).
Effect of 3-MA on (2R,6R)-HNK-Inhibited Autophagy
Infusion of 3-MA enhanced the effect of inhibiting autophagy; a higher p62 expression level was then observed (P=0.003) in the 3-MA+(2R,6R)-HNK+ECS group than in the (2R,6R)-HNK+ECS group (Figure 3A and D).
mTOR and p-mTOR Expression Levels
The p-mTOR expression level significantly decreased (P=0.003) in the ECS group when compared with that in the sham ECS group (Figure 4C). Meanwhile, the p-mTOR expression level was significantly higher in the (2R,6R)-HNK+ECS group (P=0.042) than in the ECS group (Figure 4C). There was no difference found in the p-mTOR expression level between the (2R,6R)-HNK+ECS group and the SMER28+(2R,6R)-HNK+ECS (P=1) and 3-MA+(2R,6R)-HNK+ECS groups (P=1) (Figure 4C).
Discussion
ECT is a common and effective treatment for TRD but often causes cognitive impairment in patients. In clinical studies, it has been found that ketamine alleviates the cognitive side effects when used as an anesthetic in ECT; however, the mechanism is still unclear. In this study, we found that ECS enhanced autophagy and induced spatial learning impairment, and (2R,6R)-HNK administration inhibited autophagy and offered potential protection against learning impairment. Furthermore, we revealed for the first time that mTOR activation-induced inhibition of autophagy critically contributed to the effect of (2R,6R)-HNK on ECS-induced autophagy.
Autophagy has both favorable and unfavorable consequences, as both impaired autophagy and excessive autophagy are associated with many diseases. Autophagy imbalance is involved in the pathophysiological process of many diseases. Once autophagy is overactivated, it will lead to programmed cell death.10 Studies have shown that the decline in cognitive function caused by traumatic damage is closely related to the activation of autophagy.11,12 The excitotoxicity of glutamate is an important cause of cognitive impairment in ECT. ECT stimulation induces glutamate release and enhances the glutamate signaling system. Glutamate excitotoxicity induces autophagy.19 Activation of the δ2 glutamate receptor significantly upregulates autophagy.20 In this study, ECS-induced autophagy was confirmed as demonstrated by increased expression of autophagy signals Beclin-1 and LC3-II/LC3-I and decreased expression of autophagy‐specific substrate p62. These results are consistent with those of a previous study.9
ECS has been shown to enhance autophagy signaling in the rat hippocampus.9 Excessive autophagy may induce learning impairment by decreasing the phosphorylated cAMP-response element-binding protein,21 which plays an important role in regulating hippocampal neuronal regeneration, synaptic formation, learning, and memory. In this study, we found that the escape latency for the Morris water maze in the ECS group was significantly longer than that in the sham ECS group, indicating learning impairment.
Administration of (2R,6R)-HNK inhibited the activation of ECS-induced autophagy in the hippocampus, showing decreased expression of autophagy signals Beclin-1 and LC3-II/LC3-I and increased expression of autophagy‐specific substrate p62, when compared with ECS. These results indicate that (2R,6R)-HNK inhibited autophagy. This is in line with a previous report that ketamine was involved in the protective effect on allergic airway inflammation by alleviating autophagy.22 Consistently, we observed better performance in the escape latency for the Morris water maze after (2R,6R)-HNK administration. These results indicate that (2R,6R)-HNK alleviated ECS-induced learning impairment by inhibiting the activation of ECS-induced autophagy.
To further determine the role of (2R,6R)-HNK in downregulating autophagy in the pathology of ECS-induced autophagy, we tested the effects of the autophagy enhancer SMER28. Our analysis showed that SMER28 weakened the inhibitory effect of (2R,6R)-HNK on autophagy, as demonstrated by a significant increase in the expression of autophagy markers, including higher LC3‐II/LC3-I ratio and Beclin-1 expression level and lower autophagy‐specific substrate p62 expression level in the SMER28+(2R,6R)-HNK+ECS group than in the (2R,6R)-HNK+ECS group. In addition, we tested the effects of the autophagy inhibitor 3-MA and found that autophagy inhibition was enhanced, showing that the expression level of autophagy‐specific substrate p62 was higher in the 3-MA+(2R,6R)-HNK+ECS group than in the (2R,6R)-HNK+ECS group. Consistently, we observed that the performance in the Morris water maze for the rats in the (2R,6R)-HNK+ECS and 3-MA+(2R,6R)-HNK+ECS groups was better than that in the ECS group. However, we failed to find a better protective effect on learning impairment when the autophagy inhibitor 3-MA was administered in comparison to that when (2R,6R)-HNK+ECS was administered. These results suggest that (2R,6R)-HNK exerts protective effects against ECS-induced autophagy and learning impairment.
mTOR is a kinase that plays a critical role in regulating autophagy. When mTOR is phosphorylated, autophagy is inhibited. A number of studies have reported consistent findings that ketamine rapidly activates mTOR.23–27 The activation of mTOR by ketamine may result in the inhibition of autophagy.11,22,28 In this study, we found that the ketamine metabolite (2R,6R)-HNK before ECS mimicked the effects of ketamine, showing higher p-mTOR expression levels after (2R,6R)-HNK administration; this suggests that the inhibition of autophagy by (2R,6R)-HNK was mainly attributed to the activation of mTOR. However, there are still conflicting results regarding the effect of (2R,6R)-HNK on mTOR activation. Herein, we found that (2R,6R)-HNK increases the phosphorylated form of mTOR in the hippocampus after repeated administration (once a day for 3 days in a row). Fukumoto et al observed a rapid but transient activation of mTOR in the medial prefrontal cortex at 30 min after administration.15 However, Zanos et al did not observe stimulation of mTORC1 signaling in the hippocampus at 60 min after administration.14 This could be attributed to the different regions analyzed,15 time points,15 and timing of (2R,6R)-HNK administration.
Conclusion
In summary, our study showed that (2R,6R)-HNK improves the cognitive performance of ECS-treated rats, indicating a protective effect on learning ability. (2R,6R)-HNK could inhibit excessive ECS-induced autophagy in the hippocampus, thus protecting the learning function in depressed rats after ECS. The inhibition of autophagy by (2R,6R)-HNK may be mediated by the activation of mTOR.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81701341), Guangdong Provincial Medical Science and Technology Research Foundation (No. A2020446), Science and Technology Plan Project of Guangdong Province (No. 2019B030316001), and Guangzhou Municipal Key Discipline in Medicine (2017–2019).
Disclosure
All authors declare that they have no competing interests.
References
1. Ruberto V, Jha M, Murrough J. Pharmacological treatments for patients with treatment-resistant depression. Pharmaceuticals. 2020;13(6). doi:10.3390/ph13060116
2. An X, Shi X. Effects of electroconvulsive shock on neuro-immune responses: does neuro-damage occur? Psychiatry Res. 2020;292:113289. doi:10.1016/j.psychres.2020.113289
3. Naughton M, Clarke G, O’Leary O, Cryan J, Dinan T. A review of ketamine in affective disorders: current evidence of clinical efficacy, limitations of use and pre-clinical evidence on proposed mechanisms of action. J Affect Disord. 2014;156:24–35. doi:10.1016/j.jad.2013.11.014
4. Phillips J, Jaworska N, Kamler E, et al. A randomized, crossover comparison of ketamine and electroconvulsive therapy for treatment of major depressive episodes: a Canadian biomarker integration network in depression (CAN-BIND) study protocol. BMC Psychiatry. 2020;20(1):268. doi:10.1186/s12888-020-02672-3
5. Sartorius A, Beuschlein J, Remennik D, et al. Empirical ratio of the combined use of S-ketamine and propofol in electroconvulsive therapy and its impact on seizure quality. Eur Arch Psychiatry Clin Neurosci. 2020. doi:10.1007/s00406-020-01170-7
6. Yalcin S, Aydoğan H, Selek S, et al. Ketofol in electroconvulsive therapy anesthesia: two stones for one bird. J Anesth. 2012;26(4):562–567. doi:10.1007/s00540-012-1378-6
7. Zhong X, He H, Zhang C, et al. Mood and neuropsychological effects of different doses of ketamine in electroconvulsive therapy for treatment-resistant depression. J Affect Disord. 2016;201:124–130. doi:10.1016/j.jad.2016.05.011
8. Moscrip T, Terrace H, Sackeim H, Lisanby S. A primate model of anterograde and retrograde amnesia produced by convulsive treatment. J ECT. 2004;20(1):26–36. doi:10.1097/00124509-200403000-00007
9. Otabe H, Nibuya M, Shimazaki K, et al. Electroconvulsive seizures enhance autophagy signaling in rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2014;50:37–43. doi:10.1016/j.pnpbp.2013.11.012
10. Edinger A, Thompson C. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16(6):663–669. doi:10.1016/j.ceb.2004.09.011
11. Wang C, Ye Y, Chen F, et al. Posttraumatic administration of a sub-anesthetic dose of ketamine exerts neuroprotection via attenuating inflammation and autophagy. Neuroscience. 2017;343:30–38. doi:10.1016/j.neuroscience.2016.11.029
12. Hensley K, Poteshkina A, Johnson M, et al. Autophagy modulation by lanthionine ketimine ethyl ester improves long-term outcome after central fluid percussion injury in the mouse. J Neurotrauma. 2016;33(16):1501–1513. doi:10.1089/neu.2015.4196
13. Li N, Lee B, Liu R, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. doi:10.1126/science.1190287
14. Zanos P, Moaddel R, Morris P, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533(7604):481–486. doi:10.1038/nature17998
15. Fukumoto K, Fogaça M, Liu R, et al. activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2,6)-hydroxynorketamine. Proc Natl Acad of Sci USA. 2019;116(1):297–302. doi:10.1073/pnas.1814709116
16. Kim J, Kundu M, Viollet B, Guan K. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152
17. Segi-Nishida E, Warner-Schmidt JL, Duman RS. Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc Natl Acad Sci USA. 2008;105(32):11352–11357. doi:10.1073/pnas.0710858105
18. Luo J, Min S, Wei K, Zhang J, Liu Y. Propofol interacts with stimulus intensities of electroconvulsive shock to regulate behavior and hippocampal BDNF in a rat model of depression. Psychiatry Res. 2012;198(2):300–306. doi:10.1016/j.psychres.2011.09.010
19. Kumari S, Mehta SL, Li PA. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: preventive effects of selenium. PLoS One. 2012;7(6):e39382. doi:10.1371/journal.pone.0039382
20. Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35(5):921–933. doi:10.1016/S0896-6273(02)00861-9
21. Xu Y, Tian Y, Tian Y, Li X, Zhao P. Autophagy activation involved in hypoxic-ischemic brain injury induces cognitive and memory impairment in neonatal rats. J Neurochem. 2016;139(5):795–805. doi:10.1111/jnc.13851
22. Zou H, Wang L, Wang M, et al. MTOR-mediated autophagy is involved in the protective effect of ketamine on allergic airway inflammation. J Immunol Res. 2019;2019:5879714. doi:10.1155/2019/5879714
23. Zhou X, Lv X, Zhang L, et al. Ketamine promotes the neural differentiation of mouse embryonic stem cells by activating mTOR. Mol Med Rep. 2020;21(6):2443–2451. doi:10.3892/mmr.2020.11043
24. Lin C, Yang A, Lin A. Activation of the mTOR dependent signaling pathway underlies ketamine-induced uropathy. Neurourol Urodyn. 2017;36(8):1988–1995. doi:10.1002/nau.23234
25. Harraz M, Tyagi R, Cortés P, Snyder S. Antidepressant action of ketamine via mTOR is mediated by inhibition of nitrergic Rheb degradation. Mol Psychiatry. 2016;21(3):313–319. doi:10.1038/mp.2015.211
26. Cavalleri L, Merlo Pich E, Millan M, et al. Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Mol Psychiatry. 2018;23(4):812–823. doi:10.1038/mp.2017.241
27. Abelaira H, Réus G, Ignácio Z, et al. Effects of ketamine administration on mTOR and reticulum stress signaling pathways in the brain after the infusion of rapamycin into prefrontal cortex. J Psychiatr Res. 2017;87:81–87. doi:10.1016/j.jpsychires.2016.12.002
28. Yang J, Li X, Yang C, Bu X, Shen J, Hong T. Research on ketamine in mediating autophagy and inhibiting apoptosis of astrocytes in cerebral cortex of rats through NF-κB pathway. Eur Rev Med Pharmacol Sci. 2018;22(16):5385–5393. doi:10.26355/eurrev_201808_15741
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.