Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
β2-Microglobulin Induces Mitochondrial Dysfunction Accompanied by Bronchial Epithelial Cell Senescence
Authors Gu Y ![]() , Yuan W, Xie XF, Dong HM
, Yuan W, Xie XF, Dong HM ![]() , Wei B
, Wei B ![]() , Wang JY
, Wang JY
Received 21 April 2026
Accepted for publication 15 June 2026
Published 20 June 2026 Volume 2026:21 614376
DOI https://doi.org/10.2147/COPD.S614376
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Zijing Zhou
Yu Gu,1– 3 Wei Yuan,1– 3 Xue-Fei Xie,1– 3 Hong-Meng Dong,1– 3 Bing Wei,1– 3 Jun-Yu Wang1– 3
1Emergency Medical Center, Beijing Chaoyang Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Beijing Key Laboratory of Cardiopulmonary-Cerebral Resuscitation Innovation and Translation, Beijing Chaoyang Hospital, Capital Medical University, Beijing, People’s Republic of China; 3Clinical Center for Medicine in Acute Infection, Beijing Chaoyang Hospital, Capital Medical University, Beijing, People’s Republic of China
Correspondence: Bing Wei; Jun-Yu Wang, Emergency Medical Center, Beijing Chaoyang Hospital, Capital Medical University, Beijing, People’s Republic of China, Tel +86-13810051210 ; +86-13910001172, Email [email protected]; [email protected]
Purpose: β 2-microglobulin (β 2m) is the light-chain subunit of major histocompatibility complex class I (MHC I) molecules. Our group previously showed that β 2m contributes to emphysema development by inducing epithelial cell senescence. However, the mechanism linking β 2m to epithelial senescence remains unclear. Previous studies have reported mitochondrial dysfunction in senescent lung cells from patients with emphysema, suggesting a potential mechanistic pathway. This in vitro study used BEAS-2B human bronchial epithelial cells to evaluate whether exposure to β 2m is associated with mitochondrial dysfunction and a senescent phenotype.
Methods: Human bronchial epithelial BEAS-2B cells were exposed in vitro for 48 hours to recombinant human β 2m or cigarette smoke extract (CSE). Cellular senescence was assessed by senescence-associated β-galactosidase (SA-β-gal) staining. Mitochondrial dysfunction was evaluated by measuring mitochondrial membrane potential (MMP), reactive oxygen species (ROS), mitochondrial ROS (mtROS), oxygen consumption rate (OCR), and real-time adenosine triphosphate (ATP) production rate. Cell proliferation and apoptosis were assessed using CCK-8 and Annexin V-FITC/PI assays, respectively.
Results: β 2m and CSE increased SA-β-gal staining in BEAS-2B cells, indicating enhanced cellular senescence. β 2m and CSE also decreased MMP, increased ROS and mtROS levels, and reduced OCR, indicating mitochondrial dysfunction. In addition, β 2m and CSE reduced BEAS-2B cell proliferation and increased apoptosis.
Conclusion: β 2m exposure was associated with mitochondrial dysfunction and a senescent phenotype in BEAS-2B cells, accompanied by reduced proliferation and increased apoptosis. These findings suggest that β 2m may contribute to epithelial aging in COPD/emphysema, although further mechanistic investigation and in vivo validation are required.
Keywords: β 2-microglobulin, mitochondrial dysfunction, bronchial epithelial cells, senescence
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by progressive and largely irreversible airflow limitation. Its key pathological features include chronic airway inflammation and destruction of the lung parenchyma (emphysema).1,2 COPD remains a major cause of morbidity and mortality worldwide. Although smoking is the primary risk factor, its pathogenesis is not yet fully understood. Increasing evidence suggests that COPD is an aging-related disease and that smoking accelerates lung aging.3–7 Cellular senescence is a central hallmark of this process.8–10 As the principal structural cells lining the respiratory tract, epithelial cells constitute the first barrier against inhaled toxic substances.11,12 Accelerated epithelial cell senescence has been reported in patients with COPD,8,13–16 and CSE exposure similarly promotes epithelial cell senescence in vitro.13,17,18
β2-microglobulin is the light-chain subunit of MHC class I molecules. Recent studies have identified β2m as a circulating pro-aging factor,19–21 and elevated β2m levels have been associated with multiple age-related diseases, including myocardial infarction, myocardial fibrosis, hypertension, and Alzheimer’s disease.21–23 β2m has also been proposed as a biomarker of pulmonary fibrosis in patients with COPD.24 Our previous work showed that β2m levels are increased in lung tissue from patients with emphysema and are negatively correlated with lung function, with β2m predominantly localized to pulmonary epithelial cells. In that study, plasma β2m levels were higher in patients with emphysema than in control subjects (1.89±0.12 versus 1.42±0.06 mg/L), and stimulation of β2m at 100 μg/mL induced epithelial cell senescence in vitro.17 A subsequent COPD cohort also reported serum β2m values of approximately 2.10–2.24 mg/L in patients stratified by diffusing capacity, with higher β2m associated with worse DLCO and pulmonary fibrosis-related changes.24 These clinical circulating concentrations are lower than the β2m concentrations used in the present in vitro experiments; therefore, the 100–500 μg/mL concentrations used here were selected primarily based on our previous cell-based work and should be interpreted as experimental high local/pathological exposure rather than as a direct replication of circulating plasma concentrations.
In vitro, CSE exposure increased β2m secretion by alveolar epithelial cells, and elevated β2m promoted alveolar epithelial cell senescence; anti-β2m antibodies partially reversed CSE-induced epithelial senescence. In animal models, cigarette smoke exposure increased β2m levels in lung tissue from mice with emphysema.25 Together, these findings suggest that smoking-induced β2m upregulation may contribute to emphysema progression by promoting epithelial cell senescence. The present study extends this prior work by focusing on mitochondrial injury, bioenergetic impairment, oxidative stress, proliferation, and apoptosis in bronchial epithelial cells.
Mitochondria are central to cellular energy metabolism and aerobic respiration. Mitochondrial membrane potential, ROS generation, and fusion-fission dynamics are critical for mitochondrial and cellular homeostasis.26–28 As a major intracellular source of ROS, mitochondria play a key role in cellular senescence, and mitochondrial dysfunction is considered a core mechanism of this process.29–31 Multiple studies have demonstrated mitochondrial dysfunction in aged lung cells from patients with COPD,32–37 and smoking can induce mitochondrial dysfunction in emphysematous lung cells.38 Mechanistically, mitochondrial injury and oxidative stress can converge on senescence-associated pathways such as p53/p21 and p16INK4a signaling, while persistent oxidative and inflammatory stress may also activate NF-κB-dependent senescence-associated inflammatory responses.10,14,16 Altered mitochondrial dynamics and impaired mitophagy, including excessive mitochondrial fragmentation, have also been implicated in cigarette smoke-induced bronchial epithelial cell senescence and COPD pathobiology.35,38 Because β2m has been characterized as a pro-aging factor and has been linked to epithelial senescence in emphysema,17,19 its potential connection with mitochondrial injury, oxidative stress, and these senescence pathways deserves further investigation. Because cigarette smoke promotes epithelial senescence partly through mitochondrial dysfunction and smoking also upregulates β2m, it is important to determine whether β2m exposure is associated with similar mitochondrial changes. BEAS-2B cells were selected because they are a reproducible and widely used human bronchial epithelial cell model for studying CSE-induced airway epithelial injury, oxidative stress, mitochondrial dysfunction, and senescence. Bronchial epithelial cells form the first structural and immunologic barrier against inhaled toxicants and are central to COPD and emphysema pathogenesis through barrier dysfunction, inflammatory signaling, impaired repair, senescence, and apoptosis.11,12,18,35 Nevertheless, we recognize that BEAS-2B cells do not fully substitute for primary human airway epithelial cells. Under physiological conditions, proliferation and apoptosis are balanced to maintain tissue homeostasis. In COPD, reduced proliferation and increased apoptosis have been observed in lung tissue,39–41 and induction of epithelial apoptosis can lead to emphysema in mice.42 Therefore, we investigated whether β2m exposure is associated with mitochondrial dysfunction, epithelial senescence, reduced proliferation, and increased apoptosis in BEAS-2B cells.
Methods
Cigarette Smoke Extract (CSE) Preparation
The CSE preparation protocol was adapted from a previous study.43 Briefly, one Marlboro cigarette (0.8 mg nicotine, 11 mg carbon monoxide, and 10 mg tar) was combusted, and the smoke was drawn slowly and uniformly through a bubbling flask under negative pressure into 10 mL of sterile culture medium. The resulting suspension was filtered through a 0.22 μm filter for sterilization. This solution was defined as 100% CSE and was used immediately after preparation.
Cell Culture and Treatment
Human bronchial epithelial cells (BEAS-2B; American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle medium (DMEM; Thermo Fisher Scientific, Grand Island, NY, USA). Purified human β2m (Lee BioSolutions, Maryland Heights, MO, USA) was dissolved in phosphate-buffered saline (PBS) and then added to medium containing 2% fetal bovine serum (Thermo Fisher Scientific) to final concentrations of 100 or 500 μg/mL. These concentrations were selected based on previous in vitro β2m studies and were intended to model relatively high local/pathological exposure rather than directly reproduce circulating clinical concentrations, because the exact β2m concentration within diseased airway microenvironments is not well established. In parallel experiments, cells were incubated for 48 hours in medium containing 2% fetal bovine serum plus 1% or 5% CSE. Control cells were cultured for the same duration in medium containing 2% fetal bovine serum.
Measurement of Cell Senescence, Mitochondrial Function, and Cell Proliferation/Apoptosis
Senescence-associated β-galactosidase (SA-β-gal) activity was measured using a commercially available assay kit (Abbkine Scientific Co., Ltd., Wuhan, China). MMP, intracellular ROS, and mtROS were measured using TMRM (Thermo Fisher Scientific, Waltham, MA, USA), ROS Brite 670 (AAT Bioquest, Pleasanton, CA, USA), and MitoROS 580 probes (AAT Bioquest), respectively. For flow cytometry-based measurements, unstained and single-stained controls were used, when applicable, to set fluorescence thresholds and compensation. Viable single cells were identified using forward- and side-scatter profiles; debris and doublets were excluded using scatter and pulse-geometry parameters, and the same acquisition settings and gating strategy were applied to all groups within each experiment before fluorescence quantification. Mitochondrial function was assessed by OCR using the Cell Mito Stress Test Kit and by ATP production rate using the Real-Time ATP Rate Assay Kit (Agilent Technologies, Santa Clara, CA, USA). For Seahorse assays, cells were seeded at the same density in Seahorse XF culture microplates and treated as described above. Before measurement, the culture medium was replaced with assay medium prepared according to the manufacturers’ instructions, and cells were equilibrated in a non-CO2 incubator before loading into the analyzer. OCR was recorded after sequential compound injections according to the Cell Mito Stress Test workflow, and basal respiration, ATP-linked respiration, maximal respiration, and spare respiratory capacity were calculated using the manufacturer’s analysis workflow. Raw OCR and ATP production-rate values were first corrected using background wells without cells. To minimize the influence of well-to-well differences in cell numbers, Seahorse rate data from each well were normalized to the corresponding cell number in that well before group comparisons. Wells were excluded only for predefined technical reasons, including failed compound injection, unstable baseline caused by technical malfunction, obvious cell detachment or loss, absence of an analyzable cell monolayer, or failure of background correction. Cell proliferation was evaluated using Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan), and apoptosis was assessed using the Annexin V-FITC/PI Apoptosis Detection Kit (4A Biotech Co., Ltd., Beijing, China). For apoptosis analysis, Annexin V-FITC/PI quadrant gating was used to distinguish viable, early apoptotic and late apoptotic cells, and at least 10,000 single-cell events per sample were acquired whenever possible. Unless otherwise indicated, experiments were repeated in at least three independent biological replicates, and technical replicate wells were averaged before statistical analysis. All procedures were performed according to the manufacturers’ instructions. Formal blinding was not performed during data acquisition or analysis because this in vitro cell-based study relied mainly on objective instrument-based readouts; however, all groups were processed under the same experimental conditions and analyzed using predefined acquisition settings, gating strategies, normalization procedures, and statistical methods to reduce potential bias.
Statistical Analysis
Data were analyzed and graphed using GraphPad Prism version 8.2 (GraphPad Software, La Jolla, CA, USA). Data are presented as mean ± standard deviation (SD). Comparisons among multiple groups were performed using one-way analysis of variance (ANOVA), followed by Dunnett’s multiple-comparison test when treatment groups were compared with the control group. Normality and homogeneity of variance were evaluated before ANOVA. Technical replicates from the same independent experiment were averaged before statistical testing, and no data points were excluded solely based on statistical significance or the direction of the result. Because this was an exploratory in vitro study, no formal sample-size calculation was performed. P<0.05 was considered statistically significant.
Results
β2m and CSE Induce Senescence in BEAS-2B Cells
Because smoking is the most important risk factor for COPD, we used CSE stimulation to mimic in vivo smoke exposure. BEAS-2B cells were treated with 100 μg/mL β2m, 500 μg/mL β2m, 1% CSE, or 5% CSE for 48 hours, and cellular senescence was assessed by SA-β-gal activity. Representative staining and quantitative data are shown in Figure 1A–C. Both CSE and β2m significantly increased senescence in BEAS-2B cells (*P<0.05, **P<0.01, ***P<0.001).
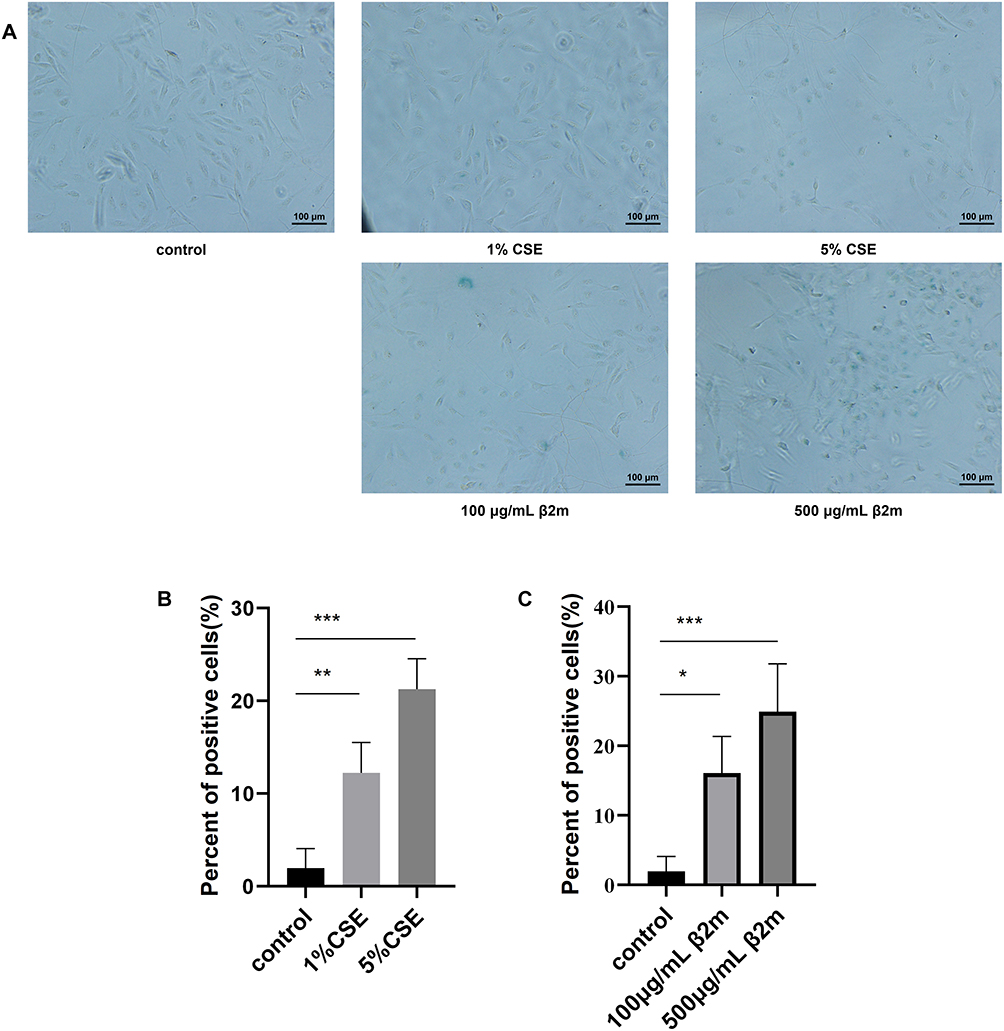 |
Figure 1 β2m and CSE increase senescence-associated β-galactosidase activity in BEAS-2B cells after 48 hours of exposure. (A) Representative SA-β-gal staining images; (B) quantification after CSE treatment; (C) quantification after β2m treatment. Data are presented as mean ± SD from three independent biological experiments (n=3). *P<0.05, **P<0.01, ***P<0.001 versus the control group. One-way ANOVA with Dunnett’s post hoc test was used for statistical analysis. |
β2m and CSE Reduced MMP and Increased Intracellular ROS and mtROS in BEAS-2B Cells
To evaluate whether β2m-associated senescence was accompanied by mitochondrial dysfunction, we measured MMP, intracellular ROS, mtROS, and OCR after β2m or CSE stimulation. Representative and quantitative data for MMP, intracellular ROS, and mtROS are shown in Figure 2A–I. Both CSE and β2m decreased MMP and increased intracellular ROS and mtROS, indicating mitochondrial dysfunction (*P<0.05, **P<0.01, ****P<0.0001).
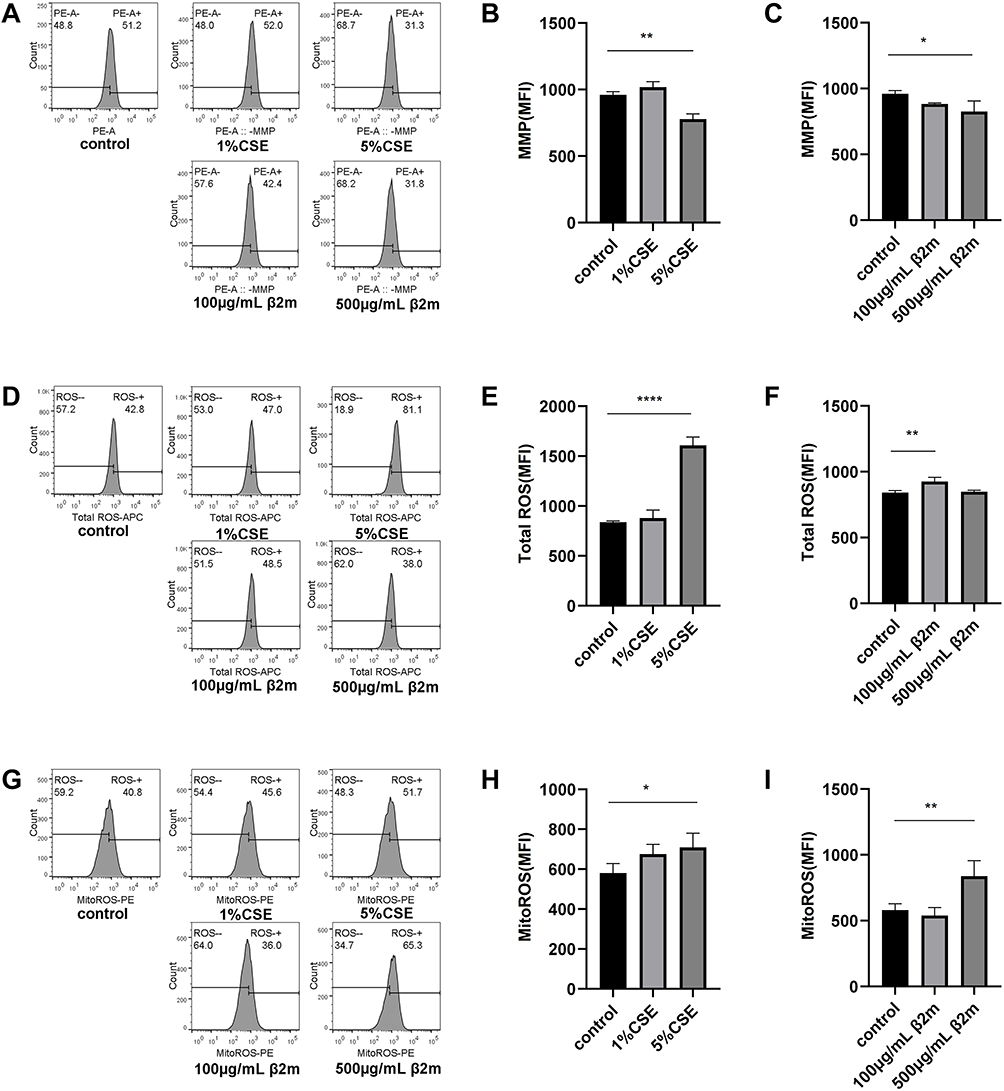 |
Figure 2 β2m and CSE induce mitochondrial dysfunction in BEAS-2B cells. (A–C) Mitochondrial membrane potential; (D–F) intracellular reactive oxygen species; (G–I) mitochondrial reactive oxygen species. Data are presented as mean ± SD from three independent biological experiments (n=3). *P<0.05, **P<0.01, ****P<0.0001 versus the control group. One-way ANOVA with Dunnett’s post hoc test was used for statistical analysis. |
β2m and CSE Reduced the Oxygen Consumption Rate (OCR) in BEAS-2B Cells
We next assessed cellular aerobic respiration by measuring OCR. Following β2m or CSE stimulation, OCR was measured using the Cell Mito Stress Test Kit. The OCR trace and respiratory parameters are shown in Figure 3A–I. β2m reduced basal respiration, maximal respiration, spare respiratory capacity, and ATP-linked respiration in BEAS-2B cells, indicating impaired aerobic metabolism. CSE similarly reduced basal respiration, maximal respiration, and ATP-linked respiration (*P<0.05, **P<0.01). These findings indicate that β2m exposure is associated with reduced mitochondrial respiratory function.
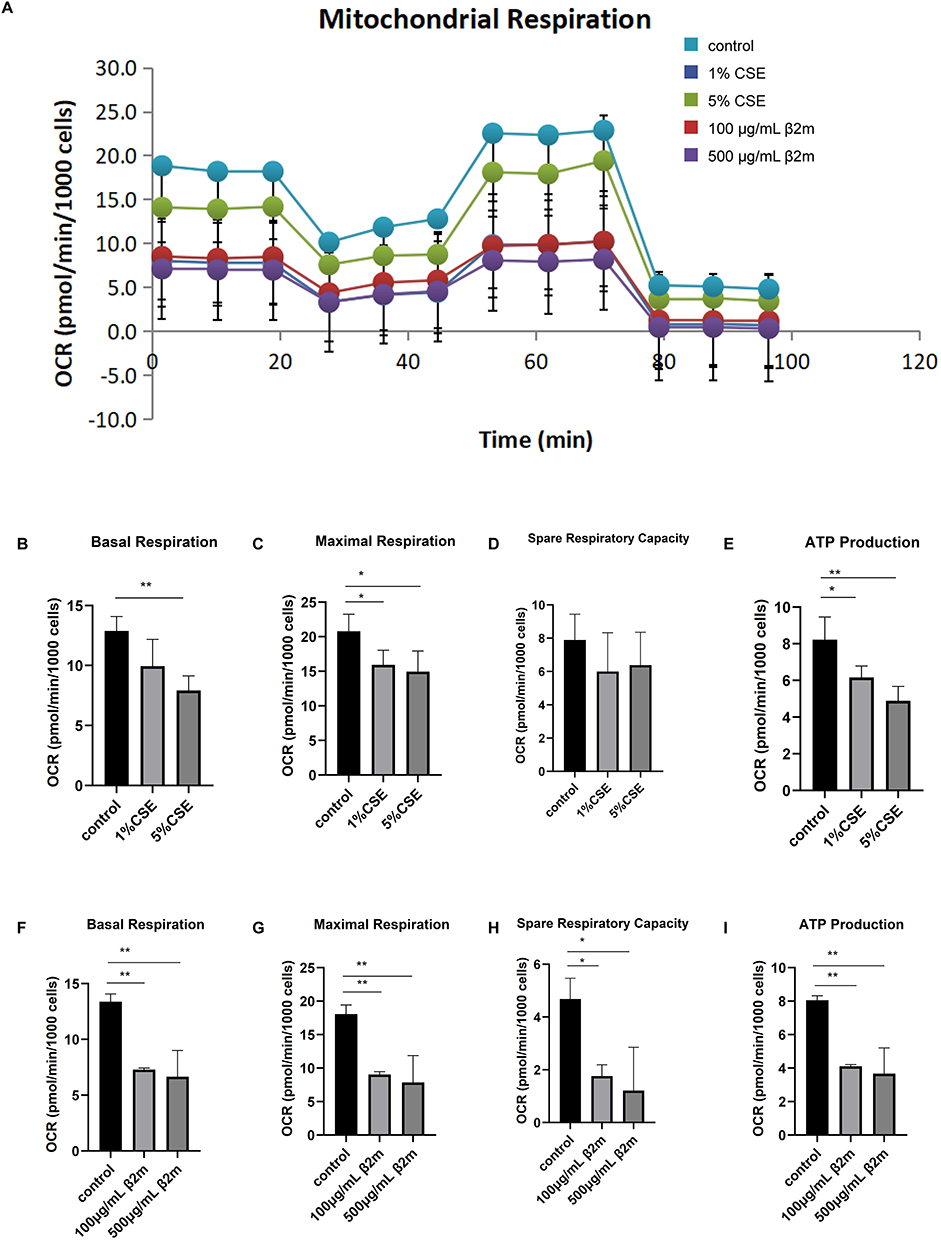 |
Figure 3 β2m and CSE reduce oxygen consumption rate and key respiratory parameters in BEAS-2B cells. (A) OCR trace; (B) basal respiration after CSE treatment; (C) maximal respiration after CSE treatment; (D) spare respiratory capacity after CSE treatment; (E) ATP-linked respiration after CSE treatment; (F) basal respiration after β2m treatment; (G) maximal respiration after β2m treatment; (H) spare respiratory capacity after β2m treatment; (I) ATP-linked respiration after β2m treatment. Data are presented as mean ± SD from three independent biological experiments (n=3). *P<0.05, **P<0.01 versus the control group. One-way ANOVA with Dunnett’s post hoc test was used for statistical analysis. |
β2m Had No Significant Effect on ATP Sources in BEAS-2B Cells, While CSE Reduced Aerobic ATP Production
Although aerobic respiration is the dominant source of cellular ATP, anaerobic metabolism also contributes. Under pathological conditions, suppression of aerobic respiration may be accompanied by compensatory enhancement of anaerobic ATP production. After observing that β2m impaired mitochondrial respiration, we examined ATP source distribution. BEAS-2B cells were treated with β2m or CSE, and ATP production rates were quantified (Figure 4A–E). β2m showed a trend toward reduced mitochondrial ATP production and increased glycolytic ATP production, but these changes were not statistically significant, suggesting that β2m did not measurably redistribute ATP sources under the basal assay conditions used here. CSE significantly reduced mitochondrial ATP production without significantly affecting glycolytic ATP production (*P<0.01).
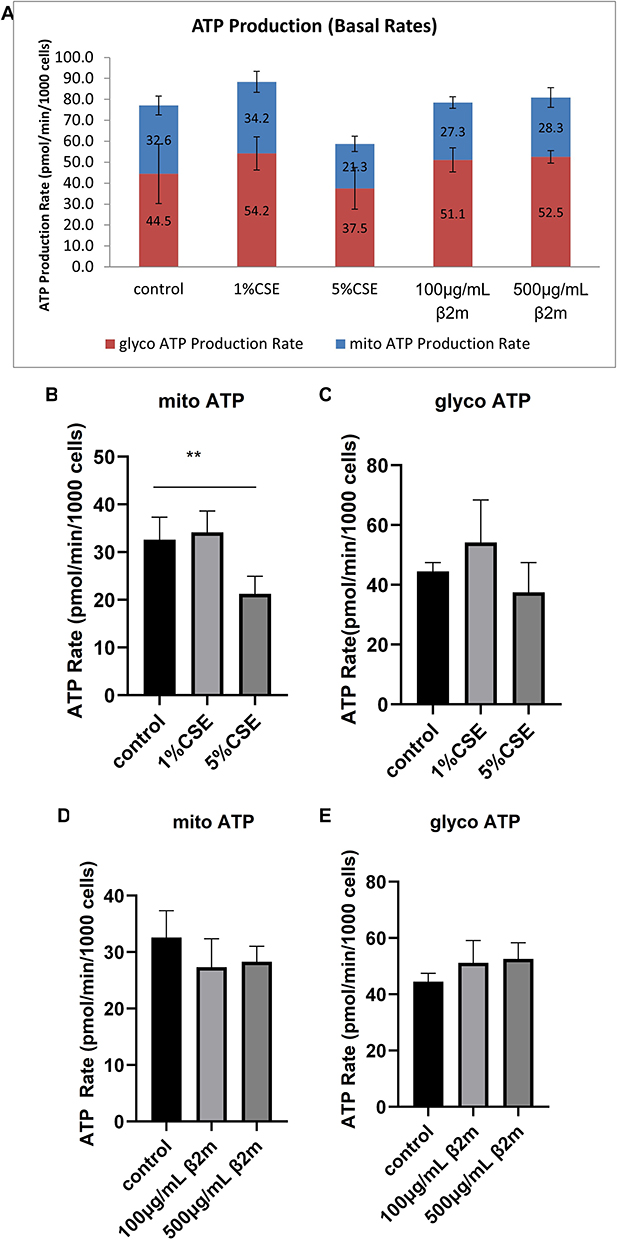 |
Figure 4 Effects of β2m and CSE on mitochondrial and glycolytic ATP production in BEAS-2B cells. (A) Real-time ATP production profile; (B) mitochondrial ATP production after CSE treatment; (C) glycolytic ATP production after CSE treatment; (D) mitochondrial ATP production after β2m treatment; (E) glycolytic ATP production after β2m treatment. Data are presented as mean ± SD from three independent biological experiments (n=3). **P<0.01 versus the control group. One-way ANOVA with Dunnett’s post hoc test was used for statistical analysis. |
β2m and CSE Inhibit BEAS-2B Cell Proliferation and Promote Apoptosis
To evaluate whether β2m affects cell-fate programs associated with tissue aging, we measured proliferation and apoptosis in BEAS-2B cells after β2m or CSE stimulation. As shown in Figure 5 and Figure 6A–C, both β2m and CSE significantly inhibited cell proliferation and increased apoptosis (*P<0.05, ****P<0.0001).
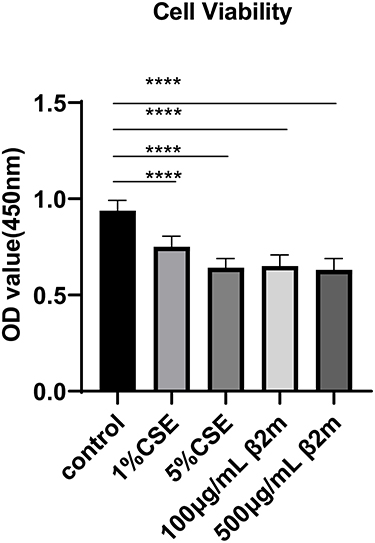 |
Figure 5 β2m and CSE suppress BEAS-2B cell proliferation as assessed by CCK-8 assay. Data are presented as mean ± SD from three independent biological experiments (n=3). ****P<0.0001 versus the control group. One-way ANOVA with Dunnett’s post hoc test was used for statistical analysis. |
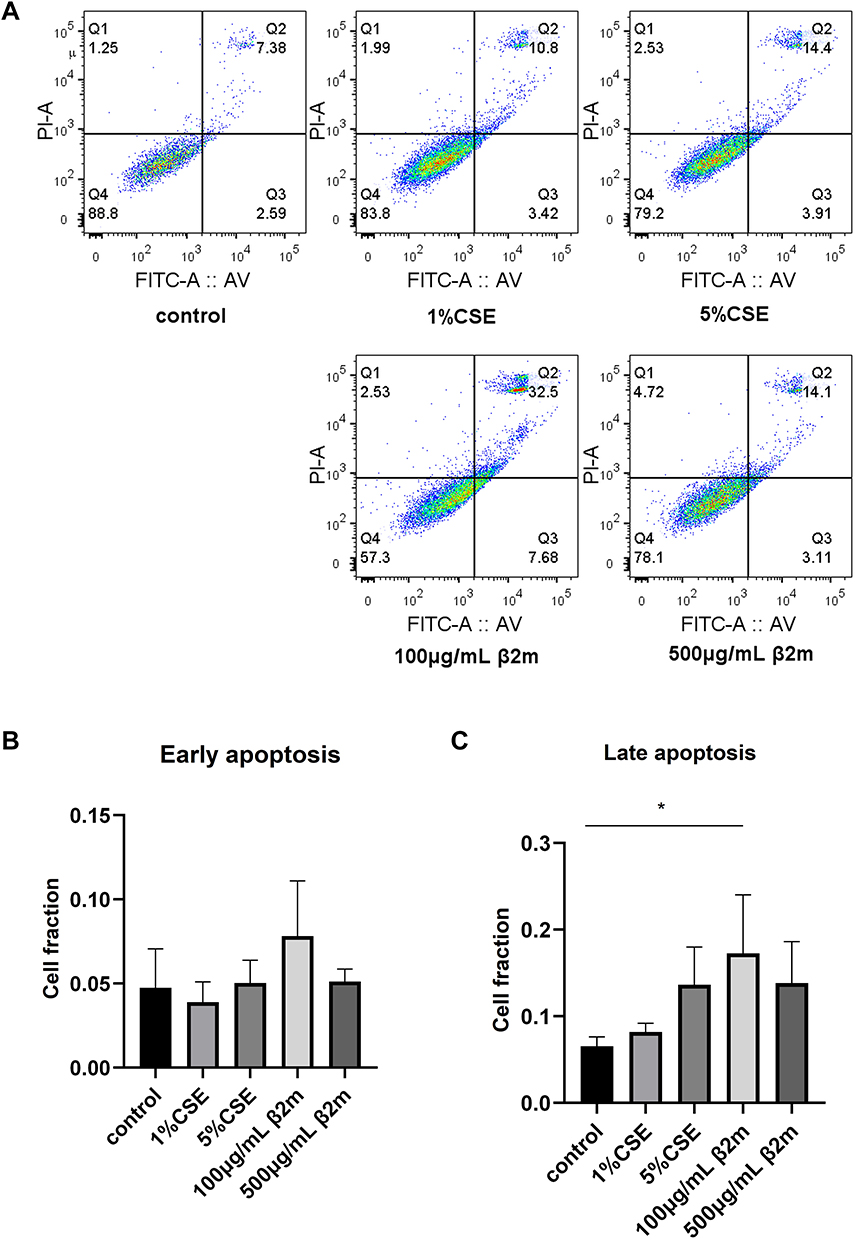 |
Figure 6 β2m and CSE promote apoptosis in BEAS-2B cells. (A) Representative Annexin V-FITC/PI flow cytometry plots; (B) quantitative analysis of early apoptotic cells after CSE and β2m treatment; (C) quantitative analysis of late apoptotic cells after CSE and β2m treatment. Data are presented as mean ± SD from three independent biological experiments (n=3). *P<0.05 versus the control group. One-way ANOVA with Dunnett’s post hoc test was used for statistical analysis. |
Discussion
COPD is characterized by accelerated pulmonary aging, and cellular senescence is a key pathological component. As the first barrier to inhaled toxicants, airway epithelial cells are central to COPD pathobiology. In patients with COPD, epithelial senescence,18,44 reduced proliferation, and increased apoptosis39,41 likely contribute to progressive lung aging. Mitochondrial dysfunction is an established mechanism underlying cellular senescence in COPD. β2m, the light-chain subunit of MHC class I molecules, is essential for adaptive immune function. Elevated β2m has been implicated in several diseases, including chronic kidney disease, rheumatoid arthritis, and cancer.45–47 More recently, β2m has been recognized as a circulating pro-aging factor associated with multiple age-related conditions.19,21,23 Despite COPD being an aging-related disease, the role of β2m in COPD has been insufficiently studied. Our previous work showed increased β2m in plasma and lung tissue from patients with emphysema, suggesting a potential contribution to epithelial senescence.17 Smoking also increases pulmonary β2m levels in emphysema mouse models.25 In this study, we further show that β2m exposure in BEAS-2B cells is associated with mitochondrial dysfunction, increased senescence, reduced proliferation, and increased apoptosis. These data extend our previous findings, but they do not by themselves establish that mitochondrial dysfunction is the sole or direct mediator of β2m-induced senescence.
Epithelial cell senescence is a major feature of lung aging during COPD progression. As the leading risk factor for COPD, smoking accelerates this process. Because cigarette smoke contains numerous toxic compounds and reactive oxygen species, CSE stimulation is widely used to model cigarette smoke exposure in vitro. Using this model, we confirmed that CSE promotes senescence in BEAS-2B cells, consistent with our previous findings.17 Importantly, we also observed that β2m, a senescence-associated factor, induces a similar pro-senescent phenotype. These observations suggest that β2m may act as a pathogenic mediator that mimics, at least in part, the cellular effects of cigarette smoke.
Mitochondria are central regulators of cellular senescence.28 They support ATP generation through aerobic respiration and regulate intracellular homeostasis through membrane potential, ROS production, and fusion-fission dynamics.48 Mitochondrial dysfunction induced by cigarette smoke has been linked to COPD progression.38,49,50 Consistent with prior reports,51,52 our data show that CSE exposure is accompanied by bronchial epithelial cell senescence and mitochondrial dysfunction. We further demonstrate that β2m produces similar mitochondrial defects and senescence phenotypes, supporting the possibility that smoking-induced β2m may contribute to epithelial aging in emphysema. However, mitochondrial rescue experiments, antioxidant interventions, or pathway-specific inhibition will be needed to confirm direct causality. We also evaluated ATP source profiles. β2m showed a non-significant trend toward reduced mitochondrial ATP production with increased glycolytic ATP production, whereas CSE significantly reduced mitochondrial ATP production without a significant change in glycolytic ATP production. These ATP findings should be interpreted together with the OCR data. OCR parameters reflect mitochondrial respiratory capacity and reserve, whereas the real-time ATP rate assay estimates ATP production under basal steady-state conditions. Therefore, β2m-induced impairment of respiratory reserve may be detectable before a statistically significant shift in basal ATP-source distribution becomes apparent. In β2m-treated cells, residual mitochondrial ATP generation together with compensatory glycolytic ATP production may have partially maintained basal ATP output, thereby masking statistically significant changes in ATP-source allocation despite reduced OCR. By contrast, CSE likely produced broader mitochondrial stress sufficient to reduce mitochondrial ATP production significantly. Thus, the ATP data do not contradict the OCR findings; rather, they suggest that β2m-associated mitochondrial impairment may affect respiratory capacity more clearly than basal ATP-source distribution in this experimental setting. Additional studies are needed to define the broader effects of β2m on anaerobic metabolism and cellular bioenergetic reprogramming.
Under physiological conditions, balanced proliferation and apoptosis maintain epithelial renewal. In COPD, this balance is disrupted, with reduced proliferation and increased apoptosis, which may accelerate lung aging. Experimental studies have shown that inducing epithelial or endothelial apoptosis can trigger emphysema-like changes in mice.42 In our study, both β2m and CSE inhibited proliferation and promoted apoptosis in BEAS-2B cells. These findings are consistent with a model in which β2m, elevated during smoke exposure and disease progression, could contribute to emphysema pathogenesis through mechanisms that overlap with cigarette smoke, including mitochondrial dysfunction and enhanced epithelial aging.
This study has several limitations. First, we did not directly test whether mitochondrial dysfunction is causally required for β2m-induced senescence; rescue experiments using mitochondrial-targeted antioxidants, metabolic modulators, or pathway-specific inhibitors will be needed. Second, the study used only the BEAS-2B cell line, and validation in primary human airway epithelial cells and in vivo models is still required. Third, although the β2m concentrations used here were selected based on previous in vitro work, the exact local β2m concentration in COPD or emphysematous airway microenvironments is not established, and translational interpretation should therefore remain cautious. Fourth, cellular senescence is a progressive phenotype, and longer exposure durations or time-course analyses would help determine whether β2m induces sustained senescence. Finally, we did not evaluate additional senescence pathways such as p16INK4a or p53/p21, nor did we directly measure NF-κB signaling or mitochondrial dynamics regulators.
In summary, β2m exposure was associated with bronchial epithelial cell senescence, mitochondrial dysfunction, reduced proliferation, and increased apoptosis. These findings provide additional evidence supporting a potential role for β2m in epithelial aging during COPD/emphysema, while highlighting the need for mechanistic rescue studies and in vivo validation.
Conclusion
This study indicates that β2m, a pathogenic factor elevated during emphysema progression, is associated with mitochondrial dysfunction, bronchial epithelial cell senescence, reduced proliferation, and increased apoptosis in vitro. These findings suggest that β2m may contribute to epithelial aging in COPD/emphysema, but direct mechanistic causality and in vivo relevance require further validation.
Abbreviations
β2m, β2-microglobulin; ATP, adenosine triphosphate; CCK-8, Cell Counting Kit-8; COPD, chronic obstructive pulmonary disease; CSE, cigarette smoke extract; DMEM, Dulbecco’s modified Eagle medium; MHC I, major histocompatibility complex class I; MMP, mitochondrial membrane potential; mtROS, mitochondrial reactive oxygen species; OCR, oxygen consumption rate; PBS, phosphate-buffered saline; ROS, reactive oxygen species; SA-β-gal, senescence-associated β-galactosidase.
Data Sharing Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval
This study was conducted exclusively in the BEAS-2B human bronchial epithelial cell line and did not involve human participants, human tissue samples, human data, or animals. Therefore, ethics committee approval and informed consent were not required for this study.
Acknowledgments
The authors thank the staff of the Department of Emergency Medicine Clinical Research Center, Beijing Chaoyang Hospital, Capital Medical University, for technical support.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or non-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–13. doi:10.1016/S0140-6736(22)00470-6
2. Celli BR, Wedzicha JA. Update on clinical aspects of chronic obstructive pulmonary disease. N Engl J Med. 2019;381(13):1257–1266. doi:10.1056/NEJMra1900500
3. Schneider JL, Rowe JH, Garcia-de-Alba C, Kim CF, Sharpe AH, Haigis MC. The aging lung: physiology, disease, and immunity. Cell. 2021;184(8):1990–2019. doi:10.1016/j.cell.2021.03.005
4. Cho SJ, Stout-Delgado HW. Aging and lung disease. Annu Rev Physiol. 2020;82:433–459. doi:10.1146/annurev-physiol-021119-034610
5. Easter M, Bollenbecker S, Barnes JW, Krick S. Targeting aging pathways in chronic obstructive pulmonary disease. Int J Mol Sci. 2020;21(18):6924. doi:10.3390/ijms21186924
6. Brandsma CA, de Vries M, Costa R, Woldhuis RR, Königshoff M, Timens W. Lung ageing and COPD: is there a role for ageing in abnormal tissue repair? Eur Respir Rev. 2017;26(146):170073. doi:10.1183/16000617.0073-2017
7. Rutten EP, Gopal P, Wouters EF, et al. Various mechanistic pathways representing the aging process are altered in COPD. Chest. 2016;149(1):53–61. doi:10.1378/chest.15-0645
8. Barnes PJ. Senescence in COPD and its comorbidities. Annu Rev Physiol. 2017;79:517–539. doi:10.1146/annurev-physiol-022516-034314
9. Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol. 2018;314(1):C73–C87. doi:10.1152/ajpcell.00110.2016
10. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: aging, cancer, and injury. Physiol Rev. 2019;99(2):1047–1078. doi:10.1152/physrev.00020.2018
11. Hiemstra PS, McCray PB, Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J. 2015;45(4):1150–1162. doi:10.1183/09031936.00141514
12. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16(1):27–35. doi:10.1038/ni.3045
13. Wu H, Ma H, Wang L, et al. Regulation of lung epithelial cell senescence in smoking-induced COPD/emphysema by microR-125a-5p via Sp1 mediation of SIRT1/HIF-1α. Int J Biol Sci. 2022;18(2):661–674. doi:10.7150/ijbs.65861
14. Subedi S, Guntipally M, Suwal N, et al. Cellular senescence in chronic obstructive pulmonary disease: molecular mechanisms and therapeutic interventions. Ageing Res Rev. 2025;110:102813. doi:10.1016/j.arr.2025.102813
15. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174(8):886–893. doi:10.1164/rccm.200509-1374OC
16. Manevski M, Muthumalage T, Devadoss D, et al. Cellular stress responses and dysfunctional mitochondrial-cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol. 2020;33:101443. doi:10.1016/j.redox.2020.101443
17. Gao N, Wang Y, Zheng CM, et al. β2-microglobulin participates in development of lung emphysema by inducing lung epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol. 2017;312(5):L669–L677. doi:10.1152/ajplung.00516.2016
18. Nyunoya T, Mebratu Y, Contreras A, Delgado M, Chand HS, Tesfaigzi Y. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am J Respir Cell Mol Biol. 2014;50(3):471–482. doi:10.1165/rcmb.2013-0348TR
19. Smith LK, He Y, Park JS, et al. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat Med. 2015;21(8):932–937. doi:10.1038/nm.3898
20. Ke Y, Chen P, Wu C, Wang Q, Zeng K, Liang M. β2-microglobulin and cognitive decline: unraveling the mediating role of the Dunedin pace of aging methylation. Front Aging Neurosci. 2025;17:1505185. doi:10.3389/fnagi.2025.1505185
21. Huang YM, Ma YH, Gao PY, et al. Plasma β2-microglobulin and cerebrospinal fluid biomarkers of Alzheimer disease pathology in cognitively intact older adults: the CABLE study. Alzheimers Res Ther. 2023;15(1):69. doi:10.1186/s13195-023-01217-6
22. Li Y, Zhang X, Li L, et al. Mechanical stresses induce paracrine β-2 microglobulin from cardiomyocytes to activate cardiac fibroblasts through epidermal growth factor receptor. Clin Sci. 2018;132(16):1855–1874. doi:10.1042/CS20180486
23. Chen F, Liu J, Li FQ, et al. β2-microglobulin exacerbates neuroinflammation, brain damage, and cognitive impairment after stroke in rats. Neural Regen Res. 2023;18(3):603–608. doi:10.4103/1673-5374.350204
24. Wu Z, Yan M, Zhang M, et al. β2-microglobulin as a biomarker of pulmonary fibrosis development in COPD patients. Aging. 2020;13(1):1251–1263. doi:10.18632/aging.202266
25. Wang HT, Gao N, Wang WJ, et al. β2-weiqiudanbai zai feiqizhong fabing guocheng zhong de zuoyong [The role of β2-microglobulin in the pathogenesis of pulmonary emphysema]. Wei Sheng Wu Xue Yu Mian Yi Xue Jin Zhan. 2019;47(2):1–8. Chinese. doi:10.13309/j.cnki.pmi.2019.02.001
26. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21(4):204–224. doi:10.1038/s41580-020-0210-7
27. Xu X, Pang Y, Fan X. Mitochondria in oxidative stress, inflammation and aging: from mechanisms to therapeutic advances. Signal Transduct Target Ther. 2025;10(1):190. doi:10.1038/s41392-025-02253-4
28. Suomalainen A, Nunnari J. Mitochondria at the crossroads of health and disease. Cell. 2024;187(11):2601–2627. doi:10.1016/j.cell.2024.04.037
29. Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest. 2022;132(13):e158447. doi:10.1172/JCI158447
30. Ferrucci L, Guerra F, Bucci C, Marzetti E, Picca A. Mitochondria break free: mitochondria-derived vesicles in aging and associated conditions. Ageing Res Rev. 2024;102:102549. doi:10.1016/j.arr.2024.102549
31. Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–957. doi:10.1172/JCI64125
32. Sanchez-Lopez E, Zhong Z, Stubelius A, et al. Choline uptake and metabolism modulate macrophage IL-1β and IL-18 production. Cell Metab. 2019;29(6):1350–1362.e7. doi:10.1016/j.cmet.2019.03.011
33. Dong T, Chen X, Xu H, et al. Mitochondrial metabolism mediated macrophage polarization in chronic lung diseases. Pharmacol Ther. 2022;239:108208. doi:10.1016/j.pharmthera.2022.108208
34. Kosmider B, Lin CR, Karim L, et al. Mitochondrial dysfunction in human primary alveolar type II cells in emphysema. EBioMedicine. 2019;46:305–316. doi:10.1016/j.ebiom.2019.07.063
35. He Q, Li P, Han L, et al. Revisiting airway epithelial dysfunction and mechanisms in chronic obstructive pulmonary disease: the role of mitochondrial damage. Am J Physiol Lung Cell Mol Physiol. 2024;326(6):L754–L769. doi:10.1152/ajplung.00362.2023
36. Ito A, Hashimoto M, Tanihata J, et al. Involvement of Parkin-mediated mitophagy in the pathogenesis of chronic obstructive pulmonary disease-related sarcopenia. J Cachexia Sarcopenia Muscle. 2022;13(3):1864–1882. doi:10.1002/jcsm.12988
37. Frankenberg Garcia J, Rogers AV, Mak JCW, et al. Mitochondrial transfer regulates bioenergetics in healthy and chronic obstructive pulmonary disease airway smooth muscle. Am J Respir Cell Mol Biol. 2022;67(4):471–481. doi:10.1165/rcmb.2022-0041OC
38. Hoffmann RF, Zarrintan S, Brandenburg SM, et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013;14(1):97. doi:10.1186/1465-9921-14-97
39. Ancer-Rodríguez J, Gopar-Cuevas Y, García-Aguilar K, et al. Cell proliferation and apoptosis—key players in the lung aging process. Int J Mol Sci. 2024;25(14):7867. doi:10.3390/ijms25147867
40. Cruz T, López-Giraldo A, Noell G, et al. Smoking impairs the immunomodulatory capacity of lung-resident mesenchymal stem cells in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2019;61(5):575–583. doi:10.1165/rcmb.2018-0351OC
41. Lu Z, Van Eeckhoutte HP, Liu G, et al. Necroptosis signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2021;204(6):667–681. doi:10.1164/rccm.202009-3442OC
42. Giordano RJ, Lahdenranta J, Zhen L, et al. Targeted induction of lung endothelial cell apoptosis causes emphysema-like changes in the mouse. J Biol Chem. 2008;283(43):29447–29460. doi:10.1074/jbc.M804595200
43. Mortaz E, Braber S, Nazary M, Givi ME, Nijkamp FP, Folkerts G. ATP in the pathogenesis of lung emphysema. Eur J Pharmacol. 2009;619(1–3):92–96. doi:10.1016/j.ejphar.2009.07.022
44. Guan R, Cai Z, Wang J, et al. Hydrogen sulfide attenuates mitochondrial dysfunction-induced cellular senescence and apoptosis in alveolar epithelial cells by upregulating sirtuin 1. Aging. 2019;11(24):11844–11864. doi:10.18632/aging.102454
45. Li S, Andoh T, Zhang Q, Uta D, Kuraishi Y. β2-microglobulin, interleukin-31, and arachidonic acid metabolites (leukotriene B4 and thromboxane A2) are involved in chronic renal failure-associated itch-associated responses in mice. Eur J Pharmacol. 2019;847:19–25. doi:10.1016/j.ejphar.2019.01.017
46. Chai D, Li K, Du H, et al. β2-microglobulin has a different regulatory molecular mechanism between ER+ and ER- breast cancer with HER2-. BMC Cancer. 2019;19(1):223. doi:10.1186/s12885-019-5410-1
47. Na HY, Park Y, Nam SK, et al. Expression of human leukocyte antigen class I and β2-microglobulin in colorectal cancer and its prognostic impact. Cancer Sci. 2021;112(1):91–100. doi:10.1111/cas.14723
48. Medini H, Cohen T, Mishmar D. Mitochondria are fundamental for the emergence of metazoans: on metabolism, genomic regulation, and the birth of complex organisms. Annu Rev Genet. 2020;54:151–166. doi:10.1146/annurev-genet-021920-105545
49. Piantadosi CA, Suliman HB. Mitochondrial dysfunction in lung pathogenesis. Annu Rev Physiol. 2017;79:495–515. doi:10.1146/annurev-physiol-022516-034322
50. Aghapour M, Remels AHV, Pouwels SD, et al. Mitochondria: at the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2020;318(1):L149–L164. doi:10.1152/ajplung.00329.2019
51. Hara H, Araya J, Ito S, et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol. 2013;305(10):L737–L746. doi:10.1152/ajplung.00146.2013
52. Cloonan SM, Choi AM. Mitochondria in lung disease. J Clin Invest. 2016;126(3):809–820. doi:10.1172/JCI81113
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.