Back to Journals » Drug Design, Development and Therapy » Volume 9
2-acetylphenol analogs as potent reversible monoamine oxidase inhibitors
Authors Legoabe L, Petzer A, Petzer J
Received 7 April 2015
Accepted for publication 19 May 2015
Published 15 July 2015 Volume 2015:9 Pages 3635—3644
DOI https://doi.org/10.2147/DDDT.S86225
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Lesetja J Legoabe,1 Anél Petzer,1 Jacobus P Petzer1,2
1Centre of Excellence for Pharmaceutical Sciences, 2Department of Pharmaceutical Chemistry, School of Pharmacy, North-West University, Potchefstroom, South Africa
Abstract: Based on a previous report that substituted 2-acetylphenols may be promising leads for the design of novel monoamine oxidase (MAO) inhibitors, a series of C5-substituted 2-acetylphenol analogs (15) and related compounds (two) were synthesized and evaluated as inhibitors of human MAO-A and MAO-B. Generally, the study compounds exhibited inhibitory activities against both MAO-A and MAO-B, with selectivity for the B isoform. Among the compounds evaluated, seven compounds exhibited IC50 values <0.01 µM for MAO-B inhibition, with the most selective compound being 17,000-fold selective for MAO-B over the MAO-A isoform. Analyses of the structure–activity relationships for MAO inhibition show that substitution on the C5 position of the 2-acetylphenol moiety is a requirement for MAO-B inhibition, and the benzyloxy substituent is particularly favorable in this regard. This study concludes that C5-substituted 2-acetylphenol analogs are potent and selective MAO-B inhibitors, appropriate for the design of therapies for neurodegenerative disorders such as Parkinson’s disease.
Keywords: monoamine oxidase, MAO, inhibition, 2-acetylphenol, structure–activity relationship
Introduction
Parkinson’s disease is a degenerative disorder of the central nervous system.1 One of the main pathological characteristics of Parkinson’s disease is the destruction of the dopamine-containing neurons of the nigrostriatal pathway, which results in the deficiency of dopamine at the nerve terminals in the corpus striatum.1 The cornerstone of Parkinson’s disease therapy is the replacement of the lost dopamine with its direct metabolic precursor, L-3,4-dihydroxyphenylalanine (L-dopa).2 To enhance the therapeutic efficacy of L-dopa and to reduce the occurrence of L-dopa-associated side effects, this drug is frequently administered in combination with inhibitors of the enzymes, peripheral dopa decarboxylase, and catechol-O-methyl transferase.3,4 The inhibition of these enzymes prevents the peripheral metabolism of L-dopa and allows for the effective L-dopa dose to be reduced.5 A second strategy to increase the efficacy of L-dopa is to inhibit the metabolism of dopamine with monoamine oxidase (MAO) type B inhibitors.6 In the central nervous system, MAO-B is a major catabolic enzyme of dopamine, and inhibition of this enzyme conserves the central dopamine supply.7 L-dopa is thus frequently also combined with MAO-B inhibitors with the aim of further enhancing central dopamine levels.8–10 In early Parkinson’s disease, monotherapy with MAO-B inhibitors may delay the necessity for l-dopa therapy.11
MAO consists of two distinct isoforms, MAO-A and MAO-B. Although both isoforms metabolize dopamine in the central nervous system, only MAO-B inhibitors are used in Parkinson’s disease, in part because MAO-B activity is higher than that of MAO-A in the affected region, the basal ganglia.6 MAO-A inhibitors have been used in the clinic as antidepressants,12,13 but their use is restricted by the occurrence of a potentially fatal hypertensive response when taken with tyramine-containing foods.14 This adverse effect is, however, mostly caused by irreversible MAO-A inhibitors, and the newer generation reversible inhibitors appear to be safe in this regard.15,16 Both reversible and irreversible MAO-B inhibitors, on the other hand, have excellent safety profiles and are not associated with a hypertensive response.17 Currently, two irreversible MAO-B inhibitors, selegiline [(R)-deprenyl] and rasagiline, are employed in the clinic for the treatment of Parkinson’s disease. Based on the patent literature, many research groups are actively developing MAO inhibitors, and many of them intended for the treatment of Parkinson’s disease.18
Since MAO-B inhibitors are considered useful agents for the management of Parkinson’s disease, the goal of this study was to discover novel MAO inhibitors. Such compounds should possess selectivity for the MAO-B isoform. In a recent study, we have shown that substituted 2-acetylphenols are promising leads for the design of MAO-B inhibitors.19 In the reported study, a limited series of six 2-acetylphenol analogs (1a–f) were synthesized, and it was found that substitution on the C5 position (eg, 1e and 1f) was particularly favorable for high potency MAO-B inhibition (Table 1). Compounds 1e and 1f thus represent exceptionally potent MAO-B inhibitors with IC50 values of 0.004 μM and 0.011 μM, respectively. Substitution on C4 (1c, 1d) and C3 (1a, 1b), on the other hand, yielded lower potency MAO-B inhibitors. The 2-acetylphenols were also selective for MAO-B over the MAO-A isoform, with 1e (selectivity index [SI] =3,950) possessing the highest selectivity of the series. Based on these results, the current study examines the MAO inhibitory properties of a series of 15 C5-substituted 2-acetylphenol analogs (2a–o) (Table 2). In the present study, the 2-acetylphenol analogs were first substituted on the C5 position with the benzyloxy, phenylethoxy, and phenylpropoxy moieties to yield compounds 2a–c. The MAO inhibitory properties of the benzyloxy-substituted 2-acetylphenol analog (2a) were further explored by substitution on the benzyloxy phenyl ring with halogens (F, Cl, I) and alkyl groups (CH3, CN, CF3) to yield 2d–m. The selection of the benzyloxy-substituted homologues for further investigation was based on the reports that the benzyloxy moiety is particularly suited for MAO inhibition by chromones19,20 and 3,4-dihydro-2(1H)-quinolinones.21 Literature also emphasizes the suitability of the benzyloxy substituent for MAO inhibition by coumarin derivatives.22,23 Furthermore, for these classes of compounds, halogen and alkyl substitution on the benzyloxy ring significantly enhances MAO inhibitory potency. Lastly, 2-acetylphenol analogs with the nonaromatic n-butoxy (2n) and n-heptyloxy (2o) substituents were also included in this study. The effect on MAO inhibition of modification of the 2-acetylphenol nucleus was investigated with acetophenone derivative 3a and 2-propanoylphenol derivative 3b (Table 3).
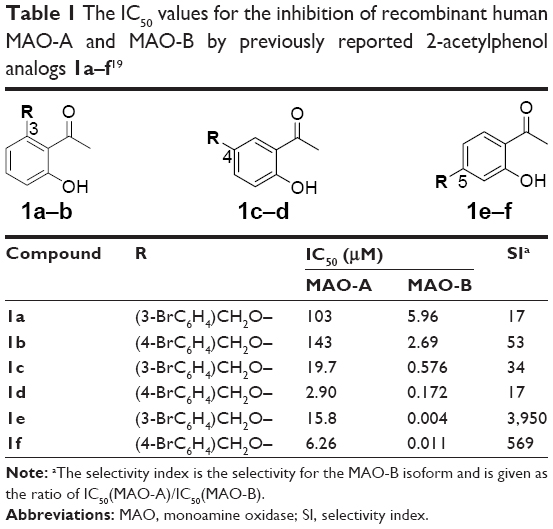 | Table 1 The IC50 values for the inhibition of recombinant human MAO-A and MAO-B by previously reported 2-acetylphenol analogs 1a–f19 |
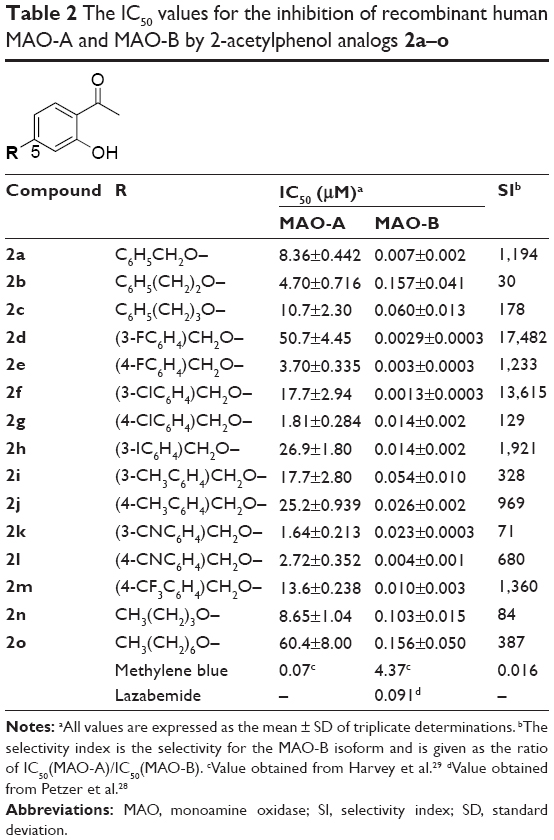 | Table 2 The IC50 values for the inhibition of recombinant human MAO-A and MAO-B by 2-acetylphenol analogs 2a–o |
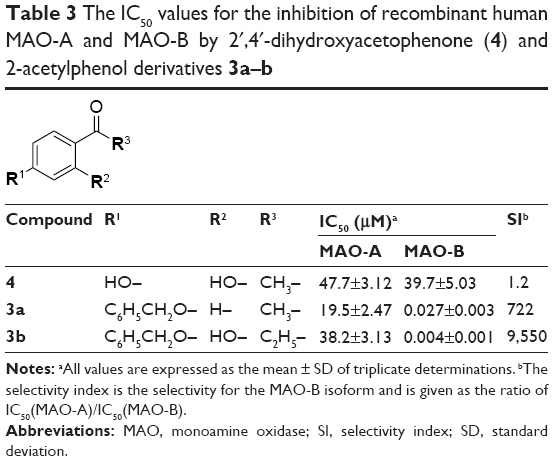 | Table 3 The IC50 values for the inhibition of recombinant human MAO-A and MAO-B by 2′,4′-dihydroxyacetophenone (4) and 2-acetylphenol derivatives 3a–b |
Materials and methods
General
Unless otherwise specified, all starting materials and reagents were obtained from Sigma-Aldrich (St Louis, MO, USA) and were used without further purification. Proton (1H) and carbon (13C) NMR spectra were recorded on a Bruker Avance III 600 spectrometer (Karlsruhe, Germany) at frequencies of 600 MHz and 151 MHz, respectively. CDCl3 served as a NMR solvent, and chemical shifts are reported in parts per million (δ). Spin multiplicities are given as singlet (s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), pentet (p), or multiplet (m). High-resolution mass spectra were recorded on a Bruker micrOTOF-Q II mass spectrometer in atmospheric-pressure chemical ionization mode. Melting points were determined with a Büchi M-545 (Büchi Labortechnik, Flawil, Switzerland) melting point apparatus and are uncorrected. Thin layer chromatography was carried out using silica gel 60 (EMD Millipore, Billerica, MA, USA) with UV254 fluorescent indicator. The mobile phase consisted of ethyl acetate (one part) and petroleum ether (three parts). Fluorescence spectrophotometry was carried out with a Varian Cary Eclipse fluorescence spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Microsomes from insect cells expressing recombinant human MAO-A and MAO-B (5 mg/mL) were obtained from Sigma-Aldrich. Kynuramine.2HBr was obtained from Sigma-Aldrich, and Slide-A-Lyzer dialysis cassettes with a molecular weight cut-off of 10,000 and a sample volume capacity of 0.5–3 mL were obtained from Thermo Fisher Scientific (Waltham, MA, USA).
Chemistry
Procedure for the synthesis of C5-substituted 2-acetylphenol analogs (2a–o and 3a–b)
To a mixture of 2′,4′-dihydroxyacetophenone (4; 1 mmol) and anhydrous K2CO3 (2 mmol) in dry acetone (15 mL), the appropriate alkyl or arylalkyl bromide (5; 1.1 mmol) was added. The mixture was stirred under reflux for 24 hours, cooled to room temperature, and filtered through a pad of Celite. The solvent was removed under reduced pressure, and the residues were recrystallized from ethanol. For the synthesis of 3a and 3b, 4′-hydroxyacetophenone (6) and 2′,4′-dihydroxypropiophenone (7), respectively, were reacted with benzyl bromide under the same conditions as above. Compounds 3a and 3b were purified by recrystallization from ethanol.19
Enzymology
IC50 value determination
For the determination of IC50 values for MAO-A and MAO-B inhibition, the recombinant human enzymes were employed. Kynuramine, a nonselective MAO substrate, served as enzyme substrate.24 The concentration of 4-hydroxyquinoline, generated upon the oxidation of kynuramine by the MAOs, was measured by fluorescence spectrophotometry (λex =310; λem =400 nm). The protocol for these experiments has been reported in detail in a recent publication.25 Employing various inhibitor concentrations (spanning at least 3 orders of magnitude), sigmoidal curves were constructed by graphing residual enzyme activity versus the logarithm of inhibitor concentration. The IC50 values were determined in triplicate from these curves and are expressed as mean ± standard deviation.
Recovery of enzyme activity after dialysis
The reversibility of MAO-B inhibition by 2e was investigated by measuring the recovery of MAO-B activity after mixtures containing the enzyme and test inhibitor were dialyzed. The protocol for these experiments has been reported in detail in recent publications.25,26 All reactions were carried out in triplicate, and the residual enzyme catalytic rates (as percentage of the negative control value) were expressed as mean ± standard deviation.
The construction of Lineweaver–Burk plots and Ki value calculations
The mode of MAO-B inhibition by 2e was investigated by constructing a set of six Lineweaver–Burk plots. The first plot was constructed in the absence of inhibitor while the remaining five plots were constructed in the presence of different concentrations of the test inhibitor. The inhibitor concentrations that were selected were 1/4× IC50, 1/2× IC50, 3/4× IC50, 1× IC50, and 11/4× IC50. Kynuramine was used at concentrations of 15–250 μM. The protocol for these experiments has been reported in detail in recent publications.25,26 A Ki value for the inhibition of MAO-B was estimated from a plot of the slopes of the Lineweaver–Burk plots versus inhibitor concentration, where the x-axis intercept equals -Ki. The Ki value may also be estimated by global (shared) fitting of the inhibition data directly to the Michaelis–Menten equation using the Prism 5 software package (GraphPad, San Diego, CA, USA).
Results and discussion
Chemistry
The C5-substituted 2-acetylphenol analogs, 2a–o, were synthesized in low to good yields (39%–93%) by reacting 2′,4′-dihydroxyacetophenone (4) with the appropriate alkyl or arylalkyl bromide (5) in the presence of K2CO3 in acetone (Figure 1). For the synthesis of 3a and 3b, 4′-hydroxyacetophenone (6) and 2′,4′-dihydroxypropiophenone (7), respectively, were reacted with benzyl bromide under the same conditions as earlier. In each instance, the structures and purities of the target compounds were verified by 1H NMR, 13C NMR, and mass spectrometry as cited in the supplementary materials.
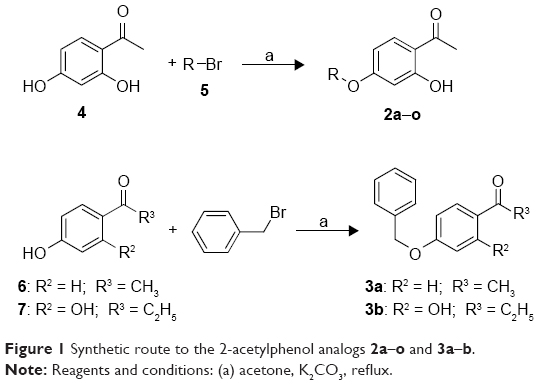 | Figure 1 Synthetic route to the 2-acetylphenol analogs 2a–o and 3a–b. |
Potencies of MAO inhibition
The 2-acetylphenol analogs were evaluated as inhibitors of the recombinant human MAO-A and MAO-B enzymes, and the inhibition potencies were expressed as the corresponding IC50 values.24 To measure the catalytic activities of the MAO enzymes, the mixed MAO-A/B substrate, kynuramine, was used. Kynuramine is oxidized by the MAOs to yield 4-hydroxyquinoline as final product. This metabolite fluoresces in alkaline media and may thus be conveniently quantified by fluorescence spectrophotometry.27 Using the appropriate control reactions, it was determined that none of the 2-acetylphenol analogs investigated here fluoresce under the specific assay conditions and thus do not interfere with the fluorescence measurement of 4-hydroxyquinoline. IC50 values were estimated from sigmoidal plots of the residual MAO activities recorded in the presence of the test inhibitors versus the logarithm of inhibitor concentration.
The human MAO inhibitory properties of the C5-substituted 2-acetylphenol analogs are shown in Tables 2 and 3. As evident from the SI values (SI >30), all of the 2-acetylphenol analogs (2a–o, 3a, b) are selective inhibitors of MAO-B. Among these, seven compounds (of 17) exhibited IC50 values <0.01 μM for the inhibition of MAO-B. These compounds (2a, 2d–f, 2l, m, 3b) may be viewed as exceptionally potent MAO-B inhibitors and possess higher potencies than the reference MAO-B inhibitor, lazabemide (IC50 =0.091 μM).28 With IC50 values in the nanomolar range (0.0013–0.157 μM), all of the 2-acetylphenol analogs may, however, be viewed as potent MAO-B inhibitors. Among the high potency inhibitors (IC50 <0.01 μM), compounds 2d, 2f, and 3b may be highlighted. These compounds exhibit SI values >9,550 and are thus the most selective MAO-B inhibitors of the present study. The finding that C5-substituted 2-acetylphenol analogs are potent and selective MAO-B inhibitors is in accordance to the previous study, which has shown that substitution on the C5 position of 2-acetylphenol (eg, 1e and 1f) yields selective MAO-B inhibitors. The IC50 values recorded for 1e and 1f are in the same range as those of the most potent inhibitors of the present study (Table 1).19
The C5-substituted 2-acetylphenol analogs (2a–o, 3a, b) also inhibited human MAO-A. With IC50 values in the micromolar range (1.64–50.7 μM), these compounds are, relative to their MAO-B inhibition potencies, weak MAO-A inhibitors. Also, compared to the reference MAO-A inhibitor, methylene blue (IC50 =0.07 μM), the 2-acetylphenol analogs are at least 23-fold weaker as MAO-A inhibitors.29 The finding that C5-substituted 2-acetylphenol analogs are relatively weak MAO-A inhibitors is in agreement to the previous study, which has shown that 1e and 1f are weak MAO-A inhibitors with IC50 values in the same range as those of the C5-substituted 2-acetylphenol analogs of the present study (Table 1).19
Structure–activity relationships for MAO inhibition
From the inhibition data, some structure–activity relationships (SARs) may be derived. The 2-acetylphenol analogs with the nonaromatic n-butoxy and n-heptyloxy substituents, compounds 2n (IC50 =0.103 μM) and 2o (IC50 =0.156 μM), respectively, are among the three weakest MAO-B inhibitors of the present study. This shows that these substituents may be less suitable for MAO-B inhibition than, for example, the benzyloxy-derived substituents (eg, 2a, 2d–m), which yield compounds with IC50 values of 0.0013–0.054 μM. It is noteworthy that substitution with the benzyloxy moiety (2a) yields more potent MAO-B inhibition than substitution with the phenylethoxy (2b) and phenylpropoxy (2c) moieties. This suggests that, among these three substituents, the benzyloxy moiety is most suitable for MAO-B inhibition by 2-acetylphenol analogs. Among the benzyloxy-derived compounds (2a, 2d–m), no SARs for the inhibition of MAO-B are apparent, and all compounds are potent MAO-B inhibitors (IC50 <0.054 μM). The effect of modification of the 2-acetylphenol nucleus was investigated with acetophenone derivative 3a and 2-propanoylphenol derivative 3b. Removal of the phenolic hydroxyl to yield 3a (IC50 =0.027 μM) resulted is a small loss of MAO-B inhibition activity when compared to its 2-acetylphenol homologue 2a (IC50 =0.007 μM), while the 2-propanoylphenol derivative 3b (IC50 =0.004 μM) was approximately equipotent to 2a. It may thus be concluded that small structural modifications to the 2-acetylphenol moiety are well tolerated and does not necessarily abolish MAO-B inhibition activity. Interestingly, 3a and 3b are significantly weaker MAO-A inhibitors than the corresponding 2-acetylphenol homologue 2a, which suggests that removal of the phenolic hydroxyl (to yield 3a) and replacing the acetyl group with propanoyl (to yield 3b) may improve selectivity of inhibition for MAO-B. This is exemplified by 3b, an equipotent MAO-B inhibitor to 2a, but with improved selectivity. Lastly, it is interesting to note that among the three most potent MAO-A inhibitors, two are the nitrile-containing compounds 2k (IC50 =1.64 μM) and 2l (IC50 =2.72 μM). This suggests that the nitrile group enhances MAO-A inhibition, an observation for which the molecular basis is not readily apparent.
As mentioned earlier, a previous study has shown that substitution on the C5 position of 2-acetylphenol (eg, 1e and 1f) is particularly favorable for high potency MAO-B inhibition and yields higher potency inhibitors than substitution on C4 (1c, 1d) and C3 (1a, 1b).19 To further evaluate the importance of the C5 substituent for the inhibition of MAO-B, 2′,4′-dihydroxyacetophenone (4) was evaluated as an inhibitor of the human MAOs. The results document that 4 is a weak MAO-B inhibitor with an IC50 value of 39.7 μM. Compound 4 is thus 250-fold weaker as a MAO-B inhibitor than the weakest inhibitor of the present series, compound 2b (IC50 =0.157 μM). This demonstrates that an appropriate C5 substituent is necessary for high potency MAO-B inhibition by 2-acetylphenol analogs. Interestingly, 4 (IC50 =47.7 μM) is a superior MAO-A inhibitor than some of the C5-substituted 2-acetylphenol analogs (eg, 2d and 2o).
Reversibility of MAO-B inhibition
Employing dialysis, the present study also investigated the reversibility of MAO-B inhibition by one representative inhibitor, compound 2e (IC50 =0.003 μM). The reversibility of inhibition was examined by measuring the recovery of enzyme activity after dialysis of enzyme-inhibitor mixtures.26 Since none of the 2-acetylphenol analogs were potent MAO-A inhibitors, only the reversibility of MAO-B inhibition was further investigated. MAO-B and 2e, at an inhibitor concentration of 4× IC50, was incubated for 15 minutes, dialyzed for 24 hours, and the residual enzyme activity was subsequently measured. The results are given in Figure 2, which show that the MAO-B inhibition by 2e is only partially reversed after 24 hours of dialysis, with the catalytic activity recovering to 52% of the negative control value (activity in the absence of inhibitor). For reversible inhibition, the enzyme activity is, however, expected to recover to 100% after dialysis. The enzyme activity of undialyzed mixtures of MAO-B and 2e is 30% of the control value. As positive control, the irreversible inhibitor, (R)-deprenyl, was similarly incubated with MAO-B and dialyzed. As expected for irreversible inhibition, enzyme activity is not recovered, with only 1.6% activity remaining. While the molecular basis for the observation that MAO-B inhibition is only partially reversed by dialysis, is not known, high potency MAO-B inhibitors such as 2e may act by tight binding. Potential tight binding of inhibitors to MAO-B has been reported on a number of occasions,30–33 and partial recovery of MAO-B activity following dialysis of enzyme-inhibitor mixtures has previously been reported.25 Further investigation is necessary to clarify this point.
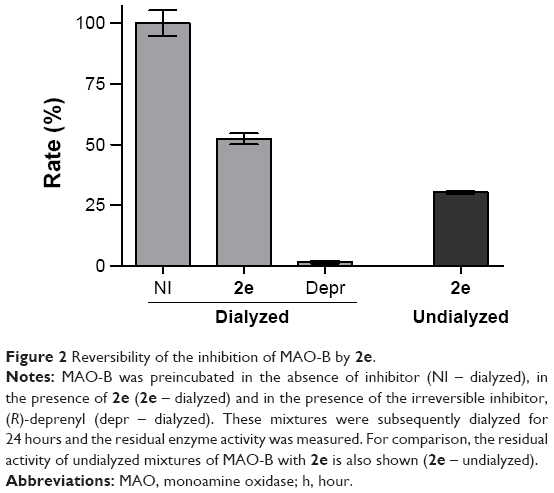 | Figure 2 Reversibility of the inhibition of MAO-B by 2e. |
Competitive inhibition
To gain further insight into the mode of MAO-B inhibition by 2-acetylphenols, a set of Lineweaver–Burk graphs were constructed for the inhibition of MAO-B by the representative inhibitor, 2e. The set consisted of six graphs, each constructed by measuring MAO-B catalytic rate at eight different kynuramine concentrations (15–250 μM). The first Lineweaver–Burk graph was constructed in the absence of inhibitor, while the remaining five graphs were constructed in the presence of different concentrations of 2e. As shown in Figure 3 the set of Lineweaver–Burk graphs is indicative of competitive inhibition since the lines are linear and intersect on the y axis. This suggests that 2e is a competitive inhibitor of human MAO-B. From a replot of the slopes of the Lineweaver–Burk plots versus the concentration of 2e, a Ki value of 0.0038 μM is estimated for the inhibition of MAO-B. Global (shared) fitting of the inhibition data directly to the Michaelis–Menten equation yielded a similar Ki value of 0.0039±0.00035 μM (r2=0.99).
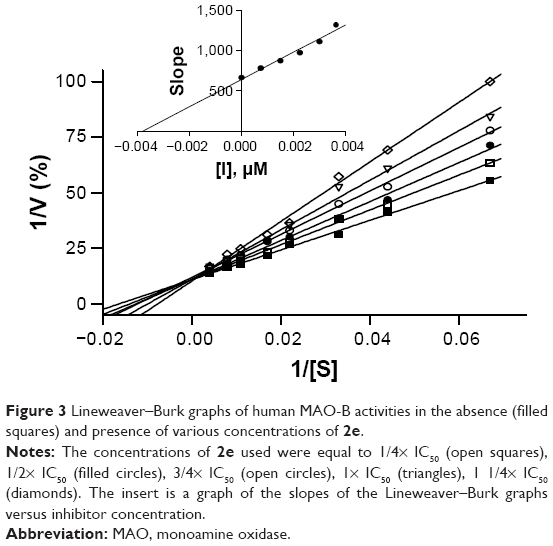 | Figure 3 Lineweaver–Burk graphs of human MAO-B activities in the absence (filled squares) and presence of various concentrations of 2e. |
Conclusion
In conclusion, this study shows that C5-substituted 2-acetylphenol analogs are highly potent MAO-B inhibitors with seven compounds (of 17) exhibiting IC50 values <0.01 μM. The 2-acetylphenol analogs also are selective for the MAO-B isoform as exemplified by compounds 2d, 2f, and 3b, which possess SI values >9,550. With relatively weak MAO-A inhibitory potencies (IC50 >17.7 μM), these three compounds may be suitable candidates for the development of MAO-B selective inhibitors for Parkinson’s disease. Such compounds are unlikely to provoke tyramine induced adverse effects as a result of MAO-A inhibition. Furthermore, the possibility that high potency 2-acetylphenol analogs bind tightly to MAO-B is not of concern since clinically used irreversible MAO-B inhibitors are reported to possess excellent safety profiles.17 Further examination of the physicochemical, biochemical, and pharmacokinetic properties is necessary to determine if promising 2-acetylphenols may be acceptable in vivo drugs.
Acknowledgments
The NMR and MS spectra were recorded by André Joubert and Johan Jordaan of the SASOL Centre for Chemistry, North-West University. This work is based on the research supported in part by the Medical Research Council and National Research Foundation of South Africa (Grant specific unique reference numbers 85642, 80647, and 80637). The Grantholders acknowledge that opinions, findings and conclusions, or recommendations expressed in any publication generated by the NRF supported research are that of the authors and that the NRF accepts no liability whatsoever in this regard.
Disclosure
The authors report no conflicts of interest in this work.
References
Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology. 2009;72(21 suppl 4):S1–S136. | ||
Carlsson A. Treatment of Parkinson’s with l-dopa. The early discovery phase, and a comment on current problems. J Neural Transm. 2002;109(5–6):777–787. | ||
Pedrosa DJ, Timmermann L. Review: management of Parkinson’s disease. Neuropsychiatr Dis Treat. 2013;9:321–340. | ||
Talati R, Baker WL, Patel AA, Reinhart K, Coleman CI. Adding a dopamine agonist to preexisting levodopa therapy vs levodopa therapy alone in advanced Parkinson’s disease: a meta analysis. Int J Clin Pract. 2009;63(4):613–623. | ||
Fahn S, Oakes D, Shoulson I, et al; Parkinson Study Group. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351(24):2498–2508. | ||
Youdim MB, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci. 2006;7(4):295–309. | ||
Youdim MB, Bakhle YS. Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness. Br J Pharmacol. 2006;147(suppl 1):S287–S296. | ||
Shoulson I, Oakes D, Fahn S, et al; Parkinson Study Group. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: a randomized placebo-controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann Neurol. 2002;51(5):604–612. | ||
Fernandez HH, Chen JJ. Monoamine oxidase-B inhibition in the treatment of Parkinson’s disease. Pharmacotherapy. 2007;27(12 pt 2):174S–185S. | ||
Finberg JP, Wang J, Bankiewicz K, Harvey-White J, Kopin IJ, Goldstein DS. Increased striatal dopamine production from L-dopa following selective inhibition of monoamine oxidase B by R(+)-N-propargyl-1-aminoindan (rasagiline) in the monkey. J Neural Transm Suppl. 1998;52:279–285. | ||
Pålhagen S, Heinonen EH, Hägglund J, et al. Selegiline delays the onset of disability in de novo parkinsonian patients. Swedish Parkinson Study Group. Neurology. 1998;51(2):520–525. | ||
Schwartz TL. A neuroscientific update on monoamine oxidase and its inhibitors. CNS Spectr. 2013;18(suppl 1):25–32. | ||
Lum CT, Stahl SM. Opportunities for reversible inhibitors of monoamine oxidase-A (RIMAs) in the treatment of depression. CNS Spectr. 2012;17(3):107–120. | ||
Da Prada M, Zürcher G, Wüthrich I, Haefely WE. On tyramine, food, beverages and the reversible MAO inhibitor moclobemide. J Neural Transm Suppl. 1988;26:31–56. | ||
Bonnet U. Moclobemide: therapeutic use and clinical studies. CNS Drug Rev. 2003;9(1):97–140. | ||
Provost JC, Funck-Brentano C, Rovei V, D’Estanque J, Ego D, Jaillon P. Pharmacokinetic and pharmacodynamic interaction between toloxatone, a new reversible monoamine oxidase-A inhibitor, and oral tyramine in healthy subjects. Clin Pharmacol Ther. 1992;52(4):384–393. | ||
Pae CU, Bodkin JA, Portland KB, Thase ME, Patkar AA. Safety of selegiline transdermal system in clinical practice: analysis of adverse events from postmarketing exposures. J Clin Psychiatry. 2012;73(5):661–668. | ||
Carradori S, Petzer JP. Novel monoamine oxidase inhibitors: a patent review (2012–2014). Expert Opin Ther Pat. 2015;25(1):91–110. | ||
Legoabe LJ, Petzer A, Petzer JP. Selected chromone derivatives as inhibitors of monoamine oxidase. Bioorg Med Chem Lett. 2012;22(17):5480–5484. | ||
Legoabe LJ, Petzer A, Petzer JP. Inhibition of monoamine oxidase by selected C6-substituted chromone derivatives. Eur J Med Chem. 2012;49:343–353. | ||
Meiring L, Petzer JP, Petzer A. Inhibition of monoamine oxidase by 3,4-dihydro-2(1H)-quinolinone derivatives. Bioorg Med Chem Lett. 2013;23(20):5498–5502. | ||
Secci D, Carradori S, Bolasco A, et al. Synthesis and selective human monoamine oxidase inhibition of 3-carbonyl, 3-acyl, and 3-carboxyhydrazido coumarin derivatives. Eur J Med Chem. 2011;46(10):4846–4852. | ||
Chimenti F, Secci D, Bolasco A, et al. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J Med Chem. 2009;52(7):1935–1942. | ||
Novaroli L, Reist M, Favre E, Carotti A, Catto M, Carrupt PA. Human recombinant monoamine oxidase B as reliable and efficient enzyme source for inhibitor screening. Bioorg Med Chem. 2005;13(22):6212–6217. | ||
Legoabe LJ, Petzer A, Petzer JP. α-Tetralone derivatives as inhibitors of monoamine oxidase. Bioorg Med Chem Lett. 2014;24(12):2758–2763. | ||
Petzer A, Pienaar A, Petzer JP. The interactions of caffeine with monoamine oxidase. Life Sci. 2013;93(7):283–287. | ||
Strydom B, Malan SF, Castagnoli N Jr, Bergh JJ, Petzer JP. Inhibition of monoamine oxidase by 8-benzyloxycaffeine analogues. Bioorg Med Chem. 2010;18(3):1018–1028. | ||
Petzer A, Pienaar A, Petzer JP. The inhibition of monoamine oxidase by esomeprazole. Drug Res (Stuttg). 2013;63(9):462–467. | ||
Harvey BH, Duvenhage I, Viljoen F, et al. Role of monoamine oxidase, nitric oxide synthase and regional brain monoamines in the antidepressant-like effects of methylene blue and selected structural analogues. Biochem Pharmacol. 2010;80(10):1580–1591. | ||
Gaspar A, Silva T, Yáñez M, et al. Chromone, a privileged scaffold for the development of monoamine oxidase inhibitors. J Med Chem. 2011;54(14):5165–5173. | ||
Gaspar A, Reis J, Fonseca A, et al. Chromone 3-phenylcarboxamides as potent and selective MAO-B inhibitors. Bioorg Med Chem Lett. 2011;21(2):707–709. | ||
Krueger MJ, Mazouz F, Ramsay RR, Milcent R, Singer TP. Dramatic species differences in the susceptibility of monoamine oxidase B to a group of powerful inhibitors. Biochem Biophys Res Commun. 1995;206(2):556–562. | ||
Mazouz F, Gueddari S, Burstein C, Mansuy D, Milcent R. 5-[4-(Benzyloxy)phenyl]-1,3,4-oxadiazol-2(3H)-one derivatives and related analogues: new reversible, highly potent, and selective monamine oxidase type B inhibitors. J Med Chem. 1993;36(9):1157–1167. |
Supplementary materials
2-Acetyl-5-benzyloxyphenol (2a)
The title compound (clear crystals) was prepared in a yield of 93%: mp 103.1°C–103.4°C (ethanol), lit. mp 101°C–103°C.1 1H NMR (600 MHz, CDCl3) δ 12.71 (s, 1H), 7.62 (d, J =8.7 Hz, 1H), 7.42–7.35 (m, 4H), 7.24 (s, 1H), 6.52–6.47 (m, 2H), 5.08 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.55, 165.13, 135.81, 132.31, 128.66, 128.27, 127.49, 114.03, 108.09, 101.82, 101.81, 70.16, 26.20; APCI-HRMS m/z: calcd for C15H15O3 (MH+), 243.1016, found 243.1001.
2-Acetyl-5-(2-phenylethoxy)phenol (2b)
The title compound (white crystals) was prepared in a yield of 51%: mp 71.0°C–72.3°C (ethanol), lit. mp 69°C.2 1H NMR (600 MHz, CDCl3) δ 12.71 (s, 1H), 7.60 (d, J =8.8 Hz, 1H), 7.31 (t, J =7.5 Hz, 2H), 7.28–7.22 (m, 3H), 6.44–6.38 (m, 2H), 4.19 (t, J =7.0 Hz, 2H), 3.09 (t, J =7.0 Hz, 2H), 2.53 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.50, 165.26, 165.15, 137.63, 132.27, 128.92, 128.53, 126.64, 107.85, 101.40, 101.39, 68.84, 35.40, 26.18; APCI-HRMS m/z: calcd for C16H17O3 (MH+), 257.1172, found 257.1153.
2-Acetyl-5-(3-phenylpropoxy)phenol (2c)
The title compound (clear flakes) was prepared in a yield of 61%: mp 76.4°C–78.1°C (ethanol), lit. mp 75°C–77°C.2 1H NMR (600 MHz, CDCl3) δ 12.73 (s, 1H), 7.61 (d, J =8.9 Hz, 1H), 7.31–7.25 (m, 2H), 7.19 (d, J =7.8 Hz, 3H), 6.43 (dd, J =8.9, 2.5 Hz, 1H), 6.37 (d, J =2.5 Hz, 1H), 3.97 (t, J =6.3 Hz, 2H), 2.82–2.76 (m, 2H), 2.54 (s, 3H), 2.14–2.07 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 202.49, 165.54, 165.18, 141.05, 132.25, 128.45, 126.03, 113.79, 107.92, 101.30, 101.29, 67.19, 31.97, 30.45, 26.18; APCI-HRMS m/z: calcd for C17H19O3 (MH+), 271.1329, found 271.1326.
2-Acetyl-5-(3-fluorobenzyloxy)phenol (2d)
The title compound (clear crystals) was prepared in a yield of 52%: mp 108.5°C–112.3°C (ethanol), lit. mp 108°C–109°C.3 1H NMR (600 MHz, CDCl3) δ 12.72 (s, 1H), 7.63 (d, J =8.9 Hz, 1H), 7.34 (q, J =7.7 Hz, 1H), 7.14 (dd, J =26.5, 8.5 Hz, 2H), 7.01 (t, J =9.1 Hz, 1H), 6.52–6.44 (m, 2H), 5.07 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.62, 165.10, 164.75, 163.75, 162.12, 138.41, 138.36, 132.39, 130.28, 130.23, 122.73, 122.71, 115.21, 115.07, 114.29, 114.19, 114.15, 108.02, 101.83, 101.81, 69.27, 69.25, 26.24; APCI-HRMS m/z: calcd for C15H14FO3 (MH+), 261.0921, found 261.0942.
2-Acetyl-5-(4-fluorobenzyloxy)phenol (2e)
The title compound (clear crystals) was prepared in a yield of 53%: mp 112.4°C–113.9°C (ethanol). 1H NMR (600 MHz, CDCl3) δ 12.72 (s, 1H), 7.62 (d, J =8.8 Hz, 1H), 7.37 (dd, J =8.4, 5.4 Hz, 2H), 7.06 (t, J =8.6 Hz, 2H), 6.51–6.45 (m, 2H), 5.03 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.60, 165.13, 164.92, 163.45, 161.81, 132.36, 131.60, 131.58, 129.45, 129.40, 115.70, 115.56, 114.12, 108.07, 101.78, 101.77, 69.49, 26.24; APCI-HRMS m/z: calcd for C15H14FO3 (MH+), 261.0921, found 261.0932.
2-Acetyl-5-(3-chlorobenzyloxy)phenol (2f)
The title compound (off-white crystals) was prepared in a yield of 67%: mp 127.1°C–129.1°C (ethanol), lit. mp 127°C–128°C.3 1H NMR (600 MHz, CDCl3) δ 12.72 (s, 1H), 7.63 (d, J =8.9 Hz, 1H), 7.40 (s, 1H), 7.32–7.22 (m, 3H), 6.49 (dd, J =8.9 Hz, 1H), 6.45 (d, J =2.5 Hz, 1H), 5.04 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.63, 165.11, 164.74, 137.88, 134.61, 132.40, 129.96, 128.41, 127.41, 125.35, 114.22, 108.02, 101.82, 69.24, 26.25; APCI-HRMS m/z: calcd for C15H14ClO3 (MH+), 277.0626, found 277.0619.
2-Acetyl-5-(4-chlorobenzyloxy)phenol (2g)
The title compound (off-white crystals) was prepared in a yield of 62%: mp 102.2°C–102.5°C (ethanol). 1H NMR (600 MHz, CDCl3) δ 12.71 (s, 1H), 7.62 (d, J =8.9 Hz, 1H), 7.37–7.30 (m, 4H), 6.50–6.43 (m, 2H), 5.03 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.60, 165.10, 164.80, 134.32, 134.11, 132.37, 128.86, 128.79, 114.16, 108.01, 101.80, 69.34, 26.24; APCI-HRMS m/z: calcd for C15H14ClO3 (MH+), 277.0626, found 277.0617.
2-Acetyl-5-(3-iodobenzyloxy)phenol (2h)
The title compound (beige crystals) was prepared in a yield of 58%: mp 143.7°C–144.6°C (ethanol). 1H NMR (600 MHz, CDCl3) δ 12.72 (s, 1H), 7.76 (s, 1H), 7.64 (dd, J =16.8, 8.5 Hz, 2H), 7.35 (d, J =8.2 Hz, 1H), 7.11 (t, J =7.8 Hz, 1H), 6.51–6.43 (m, 2H) 5.00 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.62, 165.10, 164.73, 138.13, 137.30, 136.24, 132.40, 130.35, 126.53, 114.21, 108.01, 101.80, 94.49, 69.06, 26.25; APCI-HRMS m/z: calcd for C15H14IO3 (MH+), 368.9982, found 368.9980.
2-Acetyl-5-(3-methylbenzyloxy)phenol (2i)
The title compound (clear needles) was prepared in a yield of 53%: mp 98.6°C–100.8°C (ethanol). 1H NMR (600 MHz, CDCl3) δ 12.73 (s, 1H), 7.62 (d, J =8.8 Hz, 1H), 7.27 (t, J =7.5 Hz, 1H), 7.23–7.17 (m, 2H), 7.14 (d, J =7.5 Hz, 1H), 6.53–6.47 (m, 2H), 5.03 (s, 2H), 2.54 (s, 3H), 2.36 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.55, 165.20, 165.12, 138.40, 135.69, 132.29, 129.05, 128.56, 128.24, 124.61, 113.99, 108.11, 101.78, 70.25, 26.19, 21.37; APCI-HRMS m/z: calcd for C16H17O3 (MH+), 257.1172, found 257.1157.
2-Acetyl-5-(4-methylbenzyloxy)phenol (2j)
The title compound (beige crystals) was prepared in a yield of 48%: mp 98.0°C–99.5°C (ethanol). 1H NMR (600 MHz, CDCl3) δ 12.72 (s, 1H), 7.61 (d, J =9.2 Hz, 1H), 7.29 (d, J =8.0 Hz, 2H), 7.19 (d, J =7.8 Hz, 2H), 6.51–6.45 (m, 2H), 5.03 (s, 2H), 2.53 (s, 3H), 2.35 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.54, 165.23, 165.13, 138.16, 132.76, 132.28, 129.35, 127.67, 113.97, 108.15, 101.80, 70.15, 26.20, 21.19; APCI-HRMS m/z: calcd for C16H17O3 (MH+), 257.1172, found 257.1159.
2-Acetyl-5-(3-cyanobenzyloxy)phenol (2k)
The title compound (white powder) was prepared in a yield of 65%: mp 125.0°C–125.8°C (ethanol). 1H NMR (600 MHz, CDCl3) δ 12.71 (s, 1H), 7.71 (s, 1H), 7.67–7.59 (m, 3H), 7.50 (t, J =7.8 Hz, 1H), 6.50 (dd, J =8.9, 2.5 Hz, 1H), 6.44 (d, J =2.4 Hz, 1H), 5.10 (s, 2H), 2.55 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.67, 165.09, 164.35, 137.51, 132.50, 131.83, 131.43, 130.65, 129.50, 118.44, 114.39, 112.89, 107.91, 101.78, 68.70, 26.27; APCI-HRMS m/z: calcd for C16H14NO3 (MH+), 268.0968, found 268.0971.
2-Acetyl-5-(4-cyanobenzyloxy)phenol (2l)
The title compound (white powder) was prepared in a yield of 73%: mp 145.9°C–147.6°C (ethanol), lit. mp 147°C–148°C.4 1H NMR (600 MHz, CDCl3) δ 12.70 (s, 1H), 7.66 (d, J =8.2 Hz, 2H), 7.64 (d, J =8.9 Hz, 1H), 7.51 (d, J =8.2 Hz, 2H), 6.49 (dd, J =8.9, 2.5 Hz, 1H), 6.43 (d, J =2.5 Hz, 1H), 5.13 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.65, 165.06, 164.34, 141.23, 132.48, 132.45, 127.53, 118.48, 114.38, 112.01, 107.83, 101.85, 68.91, 26.25; APCI-HRMS m/z: calcd for C16H14NO3 (MH+), 268.0968, found 268.0982.
2-Acetyl-5-[4-(trifluoromethyl)benzyloxy]phenol (2m)
The title compound (clear needles) was prepared in a yield of 39%: mp 85.6°C–87.0°C (ethanol). 1H NMR (600 MHz, CDCl3) δ 12.70 (s, 1H), 7.66 (d, J =8.2 Hz, 2H), 7.64 (d, J =8.9 Hz, 1H), 7.51 (d, J =8.2 Hz, 2H), 6.49 (dd, J =8.9, 2.5 Hz, 1H), 6.43 (d, J =2.5 Hz, 1H), 5.13 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.65, 165.11, 164.63, 139.89, 132.45, 127.39, 125.64 (q), 114.30, 107.94, 101.87, 69.19, 26.24; APCI-HRMS m/z: calcd for C16H14F3O3 (MH+), 311.0890, found 311.0866.
2-Acetyl-5-butoxyphenol (2n)
The title compound (white powder) was prepared in a yield of 47%: mp 42.7°C–43.1°C (ethanol), lit. mp 38°C–40°C.5 1H NMR (600 MHz, CDCl3) δ 12.72 (s, 1H), 7.59 (d, J =8.9 Hz, 1H), 6.43–6.36 (m, 2H), 3.97 (t, J =6.5 Hz, 2H), 2.53 (s, 3H), 1.79–1.72 (m, 2H), 1.46 (m, 2H), 0.95 (t, J =7.4 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ 202.45, 165.71, 165.21, 132.21, 113.68, 107.98, 101.22, 68.05, 30.94, 26.16, 19.12, 13.75; APCI-HRMS m/z: calcd for C12H17O3 (MH+), 209.1172, found 209.1152.
2-Acetyl-5-heptyloxyphenol (2o)
The title compound (white powder) was prepared in a yield of 65%: mp 32.8°C–33.8°C (ethanol), lit. mp oil.6 1H NMR (600 MHz, CDCl3) δ 12.73 (s, 1H), 7.60 (d, J =8.9 Hz, 1H), 6.44–6.36 (m, 2H), 3.96 (t, J =6.6 Hz, 2H), 2.53 (s, 3H), 1.79–1.72 (m, 2H), 1.42 (p, J =7.1 Hz, 2H), 1.35–1.26 (m, 6H), 0.87 (t, J =6.9 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ 202.47, 165.73, 165.23, 132.22, 113.70, 108.02, 101.24, 68.40, 31.73, 28.97, 28.94, 26.19, 25.88, 22.58, 14.07; APCI-HRMS m/z: calcd for C15H23O3 (MH+), 251.1642, found 251.1659.
4′-Benzyloxyacetophenone (3a)
The title compound (clear flakes) was prepared in a yield of 55%: mp 93.2°C–94.5°C (ethanol), lit. mp 93°C–94°C.7 1H NMR (600 MHz, CDCl3) δ 7.92 (d, J =8.9 Hz, 2H), 7.44–7.36 (m, 4H), 7.34 (d, J =7.2 Hz, 1H), 6.99 (d, J =8.9 Hz,2H), 5.11 (s, 2H), 2.54 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 196.74, 162.57, 136.13, 130.57, 130.48, 128.67, 128.22, 127.44, 114.50, 70.10, 26.34; APCI-HRMS m/z: calcd for C15H15O2 (MH+), 227.1067, found 227.1063.
2-Propanoyl-5-benzyloxyphenol (3b)
The title compound (clear needles) was prepared in a yield of 62%: mp 112.0°C–113.4°C (ethanol), lit. mp 113°C– 114°C.8 1H NMR (600 MHz, CDCl3) δ 12.81 (s, 1H), 7.65 (d, J =9.6 Hz, 1H), 7.42–7.31 (m, 5H), 6.50 (dd, J =7.0, 2.5 Hz, 2H), 5.07 (s, 2H), 2.93 (q, J =7.3 Hz, 2H), 1.21 (t, J =7.3 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ 205.34, 165.13, 164.87, 135.87, 131.44, 128.67, 128.27, 127.50, 113.46, 108.01, 101.93, 70.15, 31.15, 8.48; APCI-HRMS m/z: calcd for C16H17O3 (MH+), 257.1172, found 257.1170.
References
Yates MH, Koenig TM, Kallman NJ, Ley CP, Mitchell D. An efficient synthesis of a multipotent eicosanoid pathway modulator. Org Process Res Dev. 2009;13(2):268–275. | ||
Buckle DR, Outred DJ, Ross JW, et al. Aryloxyalkyloxy- and aralkyloxy-4-hydroxy-3-nitrocoumarins which inhibit histamine release in the rat and also antagonize the effects of a slow reacting substance of anaphylaxis. J Med Chem. 1979;22(2):158–168. | ||
Pisani L, Catto M, Nicolotti O, et al. Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur J Med Chem. 2013;70:723–739. | ||
Ma YT, Fan HF, Gao YQ, Li H, Zhang AL, Gao JM. Natural products as sources of new fungicides (I): synthesis and antifungal activity of acetophenone derivatives against phytopathogenic fungi. Chem Biol Drug Des. 2013;81(4):545–552. | ||
Arnoldi A, Merlini L, Quadri ML, Bonsignori A, Melloni P, Varasi M. Synthesis and anticonvulsant activity of some benzopyranone imino derivatives of GABA and related compounds. Farmaco. 1991;46(5):629–638. | ||
Tunoori AR, Dutta D, Georg GI. Polymer-bound triphenylphosphine as traceless reagent for mitsunobu reactions in combinatorial chemistry: Synthesis of aryl ethers from phenols and alcohols. Tetrahedron Lett. 1998;39(48):8751–8754. | ||
Zeng J, Tan YJ, Ma J, Leow ML, Tirtorahardjo D, Liu XW. Facile access to cis-2,6-disubstituted tetrahydropyrans by palladium-catalyzed decarboxylative allylation: total syntheses of (±)-centrolobine and (+)-decytospolides A and B. Chemistry. 2014;20(2):405–409. | ||
Bobik A, Holder GM, Ryan AJ. Inhibitors of hepatic mixed function oxidase. 3. Inhibition of hepatic microsomal aniline hydroxylase and aminopyrine demethylase by 2,6- and 2,4-dihydroxyphenyl alkyl ketones and related compounds. J Med Chem. 1977;20(9):1194–1199. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.