")
Back to Journals » Clinical Interventions in Aging » Volume 12
11C-PIB PET imaging reveals that amyloid deposition in cases with early-onset Alzheimer’s disease in the absence of known mutations retains higher levels of PIB in the basal ganglia
Authors Youn YC , Jang JW, Han SH , Kim HR, Seok JW, Byun JS, Park KY, An SSA , Chun IK , Kim SY
Received 20 January 2017
Accepted for publication 16 May 2017
Published 29 June 2017 Volume 2017:12 Pages 1041—1048
DOI https://doi.org/10.2147/CIA.S132884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Young Chul Youn,1 Jae-Won Jang,2 Su-Hyun Han,1 HyeRyoun Kim,3 Ju-Won Seok,4 Jun Soo Byun,5 Kwang-Yeol Park,1 Seong Soo A An,6 In Kook Chun,7 SangYun Kim8
1Department of Neurology, Chung-Ang University College of Medicine, Seoul, 2Department of Neurology, Kangwon National University Hospital, 3Department of Laboratory Medicine, 4Department of Nuclear Medicine, 5Department of Radiology, Chung-Ang University College of Medicine, 6College of Bionano Technology, Gachon Bionano Research Institute, Gachon University, 7Department of Nuclear Medicine, Kangwon National University Hospital, 8Department of Neurology, Seoul National University Bundang Hospital and Seoul National University College of Medicine, Gyeonggi-do, Republic of Korea
Purpose: Early-onset Alzheimer’s disease (EOAD) has a different pathologic burden and clinical features compared with late-onset Alzheimer’s disease (LOAD). We examined the effects of age at onset on the burden and distribution of β-amyloid in patients with EOAD, in whom well-characterized mutations associated with Alzheimer’s disease were absent.
Methods: We genotyped ApoE, APP, PSEN1 and PSEN2 in the patients with Alzheimer’s disease: 9 patients with EOAD (age <65), 11 with LOAD (age >70) and 8 normal controls (NCs), all of whom had undergone 11C-labeled Pittsburgh compound B-positron emission tomography imaging.
Results: Patients with EOAD exhibited higher z scores and larger cluster sizes, and retained higher levels of Pittsburgh compound B in the bilateral thalamus and in some parts of the globus pallidus (P<0.05, false discovery rate).
Conclusion: Distribution of amyloid deposition in EOAD outside the context of genetic mutations topographically showed some differences from that in LOAD.
Keywords: Alzheimer’s disease, early-onset Alzheimer’s disease, amyloid PET, basal ganglia, Pittsburgh compound B, amyloid
Introduction
Early-onset Alzheimer’s disease (EOAD) refers to cases of Alzheimer’s disease (AD) diagnosed before the age of 65 and accounts for 5%–10% of all AD diagnoses.1
Most studies of EOAD have focused on familial AD with mutations, even though such mutations are not common in cases of AD diagnosed before the age of 65. To evaluate the effects of symptom-onset-age alone on Aβ deposition in patients with AD and eliminate any confounding genetic effects, we excluded patients with well-known mutations, including mutations in APP, PSEN1 and PSEN2. We also performed genetic tests, including PRNP, to exclude patients with atypical prion disease mimicking AD from our study.2,3 We then analyzed Pittsburgh compound B (PIB) retention in patients with EOAD, all of whom lacked mutations in these genes, and compared its distribution to that in patients with late-onset Alzheimer’s disease (LOAD).
11C-labeled PIB position emission tomography (11C-PIB PET) scans can be used to image Aβ plaques in the brains of patients with AD.4,5
Methods
Participants
Patients with AD were divided into EOAD and LOAD groups, according to age at onset. The patient age at which clinical symptoms were first manifested was determined by information obtained from the caregiver at each patient’s first visit. Thirty-two patients with EOAD completed the genetic study between May 2010 and June 2013. We recruited 14 patients with LOAD and 9 patients with EOAD, all of whom had no mutations or polymorphisms in APP, PSEN1, PSEN2 and PRNP. Written informed consent was obtained from all patients in this study.
For each patient, history was taken and neuropsychological tests were performed to assess attention, language, visuospatial function, verbal and visual memory and frontal executive function. These tests included the Mini-Mental State Examination, Clinical Dementia Ratings, appropriate laboratory tests and brain magnetic resonance imaging (MRI) scans. All patients fulfilled the criteria for having probable AD, as proposed by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association.6 Ten healthy volunteers with no history of neurologic or psychiatric illnesses were used as controls. Control subjects also underwent neuropsychological tests, laboratory tests and brain MRI scans, and did not show any abnormalities on these neurologic examinations.
Out of the enrolled subjects, 23 subjects with AD and 10 control subjects underwent 11C-PIB PET imaging. Genetic testing was performed on ApoE, APP, PSEN1 and PSEN2 in patients with EOAD and ApoE in patients with LOAD.
Neuroimaging tests
Three-dimensional T1-weighted MRI scans (Philips 3.0T Achieva, Amsterdam, the Netherlands) were performed on all subjects. The scan parameters were: FOV, 220×220 mm; ETL, 113; FA, 8.0; sagittal slice thickness, 1 mm; no gap; TI, 800 ms; TR, 8.1 ms; TE, 3.7 ms. All subjects also underwent a 11C-PIB PET imaging with a stand-alone position emission tomography (PET) scanner system (Allegro®, Philips Healthcare, Amsterdam, the Netherlands). After intravenous bolus injection of 11C-PIB (658.6±122.1 MBq), static PET images were obtained by 20 min static PET emission and short PET transmission (duration <200 s, 137Cs) scans. Static PET emission acquisition was performed 40 min after 11C-PIB injection.
General image processing and voxel-based analysis was performed with the MATLAB (Math Works, Natick, MA, USA)-based program, Statistical Parametric Mapping version 5 (SPM5). SPM was used to compare PIB deposition between one patient in each AD group (EOAD and LOAD) with a normal control (NC), or to compare patients with EOAD to patients with LOAD.
Spatial normalization to a T1 MRI template of the parametric PIB PET images was performed using a coregistered T1 MRI scan of the same subject. Statistical comparisons between groups were performed on a voxel basis using t-statistics. Brain areas with increased PIB retention were mapped below the P-value threshold of 0.05 (false discovery rate correction) for EOAD versus NC, LOAD versus NC and EOAD versus LOAD groups. The Montreal Neurological Institute coordinates of the local maximum of each cluster were converted into Talairach coordinates.
All procedures performed in this study were approved by the Ethics Committees of Seoul National University Bundang Hospital and Chung-Ang University Hospital.
Results
Of the 23 patients with AD who were clinically diagnosed by neurologists according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association Alzheimer’s criteria, 3 subjects with LOAD were PIB-negative according to their global PIB uptake ratio values and 2 of the 10 control subjects were PIB-positive. Therefore, five subjects were excluded. The remaining cohort, therefore, consisted of 20 patients, 9 of whom exhibited EOAD (<65 years of age) without known genetic mutation and 11 exhibited LOAD (>70 years of age), and 8 control subjects. Patient demographics are presented in Table 1.
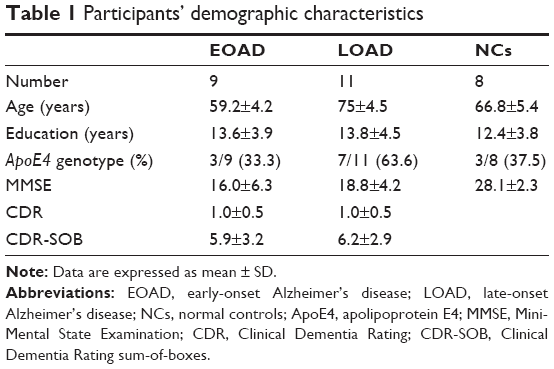 | Table 1 Participants’ demographic characteristics |
Voxel-based morphometric test results, obtained using SPM to compare PIB retention in patients with EOAD and LOAD with NC subjects, are shown in Figure 1. Morphometric test results comparing patients with EOAD to patients with LOAD are also shown in Figure 1. Patients with EOAD and LOAD exhibited diffuse and symmetric PIB retention, respectively, in the frontal, parietal and temporal lobes. Furthermore, patients with EOAD and LOAD exhibited a similar pattern of PIB deposition, which was significantly increased in the dorsolateral frontal cortex, cingulate gyrus, precuneus and bilateral temporoparietal lobes (Figure 1A and B). However, patients with EOAD exhibited higher z scores and larger cluster sizes, especially in the temporal and parietal lobes (Table 2). Furthermore, patients with EOAD had higher PIB retention in the subcortical gray matter, basal ganglia (BG) and thalamus, compared to patients with LOAD. Direct comparison of patients with EOAD and patients with LOAD revealed that patients with EOAD retained a greater amount of PIB in the bilateral thalamus and some regions of the globus pallidus (P<0.05, false discovery rate; Figure 1C; Table 2). However, no cortical regions exhibited statistically significant differences in PIB retention in patients with LOAD compared to patients with EOAD and there were also no significant subcortical regions where LOAD was greater (P<0.001, uncorrected).
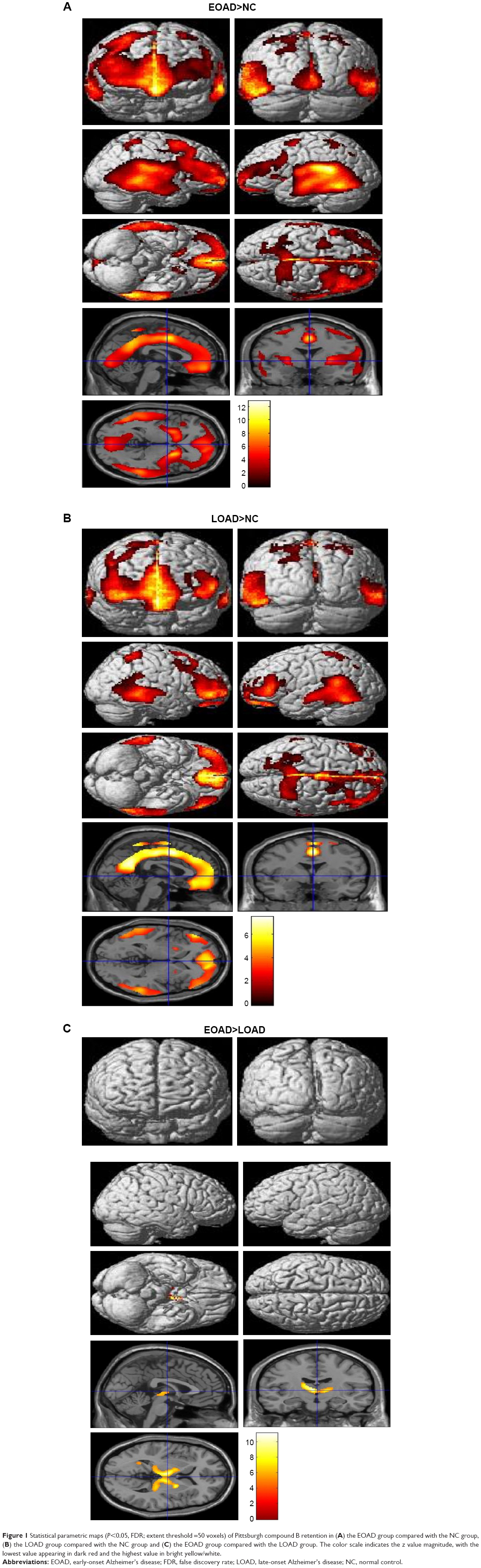 | Figure 1 Statistical parametric maps (P<0.05, FDR; extent threshold =50 voxels) of Pittsburgh compound B retention in (A) the EOAD group compared with the NC group, (B) the LOAD group compared with the NC group and (C) the EOAD group compared with the LOAD group. The color scale indicates the z value magnitude, with the lowest value appearing in dark red and the highest value in bright yellow/white. |
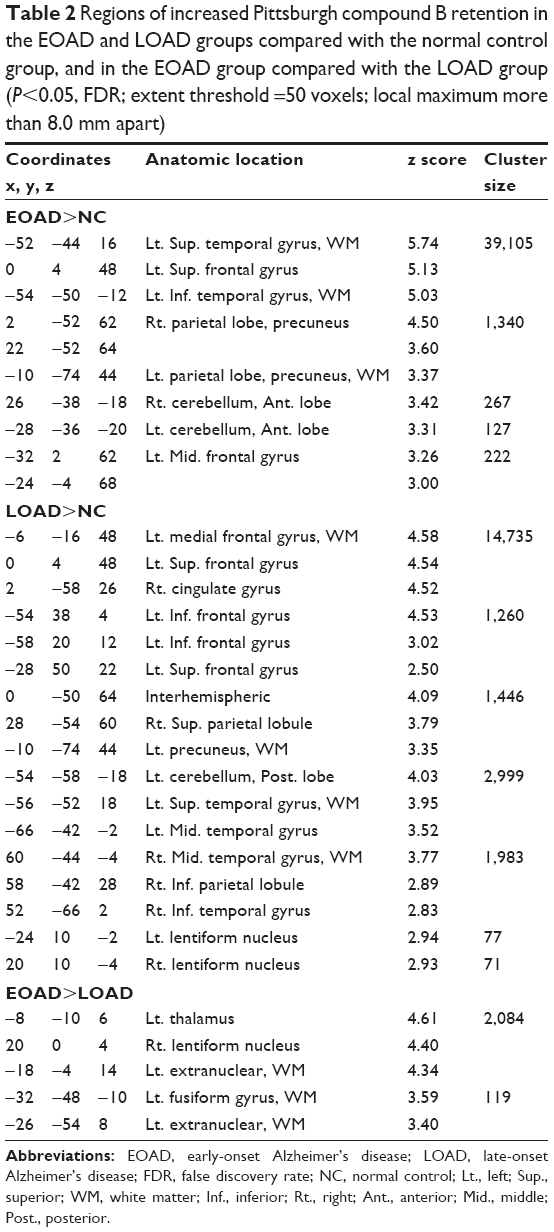 | Table 2 Regions of increased Pittsburgh compound B retention in the EOAD and LOAD groups compared with the normal control group, and in the EOAD group compared with the LOAD group (P<0.05, FDR; extent threshold =50 voxels; local maximum more than 8.0 mm apart) |
Discussion
The major findings of this study are that patients with EOAD and without any of the mutations examined in this study topographically had a similar cortical distribution of Aβ deposition as patients with LOAD, but showed higher z scores and larger cluster sizes, especially in the temporal and parietal lobes. Patients with EOAD had a statistically significant higher level of PIB retention in the bilateral thalamus and some basal ganglia, compared to patients with LOAD. Because of the possibility that the atrophic pattern difference between EOAD and LOAD could decrease BG uptake of PIB in the LOAD group, we compared ventricular volume between both groups. There were no significant volume differences in EOAD>LOAD and LOAD>EOAD in voxel-based morphometry using SPM (family-wise error rate <0.05). Previous reports, including postmortem pathologic studies, have shown that patients with EOAD exhibit a higher number of neuritic plaques and neurofibrillary tangles compared to patients with LOAD,7,8 a finding that is consistent with our results. Choo et al also reported that patients with EOAD exhibited greater PIB retention in diffuse brain areas, including the frontal, temporal, parietal lobes and the basal ganglia, compared to patients with LOAD. The same study reported an inverse relationship between age at onset and Aβ burden.9 In our study, the average Mini-Mental State Examination score of patients with EOAD was ~2.8 points lower than the average score of patients with LOAD, which may have contributed to the higher z score and amyloid burden in the EOAD group. However, since the amount of Aβ deposits in the brain has been reported not to correlate with the clinical severity of dementia in patients with AD, our cognitive results should be interpreted cautiously. Further studies are needed to clarify the relationship between cognitive level and Aβ deposits in EOAD and LOAD.
Other studies have reported that patients with EOAD exhibit higher PIB retention in the bilateral basal ganglia and the thalamus compared to patients with LOAD, whereas the total Aβ burden is not significantly different between patients with EOAD and patients with LOAD.10,11 In familial AD, the striatum is one of the first areas where amyloid deposits are observed. Even in presymptomatic or mildly affected patients, those with presenilin 1 mutations exhibit PIB deposition in the thalamus.12–15 It is not yet known why a greater amount of Aβ deposition is observed in the subcortical gray matter of patients with EOAD compared to patients with LOAD. The thalamus and striatum are not usually considered to be related to the pathogenesis of AD; however, it has long been postulated that they may be affected by AD.16 These subcortical nuclei are interconnected with the cortex through a variety of networks, serving both cognitive and behavioral functions.17 The caudate plays a role in planning and executive functions, whereas the thalamus is involved in declarative memory.18,19 Therefore, AD has been considered to be a network disease inflicting selective damage on neuronal circuits involving subcortical gray matter, cortical areas and their connecting white matter.20 Consistent with previous reports, our SPM analysis of patients with EOAD and patients with LOAD also revealed Aβ retention in the bilateral thalamus and the basal ganglia (Figure 1C). Volumetric studies have shown that patients with EOAD predominantly exhibit bilateral atrophic changes in the dorsolateral temporal lobes, inferior parietal lobules, precuneus and perisylvian cortices; in contrast, patients with LOAD exhibit atrophy in the bilateral medial temporal cortices.21 One possible explanation of these observations is that EOAD primarily damages the circuit connecting the subcortical nuclei and the corresponding cerebral cortex; we hypothesize that this network is highly vulnerable in patients with EOAD. In contrast, patients with LOAD predominantly exhibit damage in the medial and temporal areas. Contrary to our expectations, we did not observe any differences in PIB loading in the bilateral hippocampus through SPM analysis of patients with LOAD versus patients with EOAD, or of patients with LOAD versus control patients. These results can be explained by the partial volume effect, wherein atrophy, predominantly in the medial temporal lobe, leads to a relatively low PIB signal.
Some studies have reported that patients with EOAD exhibit hypometabolism in the basal ganglia, which could explain why these patients manifest more frequent extrapyramidal symptoms and frontal executive dysfunction.22–24 One limitation of our study is that we did not stratify patients with EOAD according to clinical symptoms such as extrapyramidal or frontal executive function.
In conclusion, our PIB PET amyloid imaging study showed that Aβ deposition is different in EOAD and LOAD. Comparing our results with data obtained from previous studies of familial EOAD, we found the possibility that the pattern and degree of Aβ deposition was different between EOAD and LOAD, regardless of known mutation status. However, even though we excluded the EOAD with well-known genetic mutation, there was a possibility of it being unknown mutation. More studies are needed to validate our results.
Acknowledgments
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C3331).
This study was presented at “The 12th International Conference on Alzheimer’s and Parkinson’s Diseases and Related Neurological Disorders (AD/PD 2015)” and the abstract was published in “Neurodegenerative Disease”.
Disclosure
The authors report no conflicts of interest in this work.
References
Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–670. | ||
Leuba G, Saini K, Savioz A, Charnay Y. Early-onset familial Alzheimer disease with coexisting beta-amyloid and prion pathology. JAMA. 2000;283(13):1689–1691. | ||
Jayadev S, Nochlin D, Poorkaj P, et al. Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol. 2011;69(4):712–720. | ||
Kemppainen NM, Aalto S, Wilson IA, et al. Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology. 2006;67(9):1575–1580. | ||
Edison P, Archer HA, Hinz R, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68(7):501–508. | ||
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. | ||
Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL. Early-onset Alzheimer’s disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol. 2007;20(1):29–33. | ||
Bigio EH, Hynan LS, Sontag E, Satumtira S, White CL. Synapse loss is greater in presenile than senile onset Alzheimer disease: implications for the cognitive reserve hypothesis. Neuropathol Appl Neurobiol. 2002;28(3):218–227. | ||
Choo IH, Lee DY, Kim JW, et al. Relationship of amyloid-beta burden with age-at-onset in Alzheimer disease. Am J Geriatr Psychiatry. 2011;19(7):627–634. | ||
Cho H, Seo SW, Kim JH, et al. Amyloid deposition in early onset versus late onset Alzheimer’s disease. J Alzheimers Dis. 2013;35(4):813–821. | ||
Rabinovici GD, Furst AJ, Alkalay A, et al. Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain. 2010;133(Pt 2):512–528. | ||
Knight WD, Okello AA, Ryan NS, et al. Carbon-11-Pittsburgh compound B positron emission tomography imaging of amyloid deposition in presenilin 1 mutation carriers. Brain. 2011;134(Pt 1):293–300. | ||
Villemagne VL, Ataka S, Mizuno T, et al. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol. 2009;66(12):1537–1544. | ||
Remes AM, Laru L, Tuominen H, et al. Carbon 11-labeled pittsburgh compound B positron emission tomographic amyloid imaging in patients with APP locus duplication. Arch Neurol. 2008;65(4):540–544. | ||
Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27(23):6174–6184. | ||
Braak H, Braak E. Alzheimer’s disease affects limbic nuclei of the thalamus. Acta Neuropathol. 1991;81(3):261–268. | ||
Middleton FA, Strick PL. Basal ganglia output and cognition: evidence from anatomical, behavioral, and clinical studies. Brain Cogn. 2000;42(2):183–200. | ||
Grahn JA, Parkinson JA, Owen AM. The cognitive functions of the caudate nucleus. Prog Neurobiol. 2008;86(3):141–155. | ||
Van der Werf YD, Jolles J, Witter MP, Uylings HB. Contributions of thalamic nuclei to declarative memory functioning. Cortex. 2003;39(4–5):1047–1062. | ||
Warren JD, Rohrer JD, Hardy J. Disintegrating brain networks: from syndromes to molecular nexopathies. Neuron. 2012;73(6):1060–1062. | ||
Kim S, Youn YC, Hsiung GY, et al. Voxel-based morphometric study of brain volume changes in patients with Alzheimer’s disease assessed according to the Clinical Dementia Rating score. J Clin Neurosci. 2011;18(7):916–921. | ||
Chui HC, Teng EL, Henderson VW, Moy AC. Clinical subtypes of dementia of the Alzheimer type. Neurology. 1985;35(11):1544–1550. | ||
Kim EJ, Cho SS, Jeong Y, et al. Glucose metabolism in early onset versus late onset Alzheimer’s disease: an SPM analysis of 120 patients. Brain. 2005;128(Pt 8):1790–1801. | ||
Frisoni GB, Testa C, Sabattoli F, Beltramello A, Soininen H, Laakso MP. Structural correlates of early and late onset Alzheimer’s disease: voxel based morphometric study. J Neurol Neurosurg Psychiatry. 2005;76(1):112–114. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.