Back to Journals » Drug Design, Development and Therapy » Volume 11
10H-3,6-Diazaphenothiazine induces G2/M phase cell cycle arrest and caspase-dependent apoptosis and inhibits cell invasion of A2780 ovarian carcinoma cells through the regulation of NF-κB and (BIRC6-XIAP) complexes
Authors Zhang J, Ming C, Zhang W, Okechukwu PN, Morak-Młodawska B, Pluta K, Jeleń M, Akim AM, Ang KP, Ooi KK
Received 20 June 2017
Accepted for publication 17 August 2017
Published 24 October 2017 Volume 2017:11 Pages 3045—3063
DOI https://doi.org/10.2147/DDDT.S144415
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Jianxin Zhang,1 Chen Ming,2 Wenzhi Zhang,3 Patrick Nwabueze Okechukwu,4 Beata Morak-Młodawska,5 Krystian Pluta,5 Małgorzata Jeleń,5 Abdah Md Akim,6 Kok-Pian Ang,3 Kah Kooi Ooi6,7
1Department of Gynecology and Obstetrics, Capital Medical University Affiliated Beijing Chaoyang Hospital, Beijing, 2Department of Gynecologic Oncology, Taizhou People’s Hospital, Jiangsu, People’s Republic of China; 3Innoresearch, Subang Jaya, 4Department of Biotechnology, Faculty of Applied Sciences, UCSI University, Kuala Lumpur, Malaysia; 5Department of Organic Chemistry, School of Pharmacy with the Division of Laboratory Medicine, The Medical University of Silesia, Sosnowiec, Poland; 6Department of Biomedical Science, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, Serdang, 7Research Centre for Crystaline Materials, School of Science and Technology, Sunway University, Petaling Jaya, Malaysia
Abstract: The asymptomatic properties and high treatment resistance of ovarian cancer result in poor treatment outcomes and high mortality rates. Although the fundamental chemotherapy provides promising anticancer activities, it is associated with severe side effects. The derivative of phenothiazine, namely, 10H-3,6-diazaphenothiazine (PTZ), was synthesized and reported with ideal anticancer effects in a previous paper. In this study, detailed anticancer properties of PTZ was examined on A2780 ovarian cancer cells by investigating the cytotoxicity profiles, mechanism of apoptosis, and cell invasion. Research outcomes revealed PTZ-induced dose-dependent inhibition on A2780 cancer cells (IC50 =0.62 µM), with significant less cytotoxicity toward HEK293 normal kidney cells and H9C2 normal heart cells. Generation of reactive oxygen species (ROS) and polarization of mitochondrial membrane potential (Δψm) suggests PTZ-induced cell death through oxidative damage. The RT2 Profiler PCR Array on apoptosis pathway demonstrated PTZ-induced apoptosis via intrinsic (mitochondria-dependent) and extrinsic (cell death receptor-dependent) pathway. Inhibition of NF-κB and subsequent inhibition of (BIRC6-XIAP) complex activities reduced the invasion rate of A2780 cancer cells penetrating through the Matrigel™ Invasion Chamber. Lastly, the cell cycle analysis hypothesizes that the compound is cytostatic and significantly arrests cell proliferation at G2/M phase. Hence, the exploration of the underlying anticancer mechanism of PTZ suggested its usage as promising chemotherapeutic agent.
Keywords: anticancer, programmed cell death, ovarian cancer, oxidative damage, mitochondrial function disruption, cancer cell invasion
Introduction
Cancer ranked the top three leading cause of death worldwide after cardiovascular diseases and infectious diseases.1 Among the female genital cancer such as endometrium cancer, cervical cancer, and ovarian cancer, the later gives the highest mortality rate because of being asymptomatic and due to metastasis.2,3 Ovarian cancer is highly associated in female with BRCA1 and BRCA2 mutation, either inherited or acquired, although it is also associated with other risk factors such as obesity, late menopause, cigarette smoking, and nulliparity.4–8 The most common type of ovarian cancer is derived from epithelial cells through a series of mutation on tumor suppressor genes (such as PTEN, BRCA1, BRCA2, and p53), which result from silencing of the genes;4–6 meanwhile mutation of oncogenes (such as Bcl-2, EFGR, and VEGF) leads to over-activation, thus making the cancer cells autonomous on self-growing and providing resistance to cell death signals or escape from cell cycle checkpoints, ability to invade, and metastasis to distant location which further yields the occurrence of secondary tumor.9,10 As mentioned, ovarian cancer cells are reported with high invasion and metastasis rate, because of high activities from NF-κB gene and subsequent complex formation by several survival and proliferation factors, such as X-linked inhibitor of apoptosis (XIAP) and baculoviral inhibitor of apoptosis repeat-containing gene (BIRC) families (mainly BIRC 1–6).11 The XIAP formed multiple complexes with respective BIRC; each complex played different function in the development of cancer. For example, (BIRC2/3-XIAP) complex inhibits checkpoints and thus promotes cell cycle activities; (BIRC5-XIAP) complex inhibits caspase activation and thus negatively regulates apoptosis; (BIRC6-XIAP) complex promotes the activity of matrix metalloproteinase-2 or -9 (MMP-2 or MMP-9) to degrade the basement membrane of endothelial cells of the blood vessels, thus enhancing the entry of cancer cells into blood stream.11–13
Therapy of ovarian cancer includes surgical removal, radiotherapy, and chemotherapy.14 Chemotherapy was reported the most efficient therapy although it is commonly associated with side effects on normal cells.15 Thus, there is a need to find a new potent anticancer agent that have greater selectivity toward cancer cells. Based on previous studies, the nitrogen- and sulfur-containing heterocyclic ring system such as phenothiazine derivatives was synthesized and reported with promising pharmaceutical properties.16–21 It was originally synthesized as 10H-dibenzo-1,4-thiazine by Bernthsen in 1883, and later, up to 5,000 derivatives of phenothiazine had been synthesized by researchers. They have been valuable drugs possessing antipsychotic, antihistaminic, antitussive, and antiemetic activities.16 Recently, their activities are expanded to show anticancer, antiviral, antibacterial, anti-inflammatory, and antimalarial properties due to the modifications in chemical structure such as substitution of new substituent group at the thiazine nitrogen atom in position 10, oxidation of sulfur atom, substitution of the benzene ring with other aromatic rings, and addition of new substituent groups in the benzene ring.16–23 These classical phenothiazines are low toxic, easy to obtain, and less expensive.16
One of the modifications of the chemical structure of the phenothiazine is replacement of benzene ring with azine ring to form azaphenothiazines. As reported, azaphenothiazines of the dipyridothiazine structure were synthesized by the introduction of the pyridine ring instead of the benzene ring, namely, 10H-1,6-diazaphenothiazine, 10H-1,8-diazaphenothiazine, 10H-2,7-diazaphenothiazine, and 10H-3,6-diazaphenothiazine (PTZ).17–21 These organic compounds exhibited promising anticancer activity against several in vitro cancer cell lines (breast, ovarian, lung, colorectal, prostate, leukemia, melanoma, and renal). Among the diazaphenothiazine series, the PTZ20 had been reported effective in killing breast cancer cells (MCF-7) but less toxic toward normal human fibroblast cells (HFF-1). This compound induces apoptosis on MCF-7 cells through the upregulation of pro-apoptotic genes such as bax, p53, and CDKN1A (p21) and downregulates anti-apoptotic gene such as Bcl-2 and H3 (a histone indicator for the proliferation of cellular DNA).19 Based on these promising results, the aims of the current study were to investigate anticancer activities with detailed apoptosis pathway induced by PTZ toward ovarian cancer cell line (A2780), which was also reported with chemoresistance to cisplatin. In addition, the ability of the trial compound to inhibit ovarian cancer cells invasion was also studied through regulation on NF-κB and (BIR6-XIAP) complex through RT2 Profiler PCR Array.
Materials and methods
Trial compound
The PTZ (Figure 1; molecular weight =201 g/mol) was synthesized by our collaborators from Medical University of Silesia, Sosnowiec, Poland, in conjunction with collaboration from Dr Patrick Nwabueze Okechukwu from School of Applied Sciences, UCSI, Malaysia. It was obtained as a dark green powder as reported in the respective chemical properties.19 Dimethyl sulfoxide (DMSO; Sigma-Aldrich Co., St Louis, MO, USA) was used as a solvent to dissolve the trial compound. Meanwhile, cisplatin (cis-diamineplatinum(II) dichloride; Sigma-Aldrich Co., purity >99.9%) was chosen as a positive control. All drugs were diluted in serum-free media for the following parameters.
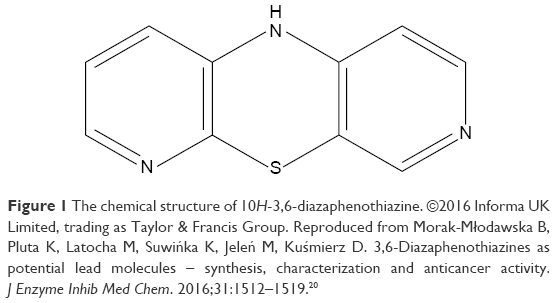 | Figure 1 The chemical structure of 10H-3,6-diazaphenothiazine. ©2016 Informa UK Limited, trading as Taylor & Francis Group. Reproduced from Morak-Młodawska B, Pluta K, Latocha M, Suwińka K, Jeleń M, Kuśmierz D. 3,6-Diazaphenothiazines as potential lead molecules – synthesis, characterization and anticancer activity. J Enzyme Inhib Med Chem. 2016;31:1512–1519.20 |
Cell culture and maintenance
The representative ovarian cancer cell line, A2780 human ovarian epithelial cancer cells were purchased from ECACC (catalog no 93112519, cisplatin-resistant type) and cultured according to the manufacturer’s instructions: RPMI-1640 media + 10% fetal bovine serum (FBS) (Sigma-Aldrich Co.). For normal cells, HEK293 human embryonic kidney epithelial cells and H9C2 rat embryonic heart myoblast cells were purchased from ATCC (catalog no CRL-1573 and CRL-1446) and cultured in DMEM + 10% FBS + 2% L-glutamine (Sigma-Aldrich Co.). All the cells were incubated at 37°C in 5% CO2 incubator with humidified atmosphere.
Cell viability assay
All the assays in the present study including cell viability assay were conducted, at least, in triplicate in contemporary A2780 cancer cells (received 1% DMSO alone) categorized as negative (untreated) control in conjunction with cisplatin as positive control for referral.
The A2780 human ovarian cancer cells were seeded in a 96-well plate with the density of 1×105 cells at final volume of 100 μL per well. The trial compound PTZ was prepared by dissolving it in DMSO to a concentration of 90 μM and further diluted in RPMI-1640 medium and then added into the well to obtain a final concentration of 0 (served as untreated control), 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, and 0.9 μM. It was then incubated for 24 hours.
To investigate the cytotoxicity value of PTZ, 20 μL of (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide, MTT) solution (5 mg/mL in PBS) was added into each well and the plate was further incubated for 4 hours at 37°C. Prior to analysis, the medium was discarded and replaced with DMSO to dissolve the formazan crystal. The absorbance was measured at 570 nm through microplate reader (Tecan Infinite-M200). The 50% inhibitory concentration of PTZ (IC50) was determined by plotting the graph of percentage of surviving cells against the concentration of PTZ. The possible toxicity of PTZ was evaluated by using two normal cell lines, HEK293 and H9C2. The protocols of assessment of cytotoxicity were as for the cell viability assay of A2780 cancer cells.
Morphological studies of apoptosis via acridine orange/propidium iodide (AO/PI) staining
A2780 cancer cells were cultured with the density of 1×105 cells/well in a 6-well plate. As for treatment, IC50 and two other reference concentrations of PTZ were used (0.3 μM, 0.62 μM [IC50], and 0.9 μM; for all parameters below). The cells were then further incubated for 24 hours. Prior to analysis, the cells were harvested and washed twice with ice-cold PBS to obtain 50 μL cell suspension. Furthermore, a 1 mg/mL AO (Sigma-Aldrich Co.) and 1 mg/mL PI (Sigma-Aldrich Co.) were added into the suspension and incubated for 20 minutes in dark at room temperature. The apoptotic morphology of cells can be observed via aliquoting 10 μL of mixtures onto a glass slide and covered with a cover slip, and it was viewed under fluorescence microscope (Nikon Corporation, Tokyo, Japan) by using a bandpass filter (excitation 490 nm, emission 520 nm).
Nuclear and DNA fragmentation observation via DAPI staining
DAPI stain (4′,6-diamidine-2′-phenylindole dihydrochloride; Sigma-Aldrich Co.) is widely used to study fragmentation of DNA during apoptosis. The seeding and treatment protocols were as per AO/PI staining; the cells were harvested and washed twice with ice-cold PBS to obtain 500 μL cell suspension. Next, 1 μg/mL of DAPI stain was added into the suspension and incubated for 10 minutes in dark at room temperature. The subsequent protocol is same as AO/PI and viewed under fluorescence microscope by applying ultraviolet bandpass filter (excitation 358 nm, emission 460 nm).
Quantification of apoptotic cells via Annexin V-FITC staining
Annexin V-FITC assay is the quantitative studies of apoptosis after AO/PI staining. A2780 cancer cells were seeded at the density of 1×106 cells/well and treated with three concentrations of PTZ for 24 hours. Next, the cells were harvested and washed twice with ice-cold PBS and lastly resuspend with 500 μL of 1× binding buffer together with 10 μL of Annexin V-FITC dye and 8 μL of PI dye according to the manufacturer’s protocol (Merck Millipore, Billerica, MA, USA). The suspension was then incubated for 20 minutes in dark at room temperature. The cell apoptosis analysis was performed using flow cytometer (FACSCalibur; BD Biosciences, San Jose, CA, USA) equipped with CellQuestPro software™. Each experiment was performed in triplicate.
Intracellular reactive oxygen species (ROS) measurement
A2780 cells were seeded with the density of 1×105 cells/well in a 96-well black plate and treated with three concentrations of PTZ and further incubated for 24 hours. Next, 100 μM H2DCFDA (Sigma-Aldrich Co.) was loaded into each well and incubated for another 1 hour in dark at 37°C. The plate was then centrifuged followed by the removal of supernatant. The wells were then refilled with 100 μL pre-warmed 1× PBS. The DCF (2′,7′-dichlorofluorescein) fluorescence intensity was measured using the fluorescence microplate reader (Tecan Infinite-M200) at the excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Mitochondrial membrane potential (ΔΨm) assay
Mitochondrial played a critical role in the initiation of apoptosis anti-cancer studies, any changes in its membrane potential (ΔΨm) reflecting progression of cell death. To investigate the activity of PTZ toward mitochondrial behavior, the mitochondrial membrane potential assay was conducted by using the JC-1 Mitochondrial Membrane Potential Assay Kit (Cayman Chemical, 10009172). The A2780 ovarian cancer cells were seeded on a 6-well plate with the density of 1×106 cells/well. After attachment of cells for overnight, the three concentrations of PTZ together with cisplatin were added to the cells for another 24 hours, with the untreated group receiving only 1% DMSO. The staining procedure was conducted according to the manufacturer’s protocol, and lastly, the cells were observed under the fluorescence microscope by implying a bandpass filter (excitation 490 nm, emission 520 nm).
Quantification of caspase activities
Caspase activity was assayed by measuring the fluorescence intensity via CaspaTag™ Caspase-3/7 in situ assay kit, CaspaTag™ Caspase-8 in situ assay kit, and CaspaTag™ Caspase-9 in situ assay kit (Merck Millipore). For the quantification of each caspase activities, the A2780 cancer cells were cultured with the density of 1×105 cells/well in a 96-well black plate followed by the application of treatment and incubated for 24 hours. Then, 5 μL of caspase reaction buffer was added into each well according to the manufacturer’s protocols and incubated for 1 hour in dark at 37°C. The contents of the plate were evaluated at 490 nm (excitation) and 520 nm (emission) by using fluorescence microplate reader (Tecan Infinite-M200).
RT2 Profiler PCR Array (human apoptosis pathway)
A2780 ovarian cancer cells were plated at the density of 3×106 cells per T-25 cm2 flask followed by the application of PTZ at IC50 dosage (0.62 μM) for 24 hours. Total RNA was extracted from the cultured A2780 cells using RNeasy Mini Kit (Qiagen NV, Venlo, the Netherlands) according to the manufacturers’ protocols; the concentration and quality of the extracted RNA was determined immediately by using spectrophotometer (Nanodrop 1000; Thermo Fisher Scientific, Waltham, MA, USA). As per requirement of the experiment, the RNA was further converted into cDNA by using the RT2 First Strand Kit (Qiagen NV). Real-time PCR was performed by using RT2 Profiler PCR Array (Human Apoptosis, PAHS-012ZA). The array illustrates the expression of 84 key genes which either upregulated or downregulated in programed cell death particularly apoptosis. The data analysis was performed according to the software associated, and the gene expression was compared based on the threshold level (CT) value.
Cell cycle analysis
A2780 cells were seeded at the density of 1×106 cells/well in a 6-well plate and treated with three concentrations of PTZ for 24 hours. Then, the cells were harvested, washed twice with ice-cold PBS, and fixed in 70% ethanol at −20°C. Prior to analysis, the samples were washed again with ice-cold PBS to remove excess ethanol and rehydrate the cells. The cell pellet was resuspended by adding 425 μL of PBS along with 50 μL of RNase A (1 mg/mL; Sigma-Aldrich Co.) and 25 μL of PI (1 mg/mL, Sigma-Aldrich Co.) to make a total 500 μL cell suspension mixture and incubated for 30 minutes in dark at room temperature. The cell cycle phase analysis was performed using flow cytometer (FACSCalibur; BD Biosciences) equipped with CellQuestPro software™. Each experiment was performed in triplicate.
Cell invasion assay
The cell invasion assay was studied by using the BioCoat™ Matrigel™ Invasion Chamber (BD Biosciences). Generally, 0.5 mL of complete medium was added to the bottom of the well; At the same time, 5×104 cells were resuspended in 0.5 mL complete medium with or without PTZ and then added to the insert of the well according to the manufacturer’s protocol. The cells were then incubated for 24 hours. Prior to analysis, non-invaded cells from the inner insert were removed with cotton tips. The inserts were fixed with methanol followed by staining with hematoxylin and eosin and lastly washed with PBS. The membrane of the insert was finally observed under an inverted microscope at 200× magnification. Five fields were randomly chosen, and the number of invaded cells were counted. The date is expressed in percentage (%) of invaded cells ± standard deviation (SD).
Data calculation
Results (quantitative data) are presented as mean ± SD. The statistical significance was calculated by using one-way analysis of variance followed by Duncan’s multiple comparison test as post hoc test. p<0.05 was considered as significant.
Results
Cell viability assay
The anti-proliferation activity of PTZ was investigated through MTT assay on A2780 ovarian carcinoma cells at various concentrations, starting from 0, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, and 0.9 μM, with 0 μM served as untreated (negative) control. On the basis of the dose–response graph in Figure 2, it was found that PTZ demonstrated significant inhibitory effects on proliferation of A2780 cells and that the 50% inhibitory concentration (IC50) value obtained is 0.62 μM.
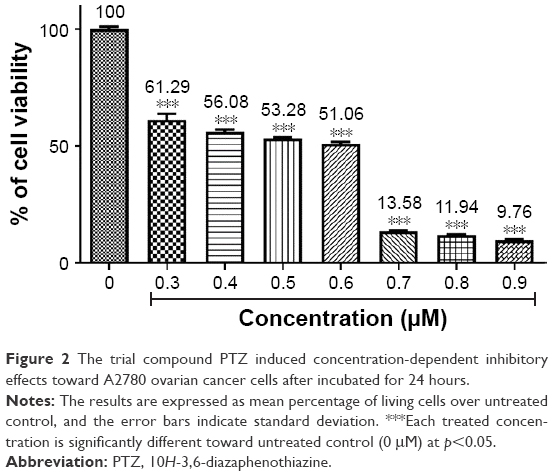 | Figure 2 The trial compound PTZ induced concentration-dependent inhibitory effects toward A2780 ovarian cancer cells after incubated for 24 hours. |
To investigate the possible cytotoxic effects of PTZ toward normal cells, the HEK293 human embryonic kidney epithelial cells and H9C2 rat embryonic heart myoblast cells were selected as representative in vitro normal cell model. By employing the same protocol as A2780 cells, the toxicity effects of PTZ toward HEK293 and H9C2 cells give the results in Figure 3. Noted from the results in Figure 3, PTZ exhibited less potency toward HEK293 and H9C2 cells, indicating it is more selective toward A2780 cancer cells.
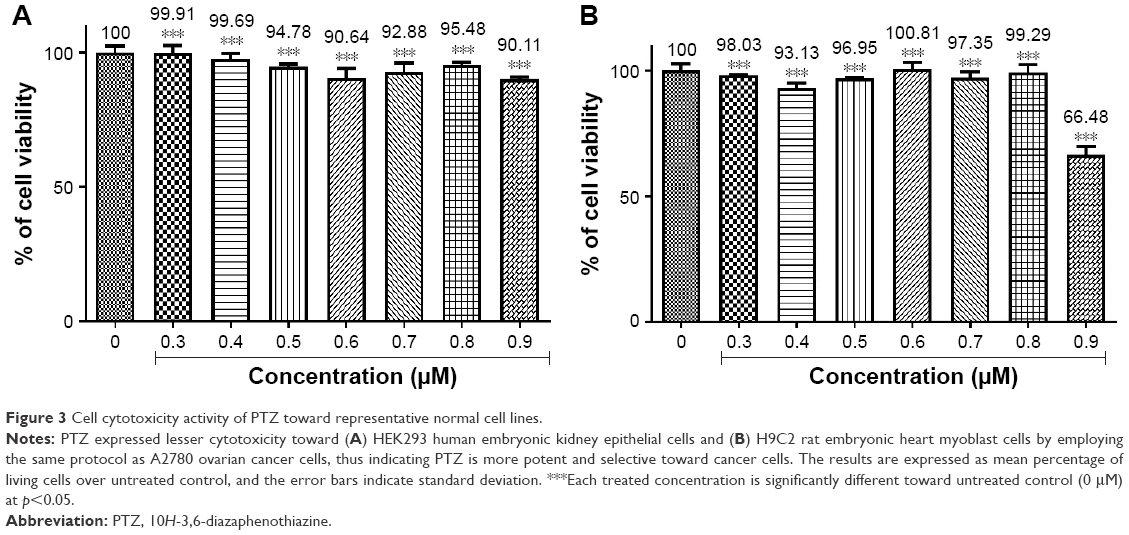 | Figure 3 Cell cytotoxicity activity of PTZ toward representative normal cell lines. |
Investigation on the morphology of apoptosis
AO/PI double staining
The AO and PI double staining was applied to investigate the morphological features of apoptosis induced by PTZ toward A2780 cancer cells at three reference concentrations (0.3, 0.62, and 0.9 μM). Generally, AO stain forms a green fluorescence on apoptotic cells with red color-stained nuclei matter and greenish yellow dots of the condensed nuclei matter. In contrast, necrotic cells exhibit red fluorescence due to their weakened cell membrane integrity, thus the PI stain was able to penetrate the cell membrane and nuclear membrane and intercalated with nuclear DNA. On the basis of the observation in Figure 4, it is found that upon the application of PTZ onto A2780 ovarian cancer cells, the trial compound induces apoptosis raising the morphology of apoptotic cells such as shrinking of size of cells, condensed chromatin, and release of DNA contents into cytoplasm.
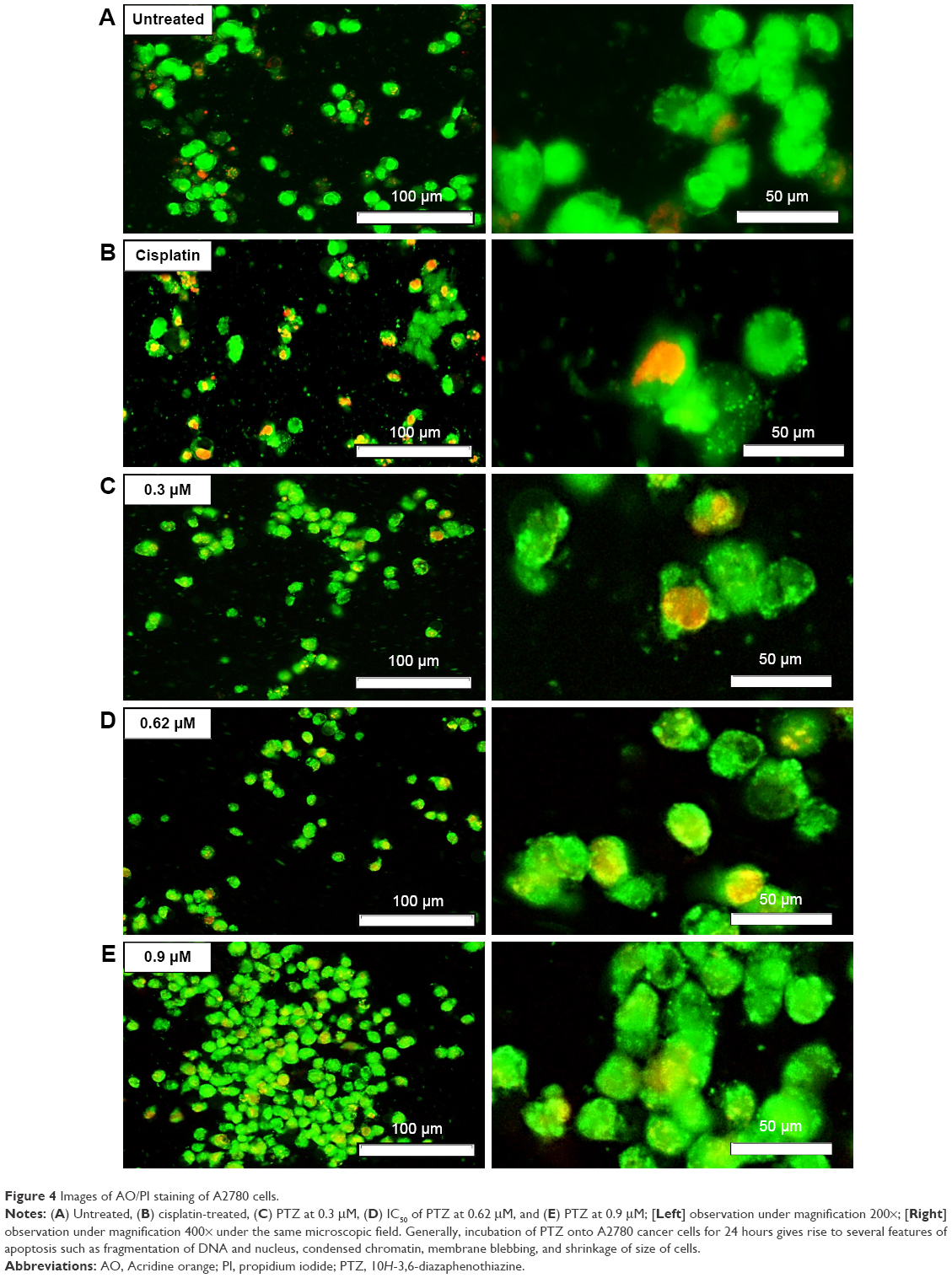 | Figure 4 Images of AO/PI staining of A2780 cells. |
DAPI staining
One of the mechanisms of action of a chemotherapeutic drug is directly bound to cancer cells DNA, either via induction of oxidative stress or adduction, leading to the fragmentation of DNA and eventually initiated apoptosis events. The DAPI dye is a blue fluorescent stain which was able to penetrate through the plasma membrane and nuclear membrane of apoptotic cells and bind strongly to the fragmented DNA. Thus, it acts as a useful indicator to study DNA fragmentation. As seen in Figure 5A, the untreated group gave less blue fluorescence intensity compared with positive control (Figure 5B) and treatment groups (Figure 5C–E); with the minimal damage and fragmentation of DNA of living cells, there was lesser probability of DAPI stain intercalating onto DNA fragments, hence generated lesser blue fluorescence. In contrast to the treatment groups (PTZ-treated and cisplatin-treated), the blue fluorescence produced by DAPI stain gives a clear and significant morphology regarding DNA fragmentation.
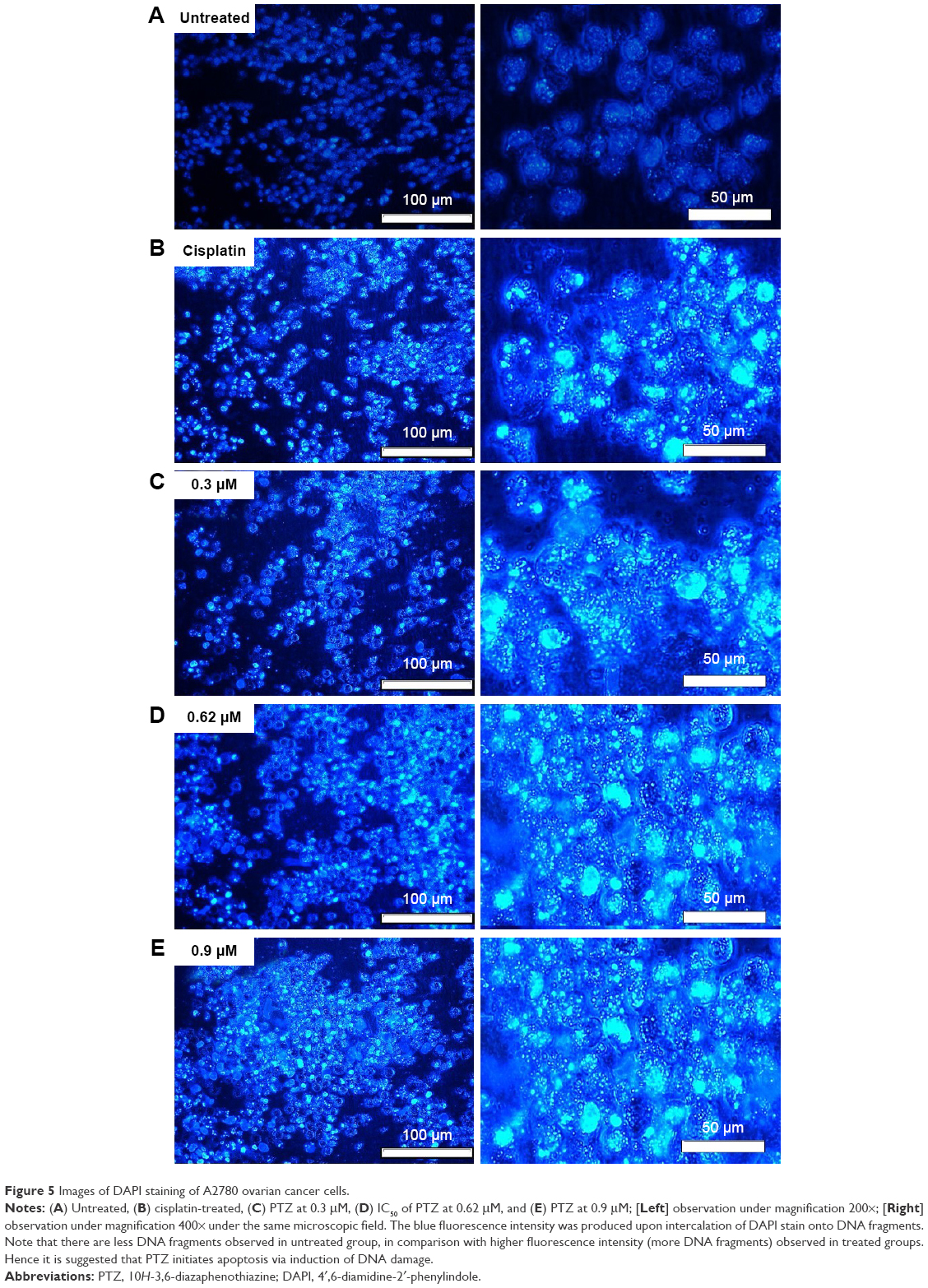 | Figure 5 Images of DAPI staining of A2780 ovarian cancer cells. |
Quantification of apoptotic cells
The results in AO/PI staining demonstrated that PTZ induces cell death on A2780 ovarian cancer cells via apoptosis. Furthermore, the quantitative analysis of apoptosis was conducted by staining the cells with Annexin V-FITC and PI after the application of PTZ at increased concentration for 24 hours. The quadrant graphs of cell apoptosis analysis are summarized in Figure 6. More than 80% of the cells (Annexin V−PI− marked) are reported to be living in the untreated group, in contrast with treatment groups (0.3, 0.62, and 0.9 μM), and there was a significant increase in the population of cells in late apoptosis (Annexin V+PI+ marked) after being treated, thus indicating that PTZ induces cell death via apoptosis pathway.
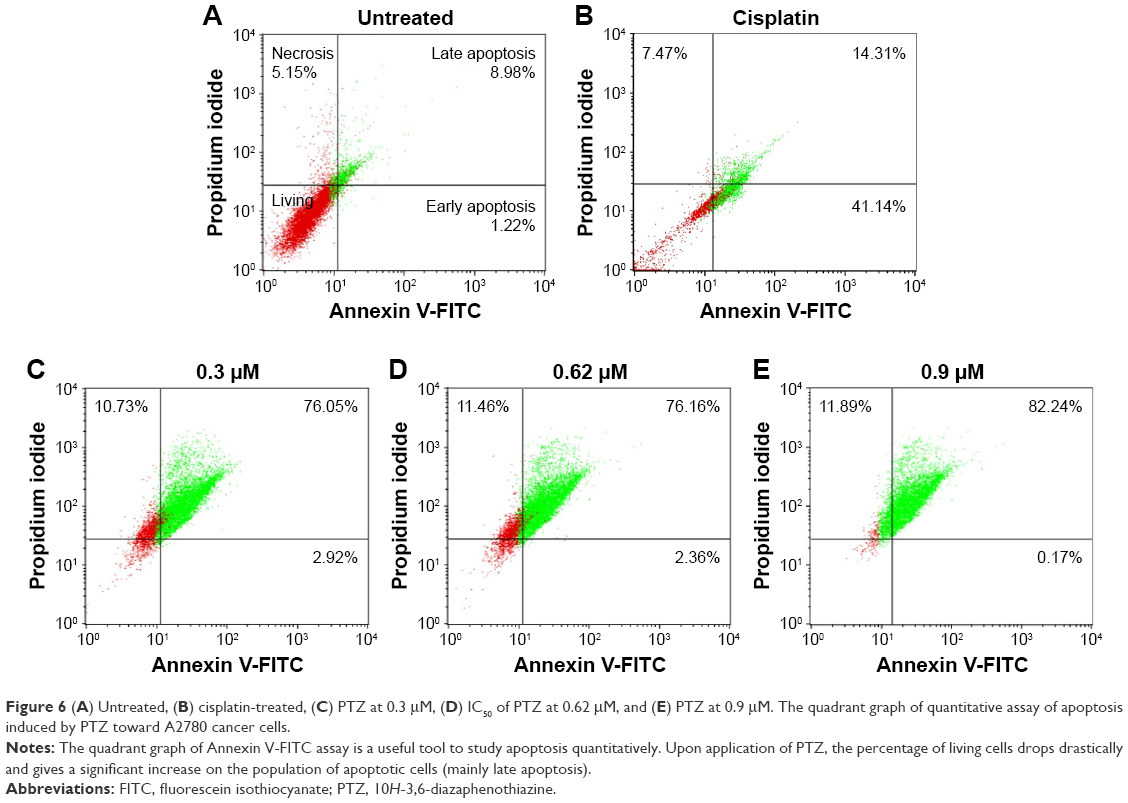 | Figure 6 (A) Untreated, (B) cisplatin-treated, (C) PTZ at 0.3 μM, (D) IC50 of PTZ at 0.62 μM, and (E) PTZ at 0.9 μM. The quadrant graph of quantitative assay of apoptosis induced by PTZ toward A2780 cancer cells. |
Intracellular ROS measurement
ROS is an essential factor to trigger apoptosis. To investigate the relations of PTZ in the generation of ROS, the bio-reagents 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) was used to detect the generation of ROS, particularly H2O2. In the presence of PTZ at increased concentration, the cellular ROS generation increased up to 129.54% compared to untreated group (normalized to 100%; Figure 7).
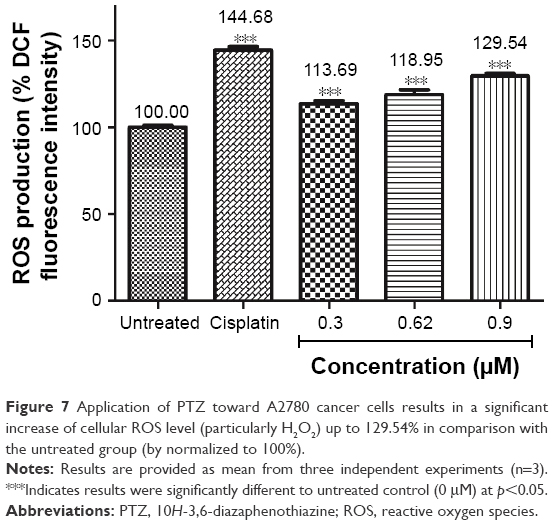 | Figure 7 Application of PTZ toward A2780 cancer cells results in a significant increase of cellular ROS level (particularly H2O2) up to 129.54% in comparison with the untreated group (by normalized to 100%). |
Mitochondrial membrane potential (ΔΨm) assay
One of the targeted programmed cell death pathways in the current study is the mitochondrial-dependent apoptosis pathway. The mitochondrial membrane potential (v) acts as an important measurement indicator for cellular mitochondrial functions, and their respective polarization activity will determine the initiation or prevention of apoptosis.24 The mitochondrial membrane potential was measured through JC-1 assay (Cayman) according to the manufacturer’s protocol, and the results are summarized in Figure 8.
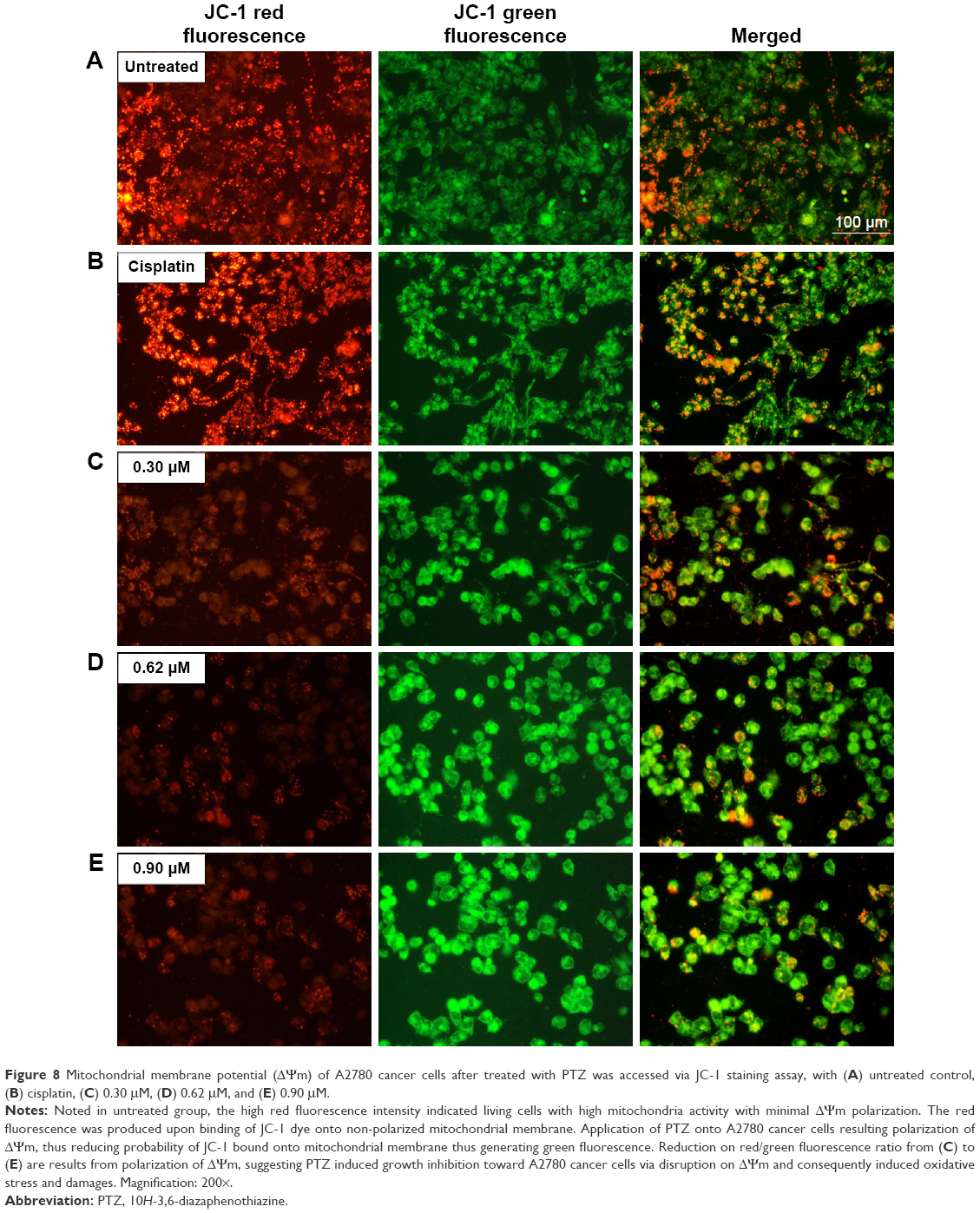 | Figure 8 Mitochondrial membrane potential (ΔΨm) of A2780 cancer cells after treated with PTZ was accessed via JC-1 staining assay, with (A) untreated control, (B) cisplatin, (C) 0.30 μM, (D) 0.62 μM, and (E) 0.90 μM. |
The fluorescent cationic dye, JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimi-dazolylcarbocyanine iodide) bound to mitochondria, gives rise to red or green fluorescence, depending on the degree of ΔΨm. Prior to the application of PTZ, the untreated A2780 cancer cells (Figure 8A) exhibited high red fluorescence intensity due to the aggregation of JC-1 dye into the mitochondria. Upon application of PTZ onto A2780 cancer cells, the green fluorescence intensity increased drastically concurrently associated with the reduction in red fluorescence intensity (Figure 8C–E) mainly due to polarization of ΔΨm and decreased aggregation of JC-1 dye; the green fluorescence intensity increased gradually upon increased concentration of the trial compound. Accumulation of JC-1 dye and decreased fluorescence ratio (red/green) indicated PTZ-induced polarization of ΔΨm and oxidative stress on mitochondria, thus initiated the early events of apoptosis.
Caspase activity (caspase-3/7, caspase-8, and caspase-9)
The quantitative ELISA measurement of caspase-3/7, caspase-8, and caspase-9 activities of A2780 cells were measured at 24 hours after exposure to PTZ. From the graphical data presented in Figure 9, it is found that PTZ increased the expression of caspase-3/7, caspase-8, and caspase-9 significantly, suggesting that PTZ induces apoptosis on A2780 cells via intrinsic (caspase-9) and extrinsic (caspase-8) pathways, followed by executional phase of apoptosis (caspase-3/7). The detailed mechanism of apoptosis induced by PTZ toward A2780 cancer cells is studied through RT2 Profiler PCR Array.
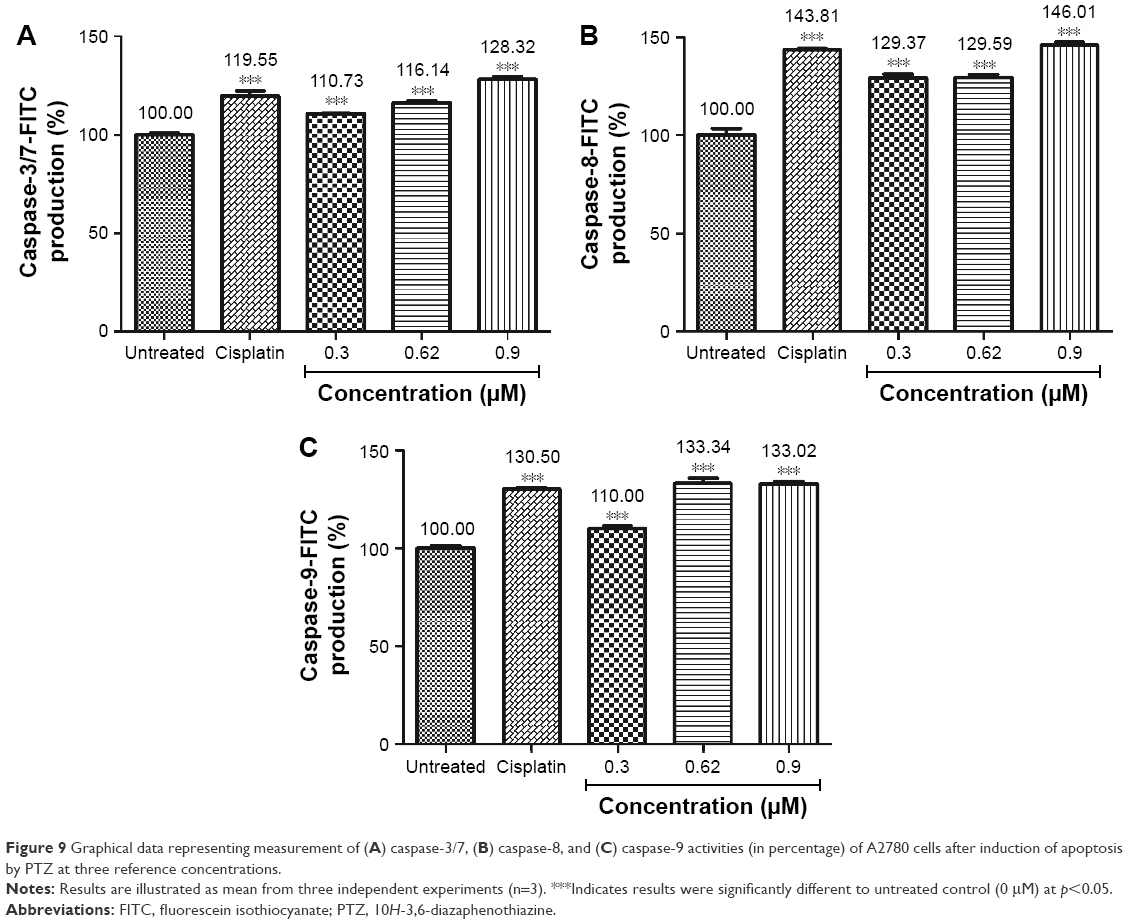 | Figure 9 Graphical data representing measurement of (A) caspase-3/7, (B) caspase-8, and (C) caspase-9 activities (in percentage) of A2780 cells after induction of apoptosis by PTZ at three reference concentrations. |
RT2 Profiler PCR Array (human apoptosis pathway)
The apoptosis-related gene expression regulated by the trial compound in A2780 ovarian cancer cells upon 24 hour incubation is summarized in Table 1.
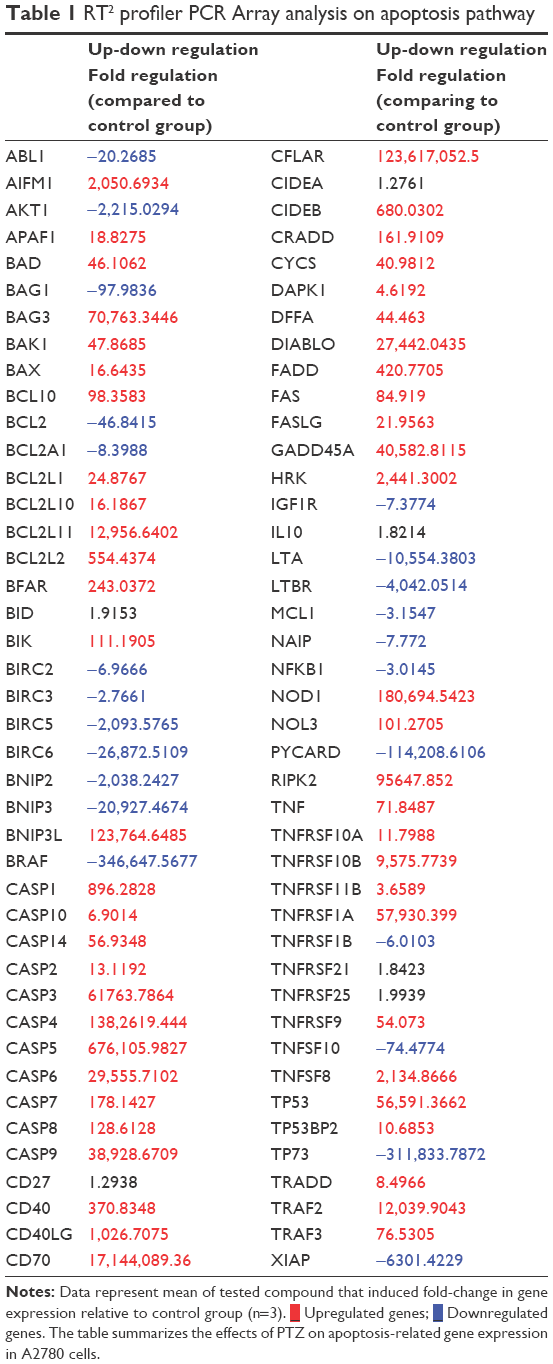 | Table 1 RT2 profiler PCR Array analysis on apoptosis pathway |
 Upregulated genes;
Upregulated genes;  Downregulated genes. The table summarizes the effects of PTZ on apoptosis-related gene expression in A2780 cells.
Downregulated genes. The table summarizes the effects of PTZ on apoptosis-related gene expression in A2780 cells.Cell cycle analysis
Activity of PTZ on cell cycle arrest was evaluated through flow cytometry, and the results are summarized in Figure 10. As seen from the results, there was a dominant population of cells accumulated in G0/G1 phase (70.18%) prior to exposure to PTZ. Upon application of PTZ on A2780 cells for 24 hours, there was a significant increase of arrested cell population from G0/G1 phase into G2/M phase as the concentration of PTZ increased.
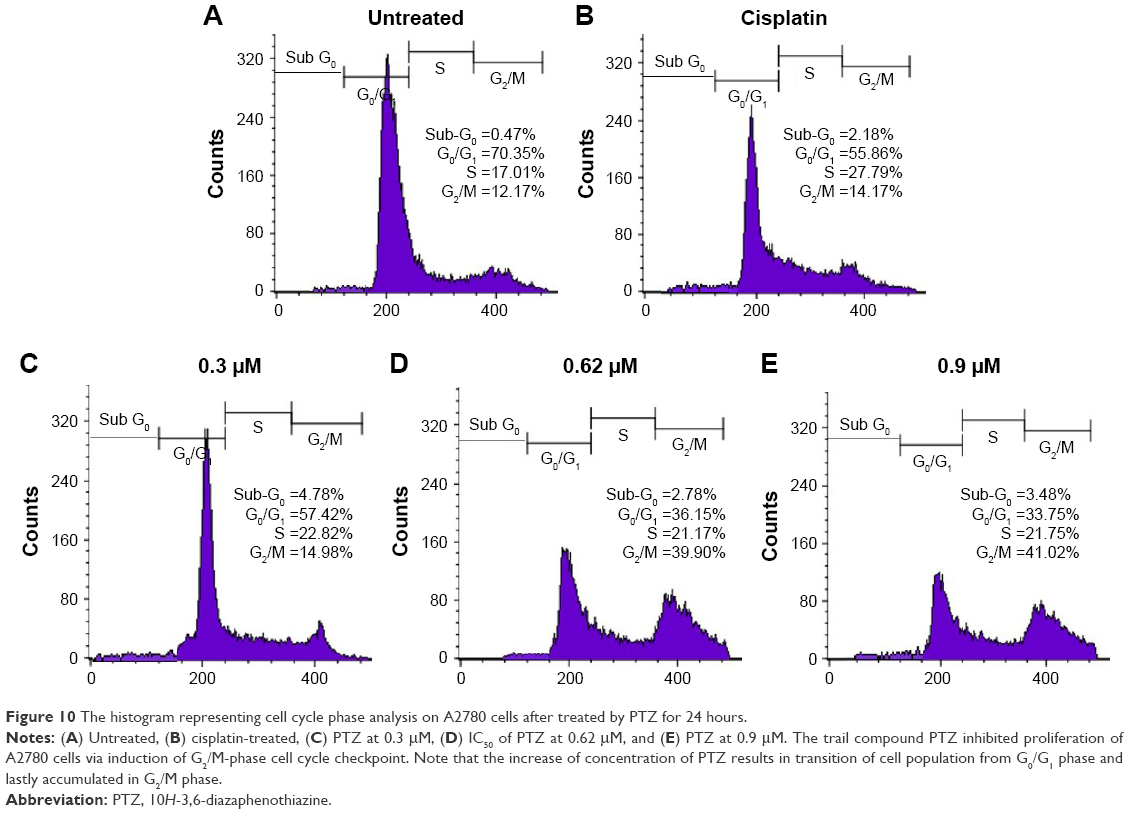 | Figure 10 The histogram representing cell cycle phase analysis on A2780 cells after treated by PTZ for 24 hours. |
Cell invasion assay
The Matrigel™ Invasion chamber provides an endothelial-like model to study the inhibition of compounds to prevent invasion of cancer cells through endothelial layer of blood vessels and metastasis to secondary location. In the present study, application of PTZ at 0.3, 0.62, and 0.9 μM showed suppression of cancer cell invasion in a concentration-dependent manner to 52.27%±3.08%, 21.73%±2.11%, and 9.14%±0.59%, respectively (Figure 11), by normalizing the invasion rate of cancer cells for untreated group to 100%.
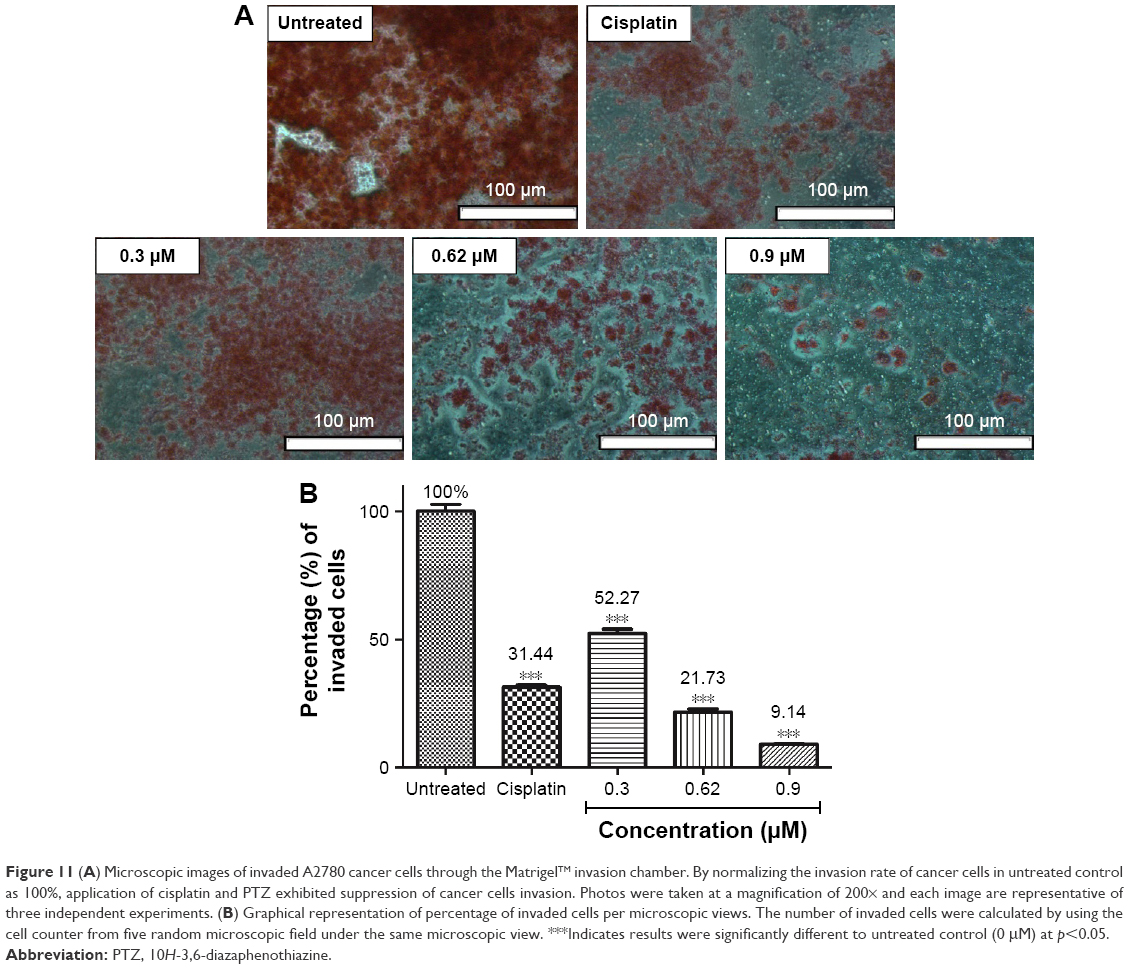 | Figure 11 (A) Microscopic images of invaded A2780 cancer cells through the Matrigel™ invasion chamber. By normalizing the invasion rate of cancer cells in untreated control as 100%, application of cisplatin and PTZ exhibited suppression of cancer cells invasion. Photos were taken at a magnification of 200× and each image are representative of three independent experiments. (B) Graphical representation of percentage of invaded cells per microscopic views. The number of invaded cells were calculated by using the cell counter from five random microscopic field under the same microscopic view. ***Indicates results were significantly different to untreated control (0 μM) at p<0.05. |
Discussion
The tricyclic phenothiazine was first reported with several important biological activities such as antipsychotic, antibacterial, antihistamic, antitussive, and antiemetic.16,25 A series of derivatives of phenothiazine were modified later to enhance their possible biological activity. Previously, the diazaphenothiazine series showed promising inhibitory activities toward several cancer cell lines.17–21 In the present study, a new diazaphenothiazine was synthesized, namely, PTZ. This compound exhibited more potent toxicity toward breast cancer MCF-7, glioblastoma SN-19, and melanoma C32 cell line compared to cisplatin, despite it also reported with their lower toxicity toward HFF-1 normal human fibroblast cell line.20 Nevertheless, the underlying molecular mechanism of PTZ particularly in the induction of apoptosis signaling cascade is still poorly understood. Among the three types of cancer tested, breast cancer shared same molecular carcinogenesis as ovarian cancer.7–10 Both the aggressive cancers generally rise from fast-growing epithelial cells through a series of mutation on BRCA1 and BRCA2 genes, inherited mutation of HNPCC gene, and acquired mutation/over-activation of Wnt-β-Catenin signaling.26–28 Ovarian cancer is often associated with poor survival rate because of being asymptomatic compared to breast tumor, indeed it is resistant to various chemotherapeutic agents, such as cisplatin.26,27
Based on the cytotoxicity test of PTZ toward A2780 ovarian cancer cell line, PTZ showed inhibitory result toward A2780 cells in a dose-dependent manner (Figure 2) and gave the IC50 value of 0.62 μM, in comparison with IC50 of cisplatin is 28.80 μM (data not shown). The lower IC50 value of PTZ compared to cisplatin suggested that it possesses higher cytotoxicity potency toward A2780 cells. Based on reviews, sulfur substituents and presence of phenyl rings effectively improved the efficacy of compound; however, detailed mechanism is recommended for future studies.16–20 Furthermore, to investigate the possible toxicity profile of PTZ toward normal cells, the HEK293 normal human kidney epithelial cells and H9C2 normal rat embryonic heart myoblast cells were selected as representative in vitro cell model. As seen in Figure 3, PTZ exhibited lesser cytotoxicity toward these two normal cells at the highest concentration (0.9 μM). Together with result reported in previous studies,20 it is suggested that PTZ is more selective toward cancer cells instead of normal cells.
Apoptosis plays important roles in physiological and pathological processes. It involves complex of signal cascades to regulate cell growth, cell division, and cell death, thus retains the cell population at a constant level.29 The hallmarks of cancers are the autonomous in self-growing, thus it can escape from cell cycle checkpoint and proceed cell division ultimately. Cancer cells are also resistant to death signals due to the silenced tumor suppressor genes and pro-apoptotic genes. To ensure PTZ-induced cell death on A2780 ovarian cancer cells by apoptosis, the AO/PI morphological staining, DAPI staining, and the apoptosis quantitative analysis (Annexin V-FITC assay) were conducted, and the activity of PTZ toward gene regulation on apoptosis toward A2780 cancer cells are discussed later.
Upon administration of chemotherapeutic agents, several reactions will be initiated to stimulate cell death responses, such as generation of ROS or direct attack onto DNA, further resulting in DNA damage and fragmentation.30 The DAPI staining results in Figure 5 demonstrate fragmentation and condensation of chromatin of A2780 cells by PTZ. The DNA fragmentation can be observed at the lowest applied dosage of 0.3 μM up to the highest dosage of 0.9 μM. Instead for untreated control, it showed less blue fluorescence intensity compared to other groups (Figure 5A) due to lower number of fragmented DNA. Referring back to the results in Figure 4C–E, it showed PTZ-induced apoptosis toward A2780 cancer cells through the illustration of several apoptosis features such as fragmented nucleus, condensed chromatin, release of DNA content into cytoplasm, shrinkage of the cells’ size due to the dissociation of plasma membrane cytoskeleton, and formation of membrane blebbing. Application of PTZ toward A2780 ovarian cancer cells results in the upregulation of caspase-6, caspase-3, and caspase-7 (Table 1); these caspases are actively involved in the formation of membrane blebbing, cell shrinkage, chromatin condensation, and fragmentation of DNA.31,32 In conjunction with the findings in Figures 4 and 5, activation of caspase-3 brings about increased enzymatic activity of DNA fragmentation factor subunit alpha (DFFA) (Table 1). The DFFA acts as substrate for caspase-3; upon induction of injury stimuli toward cancer cells, the DFFA is triggered to cleave cellular DNA into oligonucleotide DNA fragments, followed by cell death signaling cascade.32 In contrast to the untreated group, the cancer cells appeared to be clear, shiny with intact plasma membrane (Figure 4A).
Furthermore, activation of caspase-6 results in the inhibition of the LMNB1 gene which involved in encoding lamin-B1 protein. Inhibition of lamin-B1 results in the dissociation of membrane cytoskeleton matrix and loss of membrane phospholipid asymmetry.33 Hence, it gives the morphology of translocation of phosphatidylserine (PS) from inner leaflet of plasma membrane to outer leaflet.34 PS plays an important role in the last stage of apoptosis as it acts as a marker to be engulfed by phagocytes. Therefore, PS can be used as a quantitative model to study apoptosis through counting the amount of Annexin V-FITC antibodies bound to PS. The quadrant graph in Figure 6 showed that PTZ induced apoptosis toward A2780 cells from 76.05% at 0.3 μM to 82.24% at 0. 9 μM, associated with slightly increase of percentage of necrotic cells at highest concentration. To a larger extent, it is suggested that PTZ induces cell death on A2780 cells via apoptosis based on the consistent findings in AO/PI staining, DAPI staining, and Annexin V-FTIC assay.
One of the mechanisms of action of cancer therapeutic agents is induction of oxidative stress-caused cell death.34–36 Beyond the direct attack upon cellular DNA, apoptosis can be induced via increased activity of cellular ROS and the effects toward mitochondria of cancer cells.24,31 As shown in Figure 7, application of PTZ at increased concentration results in increased cellular ROS level from 113.69% to 129.54% in comparison with the untreated group (normalized to 100%), thus indicating that PTZ increased ROS level significantly, especially the H2O2. The generated ROS will then directly attack DNA and cause oxidative damage in DNA, thus inducing the expression of p53 (Table 1), the key regulator gene for apoptosis.37 If the DNA damage is irreparable, the p53 gene alternates the mitochondrial function through the upregulation of pro-apoptotic genes, at the same time it also prevents these DNA-damaged cells from further division.35 Noticed in Table 1, the mRNA levels of TP53 were upregulated 56,591.36-fold. However, PTZ exhibited a downregulatory effect toward TP73, the isomer homolog of TP53 tumor suppressor genes.31
High levels of p53 further increased the cellular levels and activities of ROS, thus it will induce oxidative stress on mitochondria to disrupt electron transport chain and mitochondrial membrane potential (ΔΨm; Figure 8).38 Polarization of ΔΨm leads to opening of the mitochondrial transmembrane pore (MTMP) and further release of its contents into cytosol. Among that, the apoptosis-inducing factor-1 (AIFM1) was delocalized from mitochondria and upregulated. As shown in Table 1, the mRNA level of AIFM1 was increased by 2,050.69-fold. The free AIFM1 was translocated into nucleus to carry out nuclear and DNA fragmentation37 which is consistent with the findings in DAPI and AOPI staining.
Apoptosis consists of intrinsic (mitochondrial-dependent) pathway and extrinsic (cell death receptor-dependent) pathway. The intrinsic pathway involves mitochondria through the regulation of mitochondrial membrane potential.34 The mitochondrial membrane is composed by inner mitochondrial membrane (IMM) and outer mitochondrial membrane (OMM). The potential of mitochondrial membrane is strictly regulated by the ratio of pro-apoptotic genes (Bax, Bad, Bid, Bik, Bcl-10) and anti-apoptotic genes (BAG1, BAG3, Bcl-2) located at OMM, respectively.39–41 Based on the PCR results, PTZ induced apoptosis pathway by the upregulation of Bax and Bcl-10 followed by the downregulation of Bcl-2, thus increasing the Bax/Bcl-2 ratio. The present studies reported the same findings, indicating that the increase of Bax levels results in the transition of Bax from OMM to mitochondrial intermembrane space and compromised inner mitochondrial membrane potential, and further disruption of outer mitochondrial membrane potential and pore-formation on mitochondrial membrane.42 As downstream events, other pro-apoptotic proteins such as cytochrome c, apoptotic activating factor 1 (APAF-1), and procaspase-9 were released from mitochondria into cytosol.43 The results in Table 1 demonstrate that application of PTZ lead to the upregulation of APAF-1, cytochrome c (CYCS), and caspase-9 by 18.82-, 40.98-, and 38,928.67-fold, respectively. The free cytochrome c further binds to APAF-1 to form a cell death complex named apoptosome, and the later initiates cleavage on procaspase-9 to caspase-9.44–46 Caspase-9 in turn activates the downstream effector caspases such as caspase-3 and the homolog, caspase-7 thus triggered the signaling cascade of apoptosis.47 Interestingly, activation of APAF-1 lead to the downregulation of NAIP by −7.77 fold (Table 1). The NAIP protein, or well known as BIRC1, played key roles in anti-apoptosis by the inhibition activity of caspase-3, caspase-7, and caspase-9.47,48 Another evidence regarding the activation of caspase-9 is through the upregulation of the NOD1 gene (Table 1). The NOD1 gene called as nucleotide-binding oligomerization domain protein 1 carried the caspase recruitment domain (CARD) and caspase recruitment death domain (CRADD) which played pro-apoptotic role by enhancing the activity of caspase-9 in the initiation of intrinsic apoptosis.49 Results in Table 1 demonstrated the application of PTZ leading to the upregulation of mRNA levels of NOD1 and CRADD by 180,694.54-fold and 161.91-fold, respectively.
On the other hand, extrinsic pathway of apoptosis is regulated by transmembrane cell death receptor, such as Fas receptor (FasR) and tumor necrosis factor (TNF) receptor (TNFR).50 Upon the binding of their respective ligands, Fas and TNF, it leads to activation of caspase-2, caspase-8, and caspase-10. Based on literature reviews, binding of Fas ligand (FasL) onto Fas receptor (Fas) leads to formation of cell death complex, namely, Fas-associated death domain (FADD);51–53 concurrently, binding of the TNF onto their receptor (TNFR) formed their respective cell death complex, named TNF-associated death domain (TRADD).52–53 In contrast to the apoptosome of mitochondria-dependent apoptosis pathway, the cell death complex of death receptor-dependent called as death-inducing signaling complex (DISC).53 The FADD and TRADD further translocate into cytosol and activate caspase-8 and caspase-10 (by FADD) or caspase-2 (by TRADD) accordingly.53 The activated caspase-2, caspase-8 and caspase-10 were then further downstream activated caspase-3 and caspase-7 to proceed the apoptosis events. Table 1 shows that the tested compound upregulated Fas, FasL, FADD, TNF, TRADD, caspase-2, caspase-8, and caspase-10 at different levels, which provided evidences on the induction of extrinsic apoptosis pathways against A2780 cancer cells. The caspase-ELISA results summarized in Figure 9 further proved the presence of caspases. Based on the findings, it was found that PTZ induced cell death on A2780 cells by the activation of both the apoptosis pathways. In addition, activation of caspase-8 and caspase-10 result in cleavage of tBid into Bid, another pro-apoptotic factor.54 As downstream response, the DIABLO was upregulated (Table 1). DIABLO played a major role in encoding mitochondrial protein which involved in apoptosis such as cytochrome c, Smac, Omi, and HtrA2.55,56 The synthesis and release of these proteins into cytosol allow upregulation activation of caspases as shown in Table 1.
NF-κB is a nuclear binding protein found in epithelial cells, which plays a key pivotal role in cellular proliferation, survival, and death.56 Mutation of NF-κB increased its activities ultimately by translocation into nucleus and direct binding to DNA resulting in the increased expression of genes that promotes cell proliferation, invasion, metastasis, and angiogenesis.57,58 Hence suppression of NF-κB pathway is targeted as one of the anticancer mechanisms. The common secondary tumor of ovarian cancer reported is tumor in pelvis and abdomen, if spreads further, it will go into liver, spleen, and lastly lungs and bones.1,9,10 Among the protein expressed in the events of mutation of NF-κB, the genes derived from Baculovial IAP repeating containing gene family (BIRC family) such as BIRC6 and XIAP (or known as BIRC4) are the causing factor for cancer cell invasion and metastasis;11,12 meanwhile, BIRC5 and IGF1R promote vascularization and angiogenesis.12,13,59,60 In the current study, the cell invasion assay was conducted to investigate the ability of PTZ to inhibit the invasion of A2780 ovarian cancer cells. The Matrigel™ Invasion Chamber provided an endothelial-like membrane which mimicked the endothelial layers of blood vessels. Referring to the results shown in Figure 11, PTZ exhibited the inhibition on invasion of A2780 cells through the membrane in a concentration-dependent manner, by assuming that the untreated group showed 100% of invasion. The results in Table 1 demonstrated downregulation of NF-κB, BIRC6, and XIAP genes by −3.01-, −26,872.51-, and −6,301.42-fold, respectively. Hence, it is hypothesized that upon inhibition of NF-κB by PTZ, it further prevented the formation of (BIRC6-XIAP) complexes and thus reduced the invasion rate of cancer cells.50 On the other hand, results in Table 1 show that PTZ downregulated mRNA levels of BIRC5 and IGF1R by −2,093.58- and −7.37-fold, respectively, thus it suggested PTZ to possess the anti-metastatic capability. Another supportive result was issued from the downregulation of AKT1 gene by −2,215.03-fold. During carcinogenesis, activation of phosphatidylinositol 3-kinase results in the synthesis of serine-threonine protein kinase encoded by AKT1 gene.61 This enzyme mediates several growth effects on cancer cells by the activation of vascular endothelial growth factor pathway, platelet-derived growth factor pathway, insulin-like growth factor 1 (IGF1R) pathway, and NF-κB pathway.62–64 Therefore, downregulation of AKT1 increased the possibility of this tested compound on the inhibition of cancer cells angiogenesis, in conjunction with the downregulation of IGF1R and NF-κB genes. However, future studies are recommended to focus on how PTZ inhibits invasion and metastasis of cancer cells.
A previous study demonstrated that PTZ upregulated mRNA levels of CDKN1A.20 The CDKN1A played an important role in regulating cell cycle by encoding the p21 proteins.65,66 Based on this finding, the cell cycle analysis was conducted in the current study. As seen in Figure 10, PTZ induce cell cycle checkpoint on A2780 cancer cells at G2/M phase. Accumulation of cells in G2/M phase decreased the population of cells in G0/G1 phase; increase of concentration of PTZ showed a significant transition of cell population from G0/G1 phase to S phase and lastly arrested G2/M phase; also, there was an increase in the cell population in sub-G0 phase. In the event of DNA being damaged by PTZ, p53 was expressed as response to DNA damage, thus shortens the duration of G0/G1 phase. Followed by the expression of p53, it will activate CDKN1A to encode p21, resulting in cell cycle arrest via inhibition of cyclins and their cyclin-dependent kinases (CDKs).67 Upon PTZ-induced damage on DNA of A2780 cells, p53 and p21 were activated to prevent these DNA damaged cells to proceed to cell division, thus the cells are arrested at G2/M phase and are ready to be eliminated via apoptosis.67,68 Beyond the p53 and p21 genes, the gene growth arrest and DNA damage 45a gene (GADD45a) were also upregulated (Table 1) in response to DNA damage. GADD45a directly inhibits the activities of cyclins at each cellular check point and permanently prevent transition of cells into mitotic (M) phase.69
In addition, as seen in Table 1, there is a significant downregulation of mRNA levels of AKT1 by −2,215.02-fold. The RAC-protein kinase Alpha 1 (Akt1) was reported with its primary function as cell survival protector to prevent cell death; oncogenic activation of AKT1 results in aggressive pathogenic proliferation of ovarian epithelial cells in conjunction with resistance to death signals. Based on the findings, the trial compound was suggested to increase the sensitivity of ovarian cancer cells toward chemotherapeutic agents through the inhibition of AKT1 genes and further limitation of its proliferation activity at G2/M phase.70,71 Lastly, the trial compound also downregulated BIRC2 and BIRC3 genes in A2780 cancer cells (Table 1) by −6.96- and −2.76-fold, respectively. Inhibition of BIRC2 and BIRC3 activities further limited the expression of BIRC5 gene (downregulated by −2,093.57-fold), thus inhibited the activities of CDKs and cyclins, particularly CDK1 and cyclin B1,71 and subsequently arrested the cell cycle at G2/M phase.
Conclusion
The trial compound PTZ was synthesized from the reactions of pyridine derivatives, and it showed promising anticancer activity toward A2780 ovarian cancer cells. By comparing the IC50 of PTZ (0.62 μM) and positive control, cisplatin (28.80 μM), the trial compound is much potent compared with cisplatin, indeed it also showed less toxicity toward HEK293 normal kidney cells and H9C2 normal heart cells. The apoptosis studies suggested that PTZ induced apoptosis on A2780 cancer cells via mitochondria-dependent and cell death receptor-dependent pathway by increased activities of caspase-9, caspase-8, caspase-10, and caspase-2, together with the downstream caspases, the caspase-6, caspase-3, and caspase-7. The proposed apoptosis signaling cascade induced by the PTZ is summarized in Figure 12. In addition, the cell cycle analysis demonstrated that PTZ induced cell cycle arrest of A2780 cells at G2/M phase, hence marked PTZ as potential cytostatic agent and cell cycle inhibitor. Besides that, PTZ also suppressed the invasion rate of A2780 cells via in vitro cell invasion model via inhibition of NF-κB and (BIRC6-XIAP) complex. Based on these primary findings, it can be concluded that PTZ possesses potential as the chemotherapeutic agent.
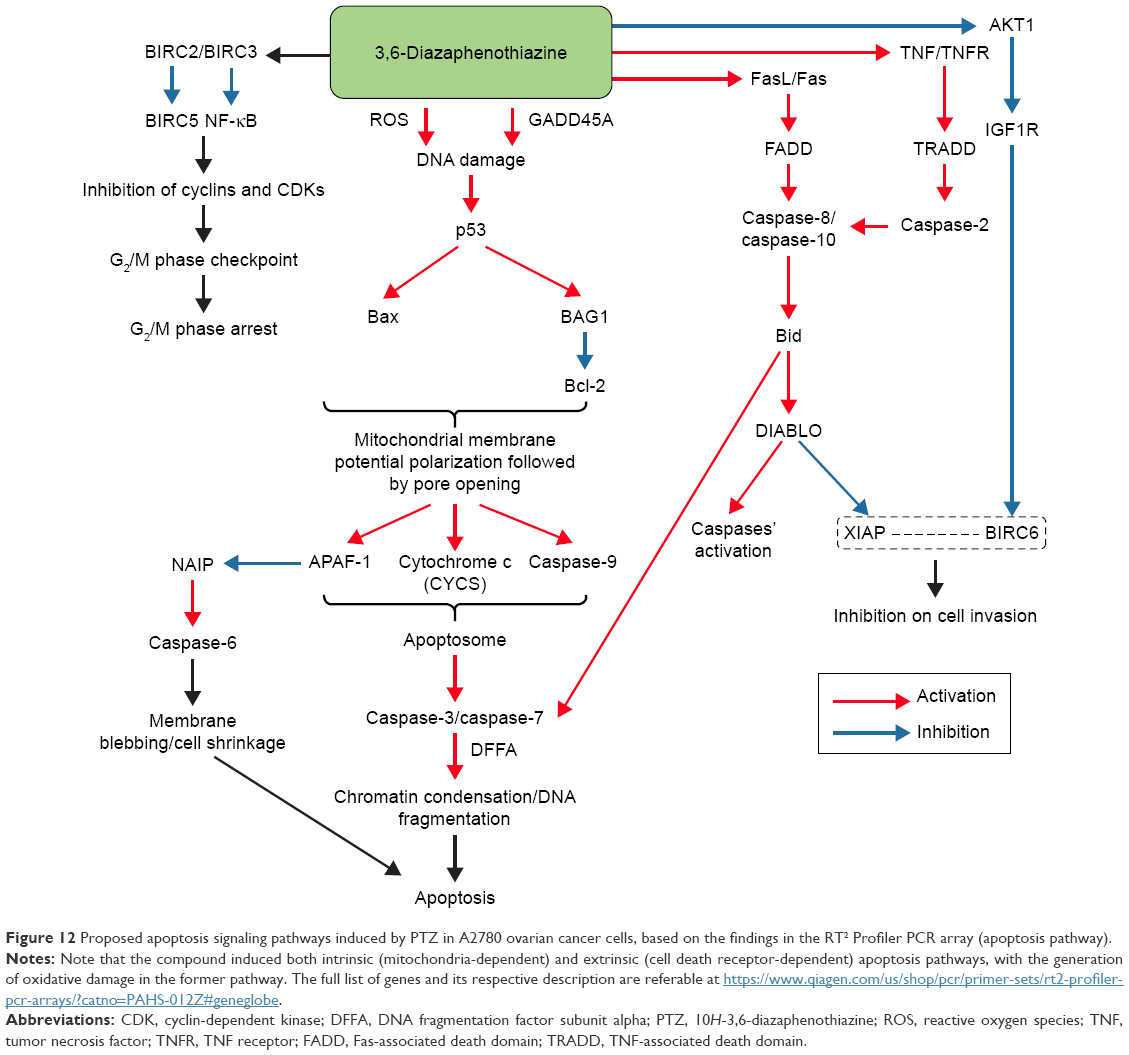 | Figure 12 Proposed apoptosis signaling pathways induced by PTZ in A2780 ovarian cancer cells, based on the findings in the RT2 Profiler PCR array (apoptosis pathway). |
Acknowledgment
The authors sincerely acknowledge the research funding support from the Innoscience Research Sdn. Bhd with the grant number ISR/RPFA/PSN/2016M002.
Author contributions
All authors contributed toward data analysis, proposing the signaling pathways, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre L, Siegel R, Jemal A. Global Cancer Facts and Figures. 3rd ed. Atlanta, GA, USA: American Cancer Society; 2015. | ||
Lee S, Whang I, Wan Q, et al. Profiles of teleost DNA fragmentation factor alpha and beta from rock bream (Oplegnathus fasciatus): molecular characterization and genomic structure and gene expression in immune stress. Genes Genom. 2016;38:193–204. | ||
Baghbani F, Moztarzadeh F. Bypassing multidrug resistant ovarian cancer using ultrasound responsive doxorubicin/curcumin co-deliver alginate nanodroplets. Col Sur B Biointerfaces. 2017;153:132–140. | ||
Rzepecka IK, Szafron LM, Stys A, et al. Prognosis of patients with BRCA1-associated ovarian carcinomas depends on TP53 accumulation status in tumor cells. Gynecol Oncol. 2017;144:369–376. | ||
Rossing MA, Wicklund KG, Cushing-Haugen KL, et al. Predictive value of symptoms for early detection of ovarian cancer. J Natl Cancer Inst. 2010;102:222–229. | ||
Ore RM, Baldwin L, Woolum D, et al. Symptoms relevant to surveillance for ovarian cancer. Diagnostics. 2017;7:18. | ||
Urban N, Hawley S, Janes H, et al. Identifying post-menopausal women at elevated risk for epithelial ovarian cancer. Gynecol Oncol. 2015;139:253–260. | ||
Leung ACY, Cook LS, Swenerton K, et al. Tea, coffee, and caffeinated beverage consumption and risk of epithelial ovarian cancers. Cancer Epid. 2016;45:119–125. | ||
Burghaus S, Fasching PA, Haberle L, et al. Genetic risk factors for ovarian cancer and their role for endometriosis risk. Gynecol Oncol. 2017;145:142–147. | ||
Chiyoda T, Hart PC, Eckert MA, et al. Loss of BRCA1 in the cells of origin of ovarian cancer induces glycolysis: a window of opportunity for ovarian cancer chemoprevention. Cancer Prev Res. 2017;10:255–266. | ||
Wang R, Kang Y, Lohr CV, et al. Reciprocal regulation of BMF and BIRC5 (Survivin) linked to Eomes overexpression in colorectal cancer. Cancer Lett. 2016;381:341–348. | ||
Lamers F, Schild L, Koster J, et al. Targeted BIRC5 silencing using YM155 causes cell death in neuroblastoma cells with low ABCB1 expression. Eur J Cancer. 2012;48:763–771. | ||
Chaudhary AK, Yadav N, Bhat TA, et al. A potential role of X-linked inhibitor of apoptosis protein in mitochondrial membrane permeabilization and its implication in cancer therapy. Drug Dis Today. 2016;21:38–47. | ||
Turner TB, Buchsbaum DJ, Straughn JM Jr, et al. Ovarian cancer and the immune system – the role of targeted therapies. Gynecol Oncol. 2016;142:349–356. | ||
Li Y, Jiang Y, Yi Y, et al. Application of auricular acupoints therapy in relieving the gastrointestinal side effects induced by chemotherapy: an integrative review. Chinese Nurs Res. 2016;3:58–61. | ||
Pluta K, Morak-Młodawska B, Jeleń M. Recent progress in biological activities of synthesized phenothiazines. Eur J Med Chem. 2011;48:3179–3189. | ||
Zimecki M, Artym J, Kocięba, M, et al. The immunosuppressive activities of newly synthesized azaphenothiazines in human and mouse models. Cell Mol Biol Lett. 2009;15:622–635. | ||
Pluta K, Jeleń M, Morak-Młodawska B, et al. Anticancer activity of newly synthesized azaphenothiazines from NCI’s anticancer screening bank. Pharmacol Rep. 2010;62:319–332. | ||
Morak-Młodawska B, Pluta K, Latocha M, et al. Synthesis, spectroscopic characterization, and anticancer activity of new 10-substituted 1,6-diazaphenothiazines. Med Chem Res. 2016;25:2425–2433. | ||
Morak-Młodawska B, Pluta K, Latocha M, Suwińka K, Jeleń M, Kuśmierz D. 3,6-Diazaphenothiazines as potential lead molecules – synthesis, characterization and anticancer activity. J Enzyme Inhib Med Chem. 2016;31:1512–1519. | ||
Kastrinsky DB, Sangodkar J, Zaware N, et al. Reengineered tricyclic anticancer agents. Bioorg Med Chem. 2015;23:6528–6534. | ||
Prinz H, Ridder AK, Vogel K, et al. N-heterocyclic (4-phenylpiperazin-1-yl)methanones derived from phenoxazine and phenothiazine as highly potent inhibitors of tubulin polymerization. J Med Chem. 2017;60:749–766. | ||
Bisi A, Meli M, Gobbi S, et al. Multidrug resistance reverting activity and antitumor profile of new phenothiazine derivatives. Bioorg Med Chem. 2008;16:6474–6482. | ||
Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304:463–470. | ||
Sappanimuthu T, Kilambi N, Sundaram S, et al. Antimicrobial activity of new dumbbell-shaped phenothiazine cinnamides. Res Chem Intermed. 2017;43:2401–2414. | ||
Lund RJ, Huhtinen K, Salmi J, et al. DNA methylation and transcriptome changes associated with cisplatin resistance in ovarian cancer. Sci Rep. 2017;7:1469. | ||
Xiao M, Cai J, Cai LQ, et al. Let-7e sensitizes epithelial ovarian cancer to cisplatin through repressing DNA double strand break repair. J Ovarian Res. 2017;10:24–36. | ||
Pai SG, Carneiro BA, Mota JM, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10:101–111. | ||
Deshmuk SK, Srivastava SK, Zubair H, et al. Resistin potentiates chemoresistance and sternness of breast cancer cells: implications for racially disparate therapeutic outcomes. Cancer Lett. 2017;396:21–29. | ||
Ooi KK, Ang KP, Abdah MA, et al. Phosphanegold(I) thiolates, Ph3PAu[SC(O-R)=NC6H4Me-4] for R = Me, Et and iPr, induce apoptosis, cell cycle arrest and inhibit cell invasion of HT-29 colon cancer cells through modulation of the nuclear factor-κB activation pathway and ubiquitination. J Biol Inorg Chem. 2015;20:855–873. | ||
Zamaraev AV, Kopeina GS, Prokhorova EA, et al. Post-translational modification of caspases: the other side of apoptosis regulation. Trends Cell Biol. 2017;27:322–339. | ||
Flach RJR, Qin H, Zhang L, et al. Loss of mitogen-activated protein kinase phosphatase-1 protects from hepatic steatosis by repression of cell death-inducing DNA fragmentation factor A (DFFA)-like effector C (CIDEC)/fat-specific protein 27. J Biol Chem. 2011;286:22195–22202. | ||
Deng Z, Gao P, Yu LL, et al. Ruthenium complexes with phenylterpyridine derivatives target cell membrane and trigger death receptors-mediated apoptosis in cancer cells. Biomaterials. 2017;129:111–126. | ||
Atkin-Smith GK, Poon IKH. Disassembly of the dying: mechanisms and functions. Trends Cell Biol. 2017;27:151–162. | ||
Holmgren A, Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem Biophys Res Commun. 2010;396:120–124. | ||
Liu Y, Duan D, Yao J, et al. Dithiaarsanes induce oxidative stress-mediated apoptosis in HL-60 cells by selectively targeting thioredoxin reductase. J Med Chem. 2014;57:5203–5211. | ||
Wei J, Zhang L, Ren LH, et al. Endosulfan induces cell dysfunction through cycle arrest resulting from DNA damage and DNA damage response signaling pathways. Sci Total Environ. 2017;589:97–106. | ||
Almada M, Fonseca BM, Amaral C, et al. Anandamide oxidative metabolism-induced endoplasmic reticulum stress and apoptosis. Apoptosis. 2017;22:816–826. | ||
Sun RWY, Che CM. The anti-cancer properties of gold (III) compounds with dianionic porphyrin and tetradentate ligands. Coord Chem Rev. 2009;253:1682–1691. | ||
Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signaling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977–2992. | ||
García-Sáez AJ, Mingarro I, Pérez-Payá E, et al. Membrane-insertion fragments of Bcl-xL, Bax, and Bid. Biochemistry. 2004;43:10930–10943. | ||
Huerta S, Heinzerling JH, Auguiano-Hernandez YM, et al. Modification of gene products involved in resistance to apoptosis in metastatic colon cancer cells: roles of Fas, Apaf-1, NFκB, IAPs, Smac/DIABLO, and AIF. J Surg Res. 2007;142:184–194. | ||
Reed JC. Apoptosis-targeted therapies for cancer. Cancer Cell. 2003;3:17–22. | ||
Attaran-Bandarabadi F, Abhari BA, Neishabouri SH, et al. Integrity of XIAP is essential for effective activity recovery of apoptosome and its downstream caspases by Smac/Diablo. Int J Biol Marc. 2017;101:283–289. | ||
Shakeri R, Kheirollahi A, Davoodi J. Apaf-1: regulation and function in cell death. Biochimie. 2017;135:111–125. | ||
Linder M, Tschernig T. Vasculogenic mimicry: possible role of effector caspase-3, caspase-6 and caspase-7. Ann Anat. 2016;204:114–117. | ||
Davoodi J, Ghahremani MH, Es-haghi A, et al. Neuronal apoptosis inhibitory protein, NAIP, is an inhibitor of procaspase-9. Int J Biochem Cell Biol. 2010;42:958–964. | ||
Kaparakis-Liaskos M. The intracellular location mechanisms and outcomes of NOD1 signalling. Cytokine. 2015;74:207–212. | ||
Kutikhin AG. Role of NOD1/CARD4 and NOD2/CARD15 gene polymorphisms in cancer etiology. Human Immunol. 2011;72:955–968. | ||
Yin D, Woodruff M, Zhang Y, et al. Morphine promotes Jurkat cell apoptosis through pro-apoptotic FADD/P53 and anti-apoptotic PI3K/Akt/NF-κB pathways. J Neuroimmunol. 2006;174:101–107. | ||
Park YH, Jeong MS, Park HH, et al. Formation of the death domain complex between FADD and RIP1 proteins in vitro. Biochim Biophys Acta. 2013;1834:292–300. | ||
Shukla K, Sharma AK, Will R, et al. MicroRNA-30c-2-3p negatively regulates NF-κB signaling and cell cycle progression through downregulation of TRADD and CCNE1 in breast cancer. Mol Oncol. 2015;9:1106–1119. | ||
Park YH, Jeong MS, Jang SB. Death domain complex of the TNFR-1, TRADD, and RIP1 proteins for death-inducing signaling. Biochem Biophys Res Commun. 2014;443:1155–1161. | ||
Veresov VG, Davidovskii AI. Activation of Bax by joint action of tBid and mitochondrial outer membrane: Monte Carlo simulations. Eur Biophys J. 2009;38:941–960. | ||
Checker R, Patwardhan RS, Sharma D, et al. Plumbagin, a vitamin K3 analogue, abrogates lipopolysaccharide-induced oxidative stress, inflammation and endotoxic shock via NF-kappa B suppression. Inflammation. 2014;37:542–554. | ||
Ahmad A, Banerjee S, Wang Z, et al. Plumbagin-induced apoptosis of human breast cancer cells is mediated by inactivation of NF-kappaB and Bcl-2. J Cell Biochem. 2008;105:1461–1471. | ||
Bao W, Zhu F, Duan Y, et al. HtrA1 resensitizes multidrug-resistant hepatocellular carcinoma cells by targeting XIAP. Biomed Pharmacother. 2015;70:97–102. | ||
McCubrey JA, Lertpiriyapong K, Fitzgerald TL, et al. Roles of TP53 in determining therapeutic sensitivity, growth, cellular senescence, invasion and metastasis. Adv Biol Regul. 2017;63:32–48. | ||
Xu L, Zhou R, Yuan L, et al. IGF1/IGF1R/STAT3 signaling-inducible IFITM2 promotes gastric cancer growth and metastasis. Cancer Lett. 2017;393:76–85. | ||
Werner H, Sarfstein R. Transcriptional and epigenetic control of IGF1R gene expression: implications in metabolism and cancer. Growth Horm IGF Res. 2014;24:112–118. | ||
Tiwari V, Kamran MZ, Ranjan A, et al. Akt1/NFκB signaling pathway activation by a small molecule DMA confers radioprotection to intestinal epithelium in xenograft model. Free Radic Biol Med. 2017;108:564–574. | ||
Sabbineni H, Alwhaibi A, Goc A, et al. Genetic deletion and pharmacological inhibition of Akt1 isoform attenuates bladder cancer cell proliferation, motility and invasion. Eur J Pharmacol. 2015;764:208–214. | ||
Zhou W, Fu X, Liu J, et al. RNAi knockdown of the Akt1 gene increases the chemosensitivity of gastric cancer cells to cisplatin both in vitro and in vivo. Regul Peptides. 2012;176:13–21. | ||
Yang X, Liu S, Kharbanda S, et al. AKT1 induces caspase-mediated cleavage of the CDK inhibitor p27Kip1 during cell cycle progression in leukemia cells transformed by FLT3-ITD. Leukemia Res. 2012;36:205–211. | ||
Lopes MR, Machado-Neto JA, Traina F, et al. Differential profile of CDKN1A and TP53 expressions in bone marrow mesenchymal stromal cells from myeloid neoplasms. Rev Bras Hematol Hemoter. 2016;38:368–370. | ||
Röhrs S, Kutzner N, Vlad A, et al. Chronological expression of Wnt target genes CCND1, MYC, CDKN1A, TFRC, PLF1 and Ramp3. Cell Biol Int. 2009;33:501–508. | ||
Suhail TV, Singh P, Manna TK. Suppression of centrosome protein TACC3 induces G1 arrest and cell death through activation of p38–p53–p21 stress signaling pathway. Eur J Cell Biol. 2015;94:90–100. | ||
Nakagawa J, Matsuoka M. Suppression of zinc-induced p53 phosphorylation and p21 expression by wortmannin in A549 human pulmonary epithelial cells. Environ Toxicol Pharmacol. 2008;26:109–112. | ||
Li D, Dai C, Zhou Y, et al. Effect of GADD45a on olaquindox-induced apoptosis in human hepatoma G2 cells: involvement of mitochondrial dysfunction. Env Toxicol Pharmacol. 2016;46:140–146. | ||
Gong HJ, Cao Y, Han G, et al. p3/microRNA-374b/AKT1 regulates colorectal cancer cell apoptosis in response to DNA damage. Int J Oncol. 2017;50:1785–1791. | ||
Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27–36. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.