Back to Journals » International Journal of Nanomedicine » Volume 15
Zinc Oxide Nanoparticles Induce Mitochondrial Biogenesis Impairment and Cardiac Dysfunction in Human iPSC-Derived Cardiomyocytes
Authors Li Y ![]() , Li F, Zhang L
, Li F, Zhang L ![]() , Zhang C, Peng H, Lan F, Peng S, Liu C, Guo J
, Zhang C, Peng H, Lan F, Peng S, Liu C, Guo J
Received 15 February 2020
Accepted for publication 1 April 2020
Published 21 April 2020 Volume 2020:15 Pages 2669—2683
DOI https://doi.org/10.2147/IJN.S249912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yan Shen
Yujie Li,1,2 Fengxiang Li,2 Lincong Zhang,2 Chi Zhang,2 Hui Peng,2 Feng Lan,3 Shuangqing Peng,2 Chao Liu,1,2 Jiabin Guo1,2
1Graduate School, Academy of Military Medical Sciences, Beijing, People’s Republic of China; 2Department of Operational Medical Protection, PLA Center for Disease Control and Prevention, Beijing, People’s Republic of China; 3Beijing Key Laboratory for Cardiovascular Precision Medicines, Anzhen Hospital, Capital Medical University, Beijing, People’s Republic of China
Correspondence: Chao Liu Tel +86-10-66948389
Email [email protected]
Jiabin Guo Tel +86-10-66948463
Email [email protected]
Background: Zinc oxide nanoparticles (ZnO NPs) are one of the most widely used nanomaterials in a variety of fields such as industrial, pharmaceutical, and household applications. Increasing evidence suggests that ZnO NPs could elicit unignorable harmful effect to the cardiovascular system, but the potential deleterious effects to human cardiomyocytes remain to be elucidated. Human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) have been increasingly used as a promising in vitro model of cardiomyocyte in various fields such as drug cardiac safety evaluation. Herein, the present study was designed to elucidate the cardiac adverse effects of ZnO NPs and explore the possible underlying mechanism using hiPSC-CMs.
Methods: ZnO NPs were characterized by transmission electron microscopy and dynamic light scattering. The cytotoxicity induced by ZnO NPs in hiPSC-CMs was evaluated by determination of cell viability and lactate dehydrogenase release. Cellular reactive oxygen species (ROS) and mitochondrial membrane potential were measured by high-content analysis (HCA). Mitochondrial biogenesis was assayed by detection of mtDNA copy number and PGC-1α pathway. Moreover, microelectrode array techniques were used to investigate cardiac electrophysiological alterations.
Results: We demonstrated that ZnO NPs concentration- and time-dependently elicited cytotoxicity in hiPSC-CMs. The results from HCA revealed that ZnO NPs exposure at low-cytotoxic concentrations significantly promoted ROS generation and induced mitochondrial dysfunction. We further demonstrated that ZnO NPs could impair mitochondrial biogenesis and inhibit PGC-1α pathway. In addition, ZnO NPs at insignificantly cytotoxic concentrations were found to trigger cardiac electrophysiological alterations as evidenced by decreases of beat rate and spike amplitude.
Conclusion: Our findings unveiled the potential harmful effects of ZnO NPs to human cardiomyocytes that involve mitochondrial biogenesis and the PGC-1α pathway that could affect cardiac electrophysiological function.
Keywords: Zinc oxide nanoparticles, ZnO NPs, human induced pluripotent stem cells-derived cardiomyocytes, hiPSC-CMs, cardiac dysfunction, mitochondrial biogenesis, PGC-1α
Introduction
Nanotechnology has emerged as a promising technique for various biomedical applications in the last decades. Zinc oxide nanoparticles (ZnO NPs) are one of the most widely used nanomaterials in industrial and household applications such as drug delivery solutions, cosmetics, food additives, clothing, sunscreens, sports, electronics, and other consumer products.1 ZnO NPs exert many excellent properties such as anticancer and antimicrobial activities, ultraviolet (UV)-absorbing properties and transparency for visible light, making these NPs superior as drug carrier systems and sunscreen agents. The annual global production of ZnO NPs has rapidly increased over recent years and is now approximately one million tons annually.2 However, the increasing production and application of ZnO NPs has raised growing concerns in the public with regard to their potential human health and environmental adverse effects. Notably, it is suggested that ZnO NPs are one of the most toxic metallic oxide NPs as compared to other NPs such as Fe3O4, Al2O3 and TiO2, due to their ion-shedding property.3
The toxicity of ZnO NPs has been investigated in many different biological systems, including various mammalian cells, bacteria, experimental animals, and clinical studies. Most of these studies are generally focused on direct target organs, such as the liver, skin and the respiratory system.4,5 However, increasing evidence suggests that ZnO NPs could also be transferred to the heart and may cross biological barriers,6–8 and so hence, could elicit harmful effects on the cardiovascular system.9 A clinical study has shown that systemic inflammatory effects and cardiovascular damage were observed in human volunteers following inhalation of ZnO NPs at concentrations below the occupational exposure limit for ZnO (5 mg/m3).10 Elevation of troponin and creatine kinase as well as induction of inflammation, DNA damage and apoptosis have been found in rat hearts after oral exposure to ZnO NPs.11 Exposure to ZnO NPs via intratracheal instillation and inhalation was also found to induce cardiac inflammation, fibrosis and necrosis of the myocardium in rats.12 Similarly, by using an in vitro gas-blood barrier model, it has been reported that ZnO NPs can induce detrimental effects via the lungs into the cardiovascular system.13,14 ZnO NPs were also found to negatively impact cardiorespiratory function and energy metabolism in freshwater fish (Catostomus commersonii).15 Many in vitro studies revealed that ZnO NPs can induce toxic effects in cardiac cells such as a decrease in survival rate, mitochondrial dysfunction, reactive oxygen species (ROS) accumulation, and release of inflammatory factors.16,17 These studies suggest that exposure to ZnO NPs via multiple routes could induce cardiovascular damage, but the exact harmful effects of ZnO NPs in cardiomyocytes remain to be elucidated.
A reliable and human-relevant platform is essential to illustrate ZnO NPs-induced deleterious effects in the cardiovascular system and to provide a good understanding of its mechanism of action.18 Currently, most of the in vitro cardiotoxicity models used in assessment of nanoparticles are based on primary cultured cardiac cells and immortalized cell lines. However, it is challenging to extrapolate the data generated from these systems to humans since these in vitro models show many limitations. For instance, primary cells are not easily obtained and are not easily maintained in culture for prolonged periods or expanded in vitro. Immortalized cell lines show some but not all characteristics of the cardiac function and cannot faithfully replicate cellular electrophysiology and biochemical function. Thus, there is a need for an effective and reliable source of cells for better investigation of nanomaterial-induced cardiac effects in vitro. As a new in vitro model of cardiomyocytes, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) are promising in diminishing the current limitations in cardiac safety evaluation.19 HiPSC-CMs have several advantages over current in vitro models such as immortalized cell lines, human cadaveric tissue and primary cultures of nonhuman animal origin. In particular, hiPSC-CMs eliminate the considerable differences that exist between traditional cell models and human cardiomyocytes, including beating rates, energetics, myofilament composition, expression of key ion channels and electrophysiology.20 Moreover, the application of high-throughput technologies, such as high-content analysis (HCA), microelectrode array (MEA) and patch-clamp analysis, enable the advantages of hiPSC-CMs to be realized in the rapid screening of cardiotoxicity.21
The present study was designed to investigate the potential cardiac adverse effects of ZnO NPs using hiPSC-CMs. Our results demonstrated that ZnO NPs concentration- and time-dependently elicited cytotoxicity. By application of high-content analysis, we showed that low-cytotoxic concentration ZnO NPs significantly promoted ROS generation with mitochondrial membrane potential loss. We further demonstrated that ZnO NPs could impair mitochondrial biogenesis and inhibit PGC-1α pathway. In addition, ZnO NPs at low concentrations were found to trigger cardiac electrophysiological alterations, using microelectrode assay analysis.
Materials and Methods
Nanoparticles and Characterization
Zinc oxide nanoparticles (ZnO NPs) were obtained from Sigma-Aldrich in the USA with the particle size ≤50 nm. The ultrastructure and mean size of 1 mM ZnO NPs were analyzed by transmission electron microscope (Hitachi H-7650, Japan). The hydrodynamic diameter and zeta potential of ZnO NPs at 1 mM in double distilled water and cell culture medium were, respectively, determined by dynamic light scattering method using Nano Brook 90Plus Zeta (Brookhaven, USA).
HiPSCs Culture and Cardiomyocyte Differentiation
HiPSCs (derived from healthy male renal epithelial cells) were purchased from the Cellapy Biological Technology Company (Cellapy, China). The 6-well plates were pre-coated with feeder-free Matrigel (1 mL per well) overnight at 37°C. HiPSCs were maintained in PSCeasy culture medium in a humidified incubator at 37°C with 5% CO2. Cells were passaged every 3 to 4 days using 0.5 mM EDTA. Thiazovivin (MedChemExpress, USA), a rho kinase inhibitor, was added into the culture media at the final concentration of 10 μM on the first day following cell passaging. Culture media were changed every day and tested for mycoplasma using a MycoEasy Kit (Cellapy).
HiPSCs were induced to cardiomyocytes by selective modulation of the wnt pathway method in monolayer culture using CardioEasy Kit (Cellapy) as previously reported.22 In brief, hiPSCs were dissociated to the single cell level by using EDTA to ensure the efficiency of differentiation. Cells were seeded uniformly into matrigel-coated 6-well plates at 7.5×105 per well and maintained for 3–4 days till 80–90% confluence. To promote the hiPSCs differentiate into cardiomyocytes, specific CardioEasy differentiation media (4 mL/well) were used every 2 days during day 0 to 10. Generally, contracting cells were observed from day 7–8 and thereafter. To obtain higher purity of cardiomyocytes, the cells were incubated with no glucose cardiomyocyte purification medium (Cellapy) on day 10–12. Afterward, the media were replaced with cardiomyocyte maintenance medium (Cellapy) and were changed every other day.
Nanoparticle Treatment
Cells were cultured to day 25–30 and used for experiments. The hiPSC‐CMs were plated on matrigel-coating plates 5 days before experimentation. Briefly, cells were dissociated using Cell Dissociation Solution (Cellapy) by two steps: step 1 for 10 min at 37°C using solution I; step 2 for 45 min at 37°C using solution II, centrifuged at 800 rpm for 5 min. The cells were plated on different plates at certain cell density (6-well 1×106/well, 24-well 4×104/well, 96-well 2~3×104/well). ZnO NPs were dispersed to a stock suspension at 100 mM in double distilled water. After sonication for 30 minutes, the suspension was diluted with cardiomyocyte maintenance medium to the indicated concentration. The dilutions of ZnO NPs were vortexed for 30 s before cell exposure to minimize nanoparticle agglomeration. All suspensions and dilution of ZnO NPs were freshly prepared for every experiment.
Cytotoxicity Assays
Cytotoxicity was measured by a cell viability assay and lactate dehydrogenase (LDH) release assay. Cell viability was analyzed by using the Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Japan) according to the manufacturer’s instructions. Briefly, after incubation with ZnO NPs in 96-well plate, CCK-8 solution was added to each well followed by 3 h incubation at 37°C. The absorbance was determined at 450 nm with a reference at 630 nm using MK-3 microplate reader (Thermo Scientific, USA). Data were normalized to control cultures which were considered as 100% cell survival. The release of LDH into the culture medium is recognized as an indicator of irreversible cell death due to cell membrane damage. LDH release was determined by using LDH Assay Kit (Beyotime, China) according to the manufacturer’s instructions. Briefly, immediately after completion of ZnO NPs treatment, 80 μL supernatant was collected and incubated with 40 μL freshly reaction mixture at room temperature in the dark for 30 min. The absorbance was determined at 490 nm with a reference at 630 nm using MK-3 microplate reader. Data were normalized to max release control cultures which were considered as 100% cell death.
High-Content Analysis (HCA)
To evaluate mitochondrial membrane potential (MMP) and cellular ROS levels, hiPSC-CMs were cultured at 2×104/well density in matrigel-coating 96-well plate with optical optimization bottom (PerkinElmer, USA). The cells were incubated with different concentrations of ZnO NPs for 6 h. Afterward, the cells were washed with PBS and labeled with co-staining include: I. nucleus probe Hoechst 33342 (1 μM), intracellular ROS probe CM-H2DCFDA (10 μM), and MMP probe TMRM (100 nM) for 15 min at 37°C; II. nuclei probe Hoechst 33342 (1 μM) and mitochondria content probe Mito-Tracker Green (100 nM) for 30 min at 37°C. All above-mentioned probes were purchased from Invitrogen (USA) and dissolved in culture medium. Cells were then washed twice with PBS to thoroughly remove the fluorescent probes. Operetta CLS™ high-content analysis system (PerkinElmer, USA) was used to capture images at specific fluorescence excitation/emission wavelength according to probe instructions (Hoechst 33342, 350/461 nm; CM-H2DCFDA, 485/530 nm, TMRM 548/574 nm, Mito-Tracker Green 490/516 nm). Each group had at least three replicate wells, and at least 9 fields of view were randomly selected for quantitative fluorescence analysis. The fluorescence intensity of each probe within the cells was performed automatically by Harmony® high-content imaging and analysis software (PerkinElmer, USA; version 4.1).
Mitochondrial DNA (mtDNA) Copy Number Quantification
mtDNA copy number was determined as a marker for mitochondrial abundance using quantitative real-time PCR (qPCR) method as we previously reported.23 Briefly, total DNA was isolated by using TIANamp Genomic DNA Kit (TIANGEN, China) according to the manufacturer’s instruction. The concentration of DNA in the extracts was measured by a NanoDrop 2000 Spectrophotometer (Thermo Scientific, USA). DNA primers were designed to detect ND1 (5ʹ-GGAGTAATCCAGGTCGGT-3ʹ and 5ʹ-TGGGTACAATGAGGAGTAGG-3ʹ) as a maker for mtDNA and GAPDH (5ʹ-AAGGTGGAGGAGTGGGTGT-3ʹ and 5ʹ-TCAAGAAGGTGGTGAAGCAG-3ʹ) for nuclear DNA. Reactions were performed on LightCycler 480 II real-time PCR machine (Roche, Switzerland) with SYBR Green I Master (Roche). The reaction conditions were 30 s at 95°C followed by 40 cycles of 5 s at 95°C and 30 s at 60°C; 1 cycle of 5 s at 95°C and 60 s at 60°C; 1 cycles of 30 s at 50°C. Relative amount of mtDNA copy number was calculated by 2−ΔΔCt method.
Western Blotting
Cells were lysed with cold RIPA buffer containing protease and phosphatase inhibitors (Beyotime, China). The lysed cells were centrifuged at 4°C with 12,000 rpm for 20 min. Total proteins were separated on 10% SDS-PAGE and then transferred to PVDF membranes (Millipore, USA). After blocking with 5% non-fat milk, the membranes were incubated with different primary antibodies (dilution 1:1000) overnight at 4°C, followed by further incubation with secondary antibodies (Thermo Fisher, USA; dilution 1:5000) for 1 h at room temperature. Primary antibodies against PGC-1α, COX IV, GAPDH were purchased from Cell Signaling Technology (USA); antibody against TFAM was purchased from Abcam (UK). The images were visualized by a super enhanced chemiluminescence detection kit (Applygen, China) and detected with Tanon Immunoassay System (Tanon, China). Relative signal intensity of bands was determined by Image J software (https://imagej.nih.gov/ij/). Protein expression levels were quantitatively analyzed by normalizing to GAPDH.
Cardiac Function Analysis by Microelectrode Assay (MEA)
The cardiac function of hiPSC-CMs was evaluated by determination of the beat rate (BPM), spike amplitude (mV) and corrected field potential duration (cFPD, ms) using MEA. In brief, hiPSC-CMs were cultured at a density of 4×104/well in matrigel-coating 24-well plate specific for MEA analysis (Axion Biosystems, USA). The field potentials signals were recorded at 37°C using Maestro EDGE MEA system (Axion Biosystems) at the end of 0, 2, and 6 h treatment of ZnO NPs. Signals were recorded with standard cardiac settings (bandpass filter: 1 Hz to 2 kHz, beat detection threshold: 300 μV). Data were outputted and analyzed with the Maestro AxIS Navigator software (Axion Biosystems; version 2.0.3). To eliminate the differences in baselines between different groups, the data were normalized to the initial values at 0 h as baseline.
Statistical Analysis
All data were expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments. Statistical significance was analyzed using Student’s t-test for two group or one-way analysis of variance (ANOVA) for comparisons of multiple groups. The values of P < 0.05 was considered statistically significant.
Results
ZnO Nanoparticle Characterization
To evaluate the physical property of ZnO NPs used in the present study, the ultrastructure, mean size, hydrodynamic diameter, and zeta potential were determined. As shown in Figure 1, TEM micrographs demonstrated ZnO NPs were spheroid and rod in shape with mild agglomeration (Figure 1A and B). The particle size was in the range between 40 and 60 nm, as consistent with the data provided by Sigma-Aldrich (≤50 nm). The hydrodynamic diameter of ZnO NPs in double distilled water was 214.16 ± 0.31 nm using Dynamic Light Scattering (DLS) analysis (Figure 1C). The surface charge (zeta potential) of ZnO NPs was −16.11 ± 0.46 mV (Figure 1D). The hydrodynamic diameter and zeta potential of ZnO NPs in cell culture medium were 266.27 ± 27.36 nm and −8.73 ± 0.68 mV, respectively. These results demonstrated that ZnO NPs had agglomeration propensity in aqueous and colloidal solution.
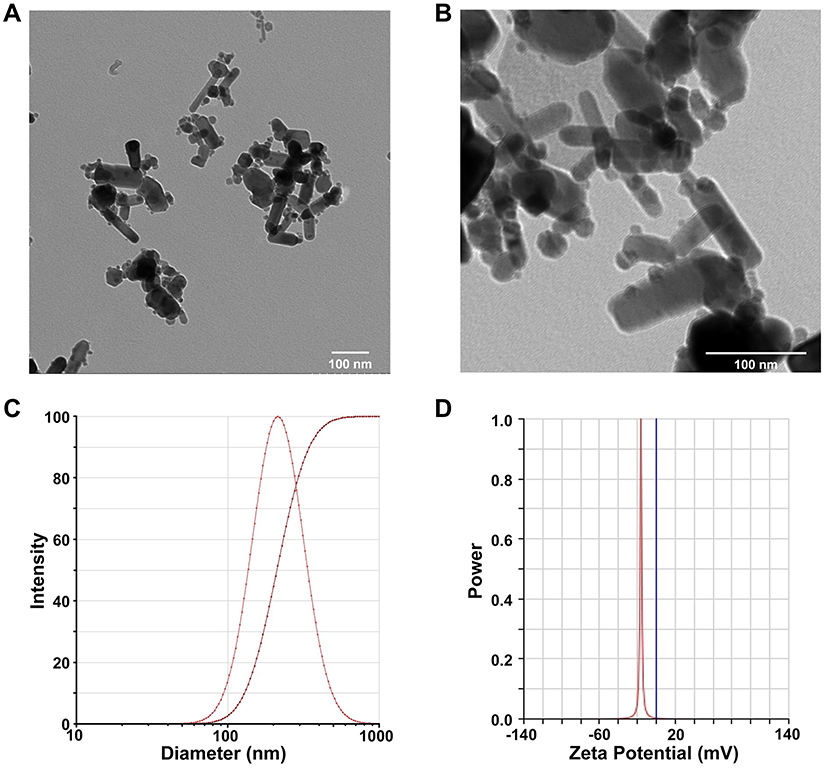 |
Figure 1 Characterization of ZnO NPs. Representative TEM images of 1mM ZnO NPs by magnification zoom of 30 k (A) and 100 k (B). The hydrodynamic diameter of 1 mM ZnO NPs in dd-water was 214.16 ± 0.31 nm, as measured by dynamic light scattering method (C). Zeta potential of 1 mM ZnO NPs in dd-water was −16.11 ± 0.46 mV (D). Data were presented as mean ± SE (n = 3). |
HiPSC-CMs Differentiation and Identification
To obtain high-purity and relatively mature cardiomyocytes, we differentiated hiPSC using small molecule-based methods and cultured to day 25–30. At the beginning of myocardial differentiation (day 0–2), the agglomerated hiPSC showed normal colony morphology with high nuclear-cytoplasmic ratio. At the end of differentiation stage (day 4–6), the cells became spindle-shaped and intertwined into a network. A large number of cells were beating with constant wavelike motions at day 8–9. At day 14, after purification for 2 days, the net-shaped cardiomyocytes were beating stronger. After reseeding and long-term culture to day 25–30, the morphologically mature cardiomyocytes with myofilament and sarcomere structure was observed to have rhythmic contraction (Figure 2A). Robust and regular baseline beating signals were detected using MEA technology. Baseline beat rate was 55.11 ± 2.51 BPM, and spike amplitude was 3.96 ± 0.57 mV (Figures 2B and 3A, C).
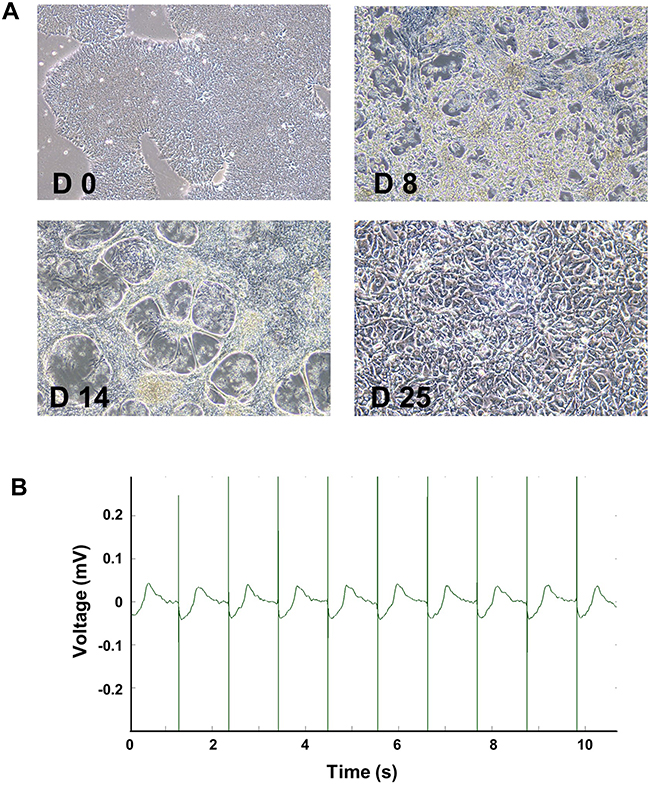 |
Figure 2 Differentiation and identification of hiPSC-CMs. Representative morphological images of hiPSCs after differentiation at day 0, 8, 14, and 25 (A, 10x). Stable beat of cardiomyocytes was confirmed by MEA (B). |
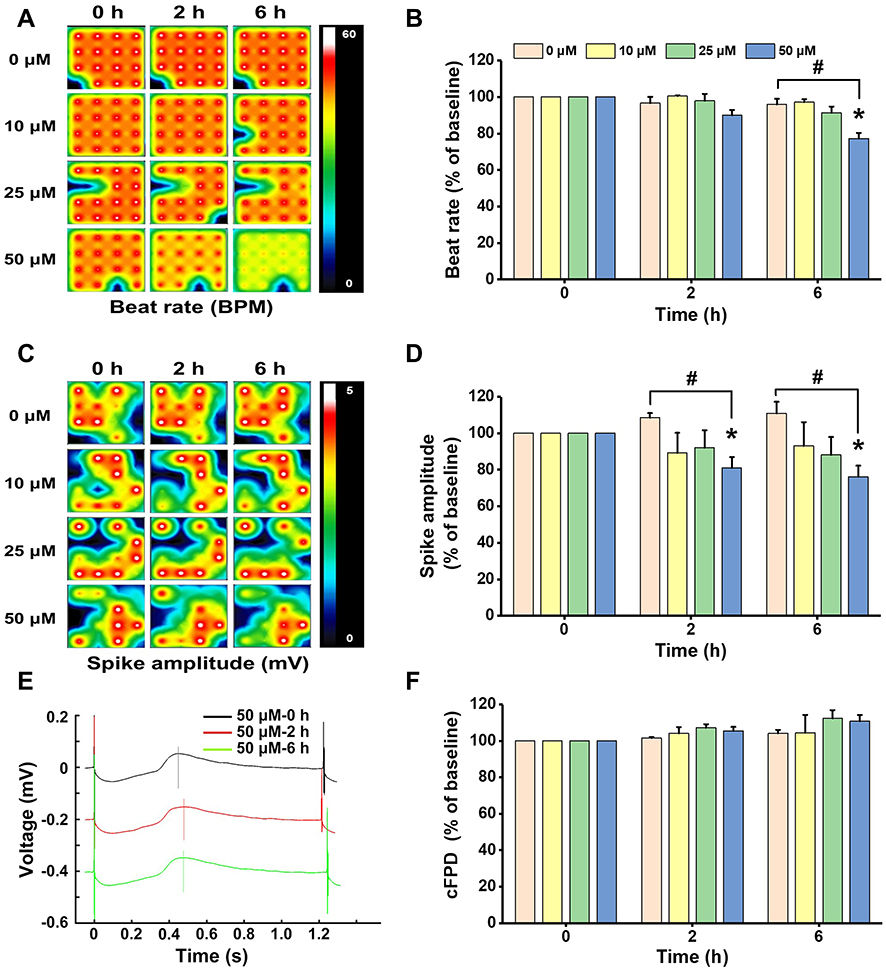 |
Figure 3 Non-cytotoxic concentrations of ZnO NPs induced electrophysiological alterations in hiPSC-CMs. HiPSC-CMs were treated with ZnO NPs at 10, 25, and 50 μM. At 0, 2, and 6 h, electrophysiological function was evaluated by analyzing beat rate, spike amplitude, and cFPD. Activity map (A, RPM) and quantitative analysis of beat rate (B). Activity map (C, mV) and quantitative analysis of spike amplitude (D). Beat waveform trace overlay to show FPD of 50 μM ZnO NPs treatment at 0, 2, and 6 h (E). Quantitative analysis of cFPD (F). The Data recorded at 0 h were used as baseline. Data were presented as mean ± SE (n = 3). *Compared with baseline (0 h), #Compared with control, P < 0.05. |
ZnO NPs Concentration/Time-Dependently Induced Cytotoxicity in hiPSC-CMs
ZnO NPs induced cytotoxicity in hiPSC-CMs was evaluated by morphological observation, cell viability and LDH leakage assays. As shown in Figure 4A, for the cells in control group, the normal spindle-shaped hiPSC-CMs were grown adherently with regular contraction. Cell morphology was affected remarkably after 24 h treatment with ZnO NPs at 62.5 μM and higher concentrations, and cell contraction became slowly or even stop. Cell shrinkage disappeared cross-linking, and many dead cells were observed at 125 and 250 μM. Cell viability was concentration-dependently decreased by ZnO NPs at the concentrations ranging from 31.25 to 500 μM (Figure 4B). As compared to the cells in control group, significant reductions of cell viability were found in the cells treated with ZnO NPs at 62.5 μM and above concentrations. Cell viability was reduced to 87% for the cells treated with 62.5 μM ZnO NPs where slower cell contraction was observed under light microscopy. Cell viability was reduced to below 40% in the cells treated 125 μM and above concentrations of ZnO NPs. The cytotoxic effects of ZnO NPs were time-dependent as shown in the time-course study. Significant reductions of cell viability were found in the cells exposed to 50 and 200 μM ZnO NPs after 12 h (86%) and 3 h (67%), respectively (Figure 4C). LDH leakage to the culture medium was determined to verify the cytotoxicity of ZnO NPs in hiPS-CMs. The cells treated with low concentration of ZnO NPs (31.25–62.5 μM) for 24 h exhibited slight but insignificant change in LDH leakage compared with control. However, at high concentration ranging from 125 to 500 μM, more than 50% significant increase of LDH leakage was found, which indicated serious cell membrane damage (Figure 4D). In the time-course study, LDH leakage was increased with ZnO NPs incubation period extended (Figure 4E).
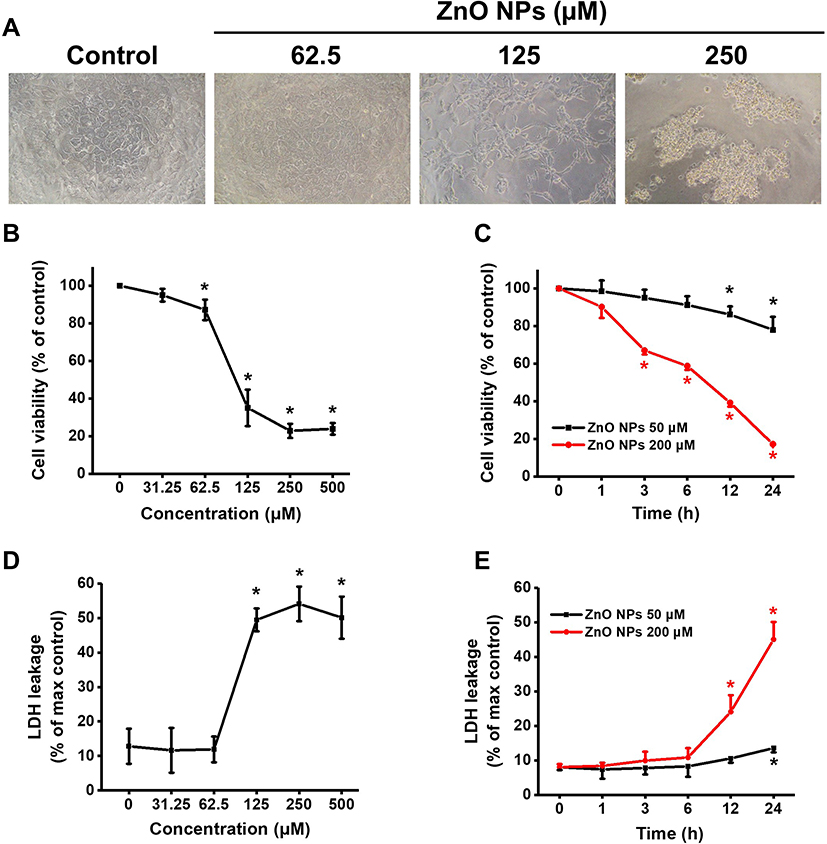 |
Figure 4 ZnO NPs concentration- and time-dependently induced cytotoxicity in hiPSC-CMs. Morphological images of hiPSC-CMs after treatment with different concentrations of ZnO NPs for 24 h (A, 10x). Cell viability was assayed after treatment with different concentrations of ZnO NPs for 24 h (B), and cell viability of the cells treated with 50, 200 μM ZnO NPs for indicated times up to 24 h (C). Concentration- and time-response of LDH release in the cells treated ZnO NPs (D, E). Data were presented as mean ± SE (n = 3). *Compared with control, P < 0.05. |
ZnO NPs Induced Cellular ROS Accumulation and Mitochondrial Dysfunction
It is well known that cardiomyocytes contain abundant mitochondria to maintain intracellular oxidative stress homeostasis and to meet the high energy requirements. Recent studies indicate mitochondria as important targets for the attack by ZnO NPs in the cardiovascular system.24 To evaluate the effect of ZnO NPs on mitochondria, we utilized high-content analysis to determine intracellular ROS levels and MMP in live-beating cells by using fluorescent probes co-staining of CM-H2DCFDA and TMRM. As shown in Figure 5, compared to the cells in control group, the intracellular ROS levels were increased by 230%, 467%, and 582%, respectively, for the cells treated with 25, 50, and 100 μM ZnO NPs. In contrast, MMP was significantly reduced to 67%, 55% respectively for the cells treated with 50 and 100 μM ZnO NPs.
 |
Figure 5 ZnO NPs induced ROS accumulation and MMP loss in hiPSC-CMs. HiPSC-CMs were treated with ZnO NPs at 0, 25, 50, and 100 μM for 6 h. Immediately after treatment, cells were stained with CM-H2DCFDA (green), TMRM (red), and Hoechst 33342 (blue) respectively for ROS, MMP and nucleus. Representative fluorescent images of the cells by high-content analysis (A, Scale bar = 50 μm). Quantitative analysis of ROS and MMP fluorescence intensity (B and C). Data were presented as mean ± SE (n = 3). *Compared with control, P < 0.05. |
Mitochondrial Biogenesis Was Impaired by ZnO NPs
A growing evidence suggests that mitochondrial biogenesis is critically involved in nanoparticles induced injury.25 To illustrate the effect of ZnO NPs on mitochondrial biogenesis, the mitochondrial density and mtDNA copy number were analyzed, respectively, by Mito-Tracker green staining and qPCR after the cells were incubated with ZnO NPs for 6 h. As compared to the cells in control group, the mitochondrial density of the cells treated with 25 μM ZnO NPs was increased slightly but not significantly (105% of control), while the mitochondrial densities of the cells treated with 50 and 100 μM ZnO NPs were decreased to 85% and 63% of control, respectively (Figure 6A and B). It has been reported that mtDNA replication transcription system was activated to counteract mitochondrial oxidative respiratory chain damage under the mild oxidative stress level.26 Our results showed that mtDNA copy number in the cells treated with 25 and 50 μM ZnO NPs was increased significantly by 290% and 180% of control, respectively. However, the increase of mtDNA copy number by ZnO NPs was dramatically declined at high dose as evidenced that the mtDNA copy number in the cells treated with 100 μM ZnO NPs was decreased to 80% of control (Figure 6C).
 |
Figure 6 Effects of ZnO NPs on mitochondrial density and mtDNA copy number in hiPSC-CMs. HiPSC-CMs were treated with ZnO NPs at 0, 25, 50, and 100 μM for 6 h. Representative fluorescence images of the cells stained with Mito-Tracker (green) for mitochondria mass and Hoechst 33342 (blue) for nucleus (A, Scale bar = 50 μm), and quantitative analysis of Mito-Tracker fluorescence intensity (B). mtDNA copy number was determined by qPCR (C). Data were presented as mean ± SE (n = 3). *Compared with control, P < 0.05. |
Effects of ZnO NPs on PGC-1α Pathway
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) has emerged as a master regulator of mitochondrial biogenesis.27 PGC-1α is also critically involved in regulating mitochondrial respiration, contractile automaticity, and oxidative stress in stem cell-derived cardiomyocyte.28 ZnO NPs were found to down-regulate the protein expression of PGC-1α by concentration-dependent manner (Figure 7). After the cells were treated with 25 μM ZnO NPs for 6 h, the protein expression of PGC-1α was slightly down-regulated to 82.1% of control. For the cells treated with 50 and 100 μM ZnO NPs, PGC-1α protein expression was significantly decreased to 65.5% and 48.6% of control, respectively. Similar results were found in the protein expression of TFAM, a downstream target of PGC-1α (significantly decreased to 47.6% and 48.9% of control, respectively, for the cells treated with 50 and 100 μM ZnO NPs). COX IV is a hetero-oligomeric enzyme localized to the inner mitochondrial membrane which is frequently used as a marker indicating mitochondrial density. ZnO NPs were found to remarkably decrease the protein expression of COX IV by 24.5% and 38.1% in the cells treated with ZnO NPs at 50 μM and 100 μM, respectively. These findings were in consistent with the results of the mitochondrial density evaluated by HCA analysis (Figure 6B).
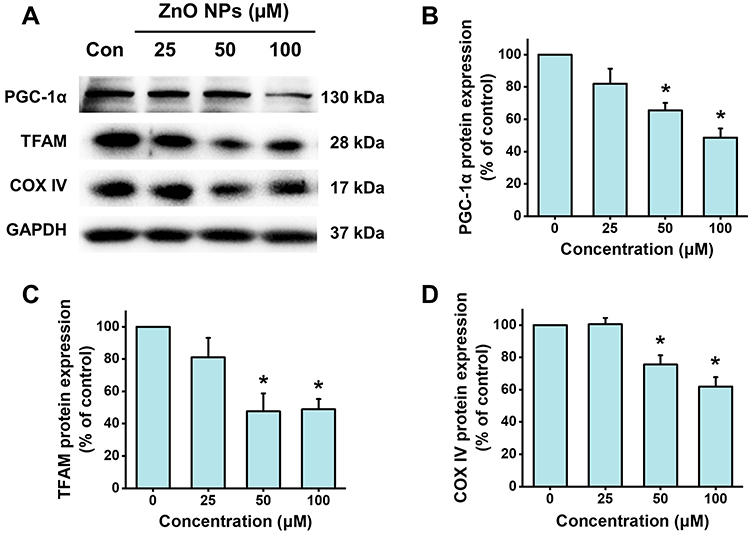 |
Figure 7 ZnO NPs inhibited PGC-1a pathway in hiPSC-CMs. HiPSC-CMs were treated with ZnO NPs at 0, 25, 50, and 100 μM for 6 h. Representative images of the protein expression of PGC-1α, TFAM, and COX IV detected by Western blot (A), and quantitative analysis (B–D). Data were presented as mean ± SE (n = 3). *Compared with control, P < 0.05. |
ZnO NPs Induced Cardiac Electrophysiological Dysfunction
As observed by light microscope in the cytotoxicity experiments, contraction of the cells was extremely weak or even ceased after treatment with 100 μM ZnO NPs for 6 h. To better investigate the effect of ZnO NPs on the electrophysiological function of hiPSC-CMs, we exposed the cells to ZnO NPs at insignificantly cytotoxic concentrations ranging from 10 to 50 μM for relative short periods (0, 2, and 6 h). The cells in all group showed similar electrophysiological properties at the beginning of MEA recording (0 h) with no significant differences were found in all parameters detected. The beat rate, spike amplitude, and cFPD of the cells in control group were 55.11 ± 2.51 BPM, 3.96 ± 0.57 mV and 401.01 ± 12.16 ms, respectively, and the values of these parameters remained stable throughout the recording. As shown in Figure 3A–D, after 2 h treatment, the beat rate of cells exposed to 50 μM ZnO NPs showed slight decrease (49.5 BPM, decreased by10.1%), but no significant differences were found in all ZnO NPs treated cells; spike amplitude was decreased in all ZnO NPs-treated cells, which was decreased by 10.9%, 8.0%, and 19.1%, respectively, for the cells treated with 10, 25, and 50 μM ZnO NPs. Significant decrease was only found in the cells treated with 50 μM ZnO NPs. After 6 h treatment, the beat rate was decreased significantly by 22.7% (42.5 BPM) in the cells treated with 50 μM ZnO NPs. Significant decrease was also found in the spike amplitude of the cells with 50 μM ZnO NPs (2.77 mV, decreased by 23.9%). No significant differences were found in cFPD between any groups and at any time point analyzed (Figure 3E and F) and no arrhythmia was observed. Collectively, these findings showed that ZnO NPs induced electrophysiological disfunction in hiPSC-CMs as manifested by decreases in beat rate and spike amplitude.
Discussion
Because of their large production volumes and broad utilization, safety concerns with respect to ZnO NPs have recently increased. Emerging evidence suggests that ZnO NPs can induce adverse effects to the cardiovascular system. Human and animal experiments have implicated ZnO NPs in triggering systemic inflammation and oxidative stress, together with myocardial injury.10,12 Many in vitro studies have shown that ZnO NPs induced toxicity in some human cardiovascular cells, such as microvascular endothelial cells, coronary artery endothelial cells, and aortic endothelial cells.16,17,29 A study by Wang et al confirmed that ZnO nanowire can affect the metabolism and decrease cell survival in HL-1 mice cardiac muscle cells and neonatal rat cardiomyocytes.30 However, there are few reports concerning the harmful effect of ZnO NPs in human cardiomyocytes.
Given the unique advantages of hiPSC-CMs, they have been increasingly used in various fields such as cardiovascular disease research and drug cardiac safety evaluation. In particular, the use of hiPSC-CMs for cardiac safety evaluation has been established by the US Food and Drug Administration (FDA) in the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative. Recently, the utility of hiPSC-CMs has been confirmed in an international multisite study as part of the CiPA initiative, supporting hiPSC-CMs as powerful in vitro models in prediction of chemical induced cardiotoxicity.31 Therefore, we utilized hiPSC-CMs in the present study to investigate the potential harmful effects of ZnO NPs in human cardiomyocytes. Our results showed that ZnO NPs can concentration- and time-dependently induce cytotoxicity in hiPSC-CMs. Similar findings were also found in previous studies showing that human endothelial cells exposed to ZnO NPs from 60 to 180 μM for 24 h induced significant cytotoxicity.16,17,29
It should be aware that the cytotoxicity of ZnO NPs is affected by multiple factors such as the size and shape of the nanoparticles and zeta potential. In order to get a better control of these factors, we have utilized single-size ZnO NPs, standardized commercial agents and cell culture medium, and prepared all particles suspensions according to a Standard Operation Procedure. Meanwhile, we have detected the hydrodynamic diameter and zeta potential of ZnO NPs in cell culture medium to confirm that these factors were under well control in our experiments. Moreover, it has been reported that the physicochemical properties of ZnO NPs can transform in suspension over time (aging) which modified the bioavailability and toxicity of ZnO NPs in mammalian cells.32,33 To avoid the cytotoxicity affected by the aging process, all suspensions and dilution of ZnO NPs used in our experiments were freshly prepared.
Although the mechanisms of ZnO NPs-mediated toxicity are still obscure, ROS production has been increasingly implicated as a crucial mechanism in various cell types for many metal oxide nanoparticles including ZnO NPs.34 Interestingly, it is suggested that ZnO NPs have their strongest ROS generation ability at lower concentrations than other metal oxide NPs such as Fe2O3, Fe3O4, MgO, Al2O3, and CuO.29 It has been proposed that ZnO NPs can promote ROS production by multiple routes.1 For instance, ZnO NPs can disrupt cellular zinc homeostasis to trigger ROS generation. ZnO NPs exposure impairs the antioxidant defense system resulting in excess ROS accumulation. By application of high-content analysis, we showed that a relative short period of exposure to ZnO NPs at low-cytotoxic concentrations could remarkably promote ROS generation in hiPSC-CMs. However, the mechanism by which ZnO NPs induce ROS production needs to be further investigated.
Mitochondria are vital organelles in cells that are critically engaged in a variety of key biological functions such as oxidative energy production and regulation of the cell survival/death.35 Mitochondria are one of the main sources of ROS, which are produced as an important by-product of energy metabolism in mitochondrial respirations. Importantly, mitochondria are the preferential target of excess ROS-mediated oxidative attack. Accumulating studies indicate that mitochondria play an essential role in ZnO NPs-induced toxicity in experimental animals as well as in vitro models.36,37 For instance, in zebrafish embryos, ZnO NPs were found to trigger excessive ROS production, reduce MMP, and induce mitochondrial pathway-mediated apoptosis.38 However, the role of mitochondria in ZnO NPs-induced cardiotoxicity and their underlying mechanisms are still poorly understood. Our results showed that ZnO NPs can induce mitochondrial dysfunction as evidenced by the decline of MMP. These findings are consistent with the increased ROS generation observed by HCA and support mitochondria as the main targets of ROS-mediated oxidative attack.
A certain number and the proper function of mitochondria are particularly important for cardiac cells to meet the high energy demand of the heart and to counteract cardiac damage. Cardiac mitochondria are highly dynamic and require mitochondrial biogenesis, which is crucial for regulation of mitochondrial density as well as function.39 As a critical regulator of mitochondrial, PGC-1α not only plays a key role in regulation of mitochondrial biogenesis by interacting with downstream targets like mitochondrial transcription factor A (TFAM) to increase the transcription of mitochondrial genes, but also plays a key role in regulation of mitochondrial oxidative stress by activating the transcription of mitochondrial antioxidants such as manganese superoxide dismutase. We previously reported that PGC-1α-mediated mitochondrial biogenesis is essential in protection against cardiomyocyte death and cardiac dysfunction.40,41 By application of human embryonic stem cell-derived cardiomyocytes, it has been shown that PGC-1α is critically involved in regulating mitochondrial respiration, contractile automaticity, and superoxide production.28 In the present study, we showed that ZnO NPs could disrupt mitochondrial biogenesis as evidenced by decreased mitochondrial density, mtDNA copy number alteration, and inhibition of the PGC-1α pathway. These findings suggested that mitochondrial biogenesis impairment is a key event and critically involved in ZnO NP-induced cytotoxicity.
Electrophysiology has been widely used in the evaluation of cardiac function both in in vitro and in in vivo tests. MEA is a label-free, non-invasive and cost-effective measurement at the high-throughput scale and with real-time observations.42 The hiPSC-CMs-based MEA assay has emerged as a stable and reliable strategy for screening of compound-induced cardiac toxicity and was recommended to be used in CiPA.43 The field potential signals are recognized as markers for depolarization and repolarization enabling the quantification of important beating parameters. In the present study, the MEA platform was applied to evaluate the cardiac electrophysiological function of hiPSC-CMs. Our results showed that beat rate and spike amplitude were decreased by 6 h treatment with ZnO NPs at insignificantly cytotoxic concentrations, which agreed with the decreased cell beating and contractility observed by optical microscopy. However, no significant changes in cFPD and arrhythmias were found in the hiPSC-CMs. Many in vivo studies have shown that ZnO NPs exposure can reduce the heart rate of zebrafish and freshwater fish Catostomus commersonii.15,44 The heart rate was also reduced in mice after injection of five nanomaterials including ZnO NPs.45 Our results showed that the electrophysiological properties of hiPSC-CMs were affected by ZnO NPs at insignificantly cytotoxic doses, suggesting that electrophysiological function is a sensitive parameter for indicating the cardiac adverse effect of nanomaterials. It is implicated that mitochondrial dysfunction crucially contributes to electrophysiological abnormalities in hiPSC-CMs.46 Presumably, the decrease of beat rate and spike amplitude in hiPSC-CMs observed in the present study might be also attributed to mitochondrial dysfunction. Clinically, more attention should be paid to detect the deleterious cardiac effects in the patients related to ZnO NPs exposure such as electrocardiogram and serum enzymes.
Conclusion
Our results demonstrated that ZnO NPs could directly induce cytotoxicity, oxidative stress, and mitochondrial dysfunction in hiPSC-CMs, and suggested that the toxicity of ZnO NPs is associated with impairment of mitochondrial biogenesis. Moreover, our findings highlighted that ZnO NPs could affect cardiac electrophysiological function. A proposed mechanism underlying ZnO NPs-induced toxicity and dysfunction in hiPSC-CMs is summarized in Figure 8. To the best of our knowledge, the present study is the first report providing clear evidence to elucidate the toxicity of ZnO NPs in human cardiomyocytes and suggesting a key role of mitochondrial biogenesis and PGC-1α in ZnO NPs-induced toxicity. These critical findings will help in the understanding of the potential harmful effects of ZnO NPs in the cardiovascular system and provide a pivotal mechanistic understanding of the involvement of mitochondrial biogenesis in nanomaterial-induced toxicity in the cardiovascular system.
 |
Figure 8 Schematic representation of the proposed mechanism underlying ZnO NPs-induced toxicity and dysfunction in hiPSC-CMs. |
Acknowledgments
This work was supported by the National Key Research and Development Project (2018YFC1602602), Beijing Nova Program (Z171100001117103) and Foundation of Beijing Key Laboratory of Toxicological Research and Risk Assessment for Food Safety (KF2019-01).
Disclosure
The authors declare no conflict of interest. This study has been approved by the Ethics Committee of PLA Center for Disease Control and Prevention.
References
1. Liu J, Feng X, Wei L, Chen L, Song B, Shao L. The toxicology of ion-shedding zinc oxide nanoparticles. Crit Rev Toxicol. 2016;46(4):348–384. doi:10.3109/10408444.2015.1137864
2. Jacobsen NR, Stoeger T, van den Brule S, et al. Acute and subacute pulmonary toxicity and mortality in mice after intratracheal instillation of ZnO nanoparticles in three laboratories. Food Chem Toxicol. 2015;85:84–95. doi:10.1016/j.fct.2015.08.008
3. Danielsen PH, Cao Y, Roursgaard M, Moller P, Loft S. Endothelial cell activation, oxidative stress and inflammation induced by a panel of metal-based nanomaterials. Nanotoxicology. 2015;9(7):813–824. doi:10.3109/17435390.2014.980449
4. Konduru NV, Murdaugh KM, Sotiriou GA, et al. Bioavailability, distribution and clearance of tracheally-instilled and gavaged uncoated or silica-coated zinc oxide nanoparticles. Part Fibre Toxicol. 2014;11:44. doi:10.1186/s12989-014-0044-6
5. Esmaeillou M, Moharamnejad M, Hsankhani R, Tehrani AA, Maadi H. Toxicity of ZnO nanoparticles in healthy adult mice. Environ Toxicol Pharmacol. 2013;35(1):67–71. doi:10.1016/j.etap.2012.11.003
6. Li CH, Shen CC, Cheng YW, et al. Organ biodistribution, clearance, and genotoxicity of orally administered zinc oxide nanoparticles in mice. Nanotoxicology. 2012;6(7):746–756. doi:10.3109/17435390.2011.620717
7. Xiaoli F, Junrong W, Xuan L, et al. Prenatal exposure to nanosized zinc oxide in rats: neurotoxicity and postnatal impaired learning and memory ability. Nanomedicine (Lond). 2017;12(7):777–795. doi:10.2217/nnm-2016-0397
8. Kielbik P, Kaszewski J, Rosowska J, et al. Biodegradation of the ZnO:Eu nanoparticles in the tissues of adult mouse after alimentary application. Nanomedicine. 2017;13(3):843–852. doi:10.1016/j.nano.2016.11.002
9. Cao Y, Gong Y, Liao W, et al. A review of cardiovascular toxicity of TiO2, ZnO and Ag nanoparticles (NPs). Biometals. 2018;31(4):457–476. doi:10.1007/s10534-018-0113-7
10. Monse C, Hagemeyer O, Raulf M, et al. Concentration-dependent systemic response after inhalation of nano-sized zinc oxide particles in human volunteers. Part Fibre Toxicol. 2018;15(1):8. doi:10.1186/s12989-018-0246-4
11. Baky NA, Faddah LM, Al-Rasheed NM, Al-Rasheed NM, Fatani AJ. Induction of inflammation, DNA damage and apoptosis in rat heart after oral exposure to zinc oxide nanoparticles and the cardioprotective role of alpha-lipoic acid and vitamin E. Drug Res. 2013;63(5):228–236. doi:10.1055/s-0033-1334923
12. Chuang HC, Juan HT, Chang CN, et al. Cardiopulmonary toxicity of pulmonary exposure to occupationally relevant zinc oxide nanoparticles. Nanotoxicology. 2014;8(6):593–604. doi:10.3109/17435390.2013.809809
13. Bengalli R, Gualtieri M, Capasso L, Urani C, Camatini M. Impact of zinc oxide nanoparticles on an in vitro model of the human air-blood barrier. Toxicol Lett. 2017;279:22–32. doi:10.1016/j.toxlet.2017.07.877
14. Yan Z, Wang W, Wu Y, et al. Zinc oxide nanoparticle-induced atherosclerotic alterations in vitro and in vivo. Int J Nanomedicine. 2017;12:4433–4442. doi:10.2147/IJN.S134897
15. Bessemer RA, Butler KM, Tunnah L, et al. Cardiorespiratory toxicity of environmentally relevant zinc oxide nanoparticles in the freshwater fish Catostomus commersonii. Nanotoxicology. 2015;9(7):861–870. doi:10.3109/17435390.2014.982737
16. Chuang KJ, Lee KY, Pan CH, et al. Effects of zinc oxide nanoparticles on human coronary artery endothelial cells. Food Chem Toxicol. 2016;93:138–144. doi:10.1016/j.fct.2016.05.008
17. Liang S, Sun K, Wang Y, et al. Role of Cyt-C/caspases-9,3, Bax/Bcl-2 and the FAS death receptor pathway in apoptosis induced by zinc oxide nanoparticles in human aortic endothelial cells and the protective effect by alpha-lipoic acid. Chem Biol Interact. 2016;258:40–51. doi:10.1016/j.cbi.2016.08.013
18. Handral HK, Tong HJ, Islam I, Sriram G, Rosa V, Cao T. Pluripotent stem cells: an in vitro model for nanotoxicity assessments. J Appl Toxicol. 2016;36(10):1250–1258. doi:10.1002/jat.3347
19. Magdy T, Schuldt AJT, Wu JC, Bernstein D, Burridge PW. Human induced pluripotent stem cell (hiPSC)-derived cells to assess drug cardiotoxicity: opportunities and problems. Annu Rev Pharmacol Toxicol. 2018;58:83–103. doi:10.1146/annurev-pharmtox-010617-053110
20. Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015;117(1):80–88. doi:10.1161/CIRCRESAHA.117.305365
21. Yoshida Y, Yamanaka S. Induced pluripotent stem cells 10 years later: for cardiac applications. Circ Res. 2017;120(12):1958–1968. doi:10.1161/CIRCRESAHA.117.311080
22. Cui N, Wu F, Lu WJ, et al. Doxorubicin-induced cardiotoxicity is maturation dependent due to the shift from topoisomerase IIalpha to IIbeta in human stem cell derived cardiomyocytes. J Cell Mol Med. 2019;23(7):4627–4639. doi:10.1111/jcmm.14346
23. Yin J, Guo J, Zhang Q, et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol in Vitro. 2018;51:1–10. doi:10.1016/j.tiv.2018.05.001
24. Bostan HB, Rezaee R, Valokala MG, et al. Cardiotoxicity of nano-particles. Life Sci. 2016;165:91–99. doi:10.1016/j.lfs.2016.09.017
25. Guo C, Wang J, Jing L, et al. Mitochondrial dysfunction, perturbations of mitochondrial dynamics and biogenesis involved in endothelial injury induced by silica nanoparticles. Environ Pollut. 2018;236:926–936. doi:10.1016/j.envpol.2017.10.060
26. Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem. 2016;85:133–160. doi:10.1146/annurev-biochem-060815-014402
27. Wenz T. Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion. 2013;13(2):134–142. doi:10.1016/j.mito.2013.01.006
28. Birket MJ, Casini S, Kosmidis G, et al. PGC-1alpha and reactive oxygen species regulate human embryonic stem cell-derived cardiomyocyte function. Stem Cell Rep. 2013;1(6):560–574. doi:10.1016/j.stemcr.2013.11.008
29. Sun J, Wang S, Zhao D, Hun FH, Weng L, Liu H. Cytotoxicity, permeability, and inflammation of metal oxide nanoparticles in human cardiac microvascular endothelial cells: cytotoxicity, permeability, and inflammation of metal oxide nanoparticles. Cell Biol Toxicol. 2011;27(5):333–342. doi:10.1007/s10565-011-9191-9
30. Wang Y, Wu Y, Quadri F, Prox JD, Guo L. Cytotoxicity of ZnO nanowire arrays on excitable cells. Nanomaterials (Basel). 2017;7(4):80. doi:10.3390/nano7040080
31. Blinova K, Dang Q, Millard D, et al. International multisite study of human-induced pluripotent stem cell-derived cardiomyocytes for drug proarrhythmic potential assessment. Cell Rep. 2018;24(13):3582–3592. doi:10.1016/j.celrep.2018.08.079
32. Wang M, Wang J, Liu Y, et al. Subcellular targets of zinc oxide nanoparticles during the aging process: role of cross-talk between mitochondrial dysfunction and endoplasmic reticulum stress in the genotoxic response. Toxicol Sci. 2019;171(1):159–171. doi:10.1093/toxsci/kfz132
33. Wang MM, Wang YC, Wang XN, et al. Mutagenicity of ZnO nanoparticles in mammalian cells: role of physicochemical transformations under the aging process. Nanotoxicology. 2015;9(8):972–982. doi:10.3109/17435390.2014.992816
34. Wang Y, Ding L, Yao C, et al. Toxic effects of metal oxide nanoparticles and their underlying mechanisms. Sci China Mater. 2017;60(2):93–108. doi:10.1007/s40843-016-5157-0
35. Pfanner N, Warscheid B, Wiedemann N. Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol. 2019;20(5):267–284. doi:10.1038/s41580-018-0092-0
36. Park YH, Bae HC, Kim J, Jeong SH, Yang SI, Son SW. Zinc oxide nanoparticles induce HIF-1alpha protein stabilization through increased reactive oxygen species generation from electron transfer chain complex III of mitochondria. J Dermatol Sci. 2018;91(1):104–107. doi:10.1016/j.jdermsci.2018.03.010
37. Babele PK, Thakre PK, Kumawat R, Tomar RS. Zinc oxide nanoparticles induce toxicity by affecting cell wall integrity pathway, mitochondrial function and lipid homeostasis in Saccharomyces cerevisiae. Chemosphere. 2018;213:65–75. doi:10.1016/j.chemosphere.2018.09.028
38. Zhao X, Ren X, Zhu R, Luo Z, Ren B. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria-mediated apoptosis in zebrafish embryos. Aquat Toxicol. 2016;180:56–70. doi:10.1016/j.aquatox.2016.09.013
39. Pohjoismaki JL, Goffart S. The role of mitochondria in cardiac development and protection. Free Radic Biol Med. 2017;106:345–354. doi:10.1016/j.freeradbiomed.2017.02.032
40. Guo J, Guo Q, Fang H, et al. Cardioprotection against doxorubicin by metallothionein Is associated with preservation of mitochondrial biogenesis involving PGC-1alpha pathway. Eur J Pharmacol. 2014;737:117–124. doi:10.1016/j.ejphar.2014.05.017
41. Yuan H, Zhang Q, Guo J, et al. A PGC-1alpha-mediated transcriptional network maintains mitochondrial redox and bioenergetic homeostasis against doxorubicin-induced toxicity in human cardiomyocytes: implementation of TT21C. Toxicol Sci. 2016;150(2):400–417. doi:10.1093/toxsci/kfw006
42. Kussauer S, David R, Lemcke H. hiPSCs derived cardiac cells for drug and toxicity screening and disease modeling: what micro- electrode-array analyses can tell us. Cells. 2019;8(11):1331. doi:10.3390/cells8111331
43. Millard D, Dang Q, Shi H, et al. Cross-site reliability of human induced pluripotent stem cell-derived cardiomyocyte based safety assays using microelectrode arrays: results from a blinded CiPA pilot study. Toxicol Sci. 2018;164(2):550–562. doi:10.1093/toxsci/kfy110
44. Du J, Wang S, You H, Jiang R, Zhuang C, Zhang X. Developmental toxicity and DNA damage to zebrafish induced by perfluorooctane sulfonate in the presence of ZnO nanoparticles. Environ Toxicol. 2016;31(3):360–371. doi:10.1002/tox.22050
45. Haberl N, Hirn S, Holzer M, Zuchtriegel G, Rehberg M, Krombach F. Effects of acute systemic administration of TiO2, ZnO, SiO2, and Ag nanoparticles on hemodynamics, hemostasis and leukocyte recruitment. Nanotoxicology. 2015;9(8):963–971. doi:10.3109/17435390.2014.992815
46. Li S, Pan H, Tan C, et al. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem Cell Rep. 2018;10(3):808–821. doi:10.1016/j.stemcr.2018.01.013
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.