Back to Journals » International Medical Case Reports Journal » Volume 18
X-Linked Autism Type 9 Caused by a Hemizygote Pathogenic Variant in the TMLHE Gene: Etiological Diagnosis in an Adult Male with Moderate Intellectual Disability
Authors Verhoeven WM ![]() , Pfundt R, Engelke UF, Kluijtmans LA, Egger JI
, Pfundt R, Engelke UF, Kluijtmans LA, Egger JI
Received 13 November 2024
Accepted for publication 6 January 2025
Published 18 January 2025 Volume 2025:18 Pages 111—116
DOI https://doi.org/10.2147/IMCRJ.S506204
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xudong Zhu
Willem MA Verhoeven,1– 3 Rolph Pfundt,4,5 Udo FH Engelke,6 Leo AJ Kluijtmans,6 Jos IM Egger3– 5
1Department of Psychiatry, Erasmus University Medical Center, Rotterdam, The Netherlands; 2Centre for Consultation and Expertise, Utrecht, The Netherlands; 3Vincent van Gogh Centre of Excellence for Neuropsychiatry, Venray, The Netherlands; 4Department of Human Genetics, Radboud University Medical Centre, Nijmegen, The Netherlands; 5Donders Institute for Brain, Cognition and Behaviour, Radboud University, Nijmegen, The Netherlands; 6Department of Human Genetics, Translational Metabolic Laboratory, Radboud University Medical Centre, Nijmegen, The Netherlands
Correspondence: Willem MA Verhoeven, Email [email protected]
Introduction: Levocarnitine is essential for brain functioning and fatty acid metabolism and stems largely from dietary sources. The Epsilon-Trimethyllysine Hydroxylase (TMLHE) gene encodes the enzyme N-Trimethyllysine hydroxylase (TMLH) which catalyses the first step in the biosynthesis of carnitine. Lack of TMLH enzyme activity is associated with developmental delay and autistic behaviours described as X-linked recessive autism, type 6 (OMIM#300872).
Patient and Methods: Here, an institutionalized adult male patient with intellectual disability, autism, and challenging behaviours is presented in whom genetic analysis disclosed a novel pathogenic variant in the TMLHE gene. Extensive somatic, neurological, psychiatric, and neuropsychological investigations were performed next to examination of hematological and biochemical parameters including plasma carnitine status. Also, Whole Exome Sequencing (WES) and Next-Generation Metabolic Screening (NGMS) were performed.
Results: Moderate intellectual disability along with obsessive and aggressive behaviour in the context of autism spectrum disorders was established as well as symptoms from the catatonic spectrum. With WES, a novel variant in the TMHLE gene was identified and using NGMS, increased concentration of trimethyllysine and decreased concentration of γ-butyrobetaine were found resulting in a significantly decreased BB/TML ratio, confirming the pathogenicity of this variant.
Conclusion: X-linked autism type 6 is characterized by moderate intellectual disability and symptoms from the autism spectrum in the absence of any dysmorphisms. To prevent regressive autistic episodes in young children, it is highly recommended to consider next-generation sequencing techniques as the first step in the differential diagnostic process of autism.
Keywords: epsilon-trimethyllysine hydroxylase gene, TMLHE, levocarnitine, autism, intellectual disability, contextual neuropsychology, psychopathology
Introduction
Levocarnitine plays a crucial role in brain function and the mitochondrial metabolism of fatty acids. It is available from both exogenous and endogenous sources. It is found in a wide variety of food groups, particularly in red meat, which is why the standard healthy human diet provides the majority of L-carnitin. In addition, L-carnitine is also synthesized in the human body from the amino acids lysine and methionine. The first step in the biosynthesis of carnitine is catalyzed by the enzyme N-trimethyllysine hydroxylase (TMLH), which converts trimethyllysine (TML) in 3-hydroxytrimethyllysine, and is encoded by the Epsilon-trimethyllysine hydroxylase (TMHLE) gene (MIM: 300777) located on the long arm of the X-chromosome (Xq28). Metabolites involved in the successive stages of carnitine formation are as follows: 3-hydroxytrimethyllysine (HTML), 4-N-trimethylaminobutyraldehyde (TMABA) and γ-butyrobetaine (γ-BB). Concentration of carnitine in plasma is dependent upon age and sex. From adolescence onwards, generally, plasma carnitine concentrations in males stabilize at a higher level than in females, suggesting regulatory involvement of sex hormones.1
In 2011, the research group of Celestina-Soper found a male patient with a hemizygous deletion comprising the TMHLE gene in a large group of probands with autism.2 TMHLE-deficiency was also found in six out of seven autistic male siblings, suggesting that it is a risk factor for autism.3 A year later, from a sample of twelve families, Nava et al reported two male patients with autism spectrum disorder (ASD) and intellectual disability in whom a pathogenic nonsense variant of the TMHLE gene was detected.4 Subsequent screening of another group of 501 male ASD patients resulted in two additional cases who had a missense variant in the reported gene, with a concomitant two-to-threefold increase in the carnitine precursor TML and a small decrease in carnitine in plasma. It was postulated that an increased concentration of TML could have toxic effects on the development of the central nervous system, hampering the typical development of neuronal networks.4 Finally, Ziats et al described a four-year-old patient with an autism spectrum disorder in whom a maternally inherited frameshift mutation in the TMLHE gene was identified, who had low levels of γ-BB and free carnitine, and who had experienced two episodes of behavioural regression that recovered from carnitine suppletion.5
The patient presented here, with intellectual disability, a diagnosis of autism spectrum disorder, and challenging behaviour, was referred for extensive examination and advice.
Patient and Methods
Ethical Aspects
Consultation was performed via the Dutch Centre for Consultation and Expertise (CCE), and assessments were performed via the Vincent van Gogh Centre of Excellence for Neuropsychiatry, Venray, the Netherlands, and partly at Sherpa, Centre for People with Intellectual Disabilities, Baarn, the Netherlands. Institutional approval was not required since both parents and his biologically unrelated healthy sister, who was also his legal representative, gave written informed consent for publication of the case history of their son and biologically unrelated brother, respectively (signed consent form dated November 2023 is provided to the Editorial Board), but also because staff members of the CCE and Sherpa Institutes attended all multidisciplinary meetings and fully agreed with publication of the patient’s case history.
Case Description
The now 46-year-old patient was originally born in Colombia to a healthy mother, after an uncomplicated pregnancy of 38 weeks. No information was available about his biological father. At birth, the weight, height, and head circumference were 2800gr, 49cm, and 34cm, respectively. Physical examination revealed no abnormalities, in particular no dysmorphisms were noticed. One day after birth, his mother brought him to an orphanage from where he was placed in a local foster family because of severe underweight. Aged almost four months, he was adopted by Dutch foster parents who readily observed developmental delay, lack of contact, and apathetic behaviour. He has a biologically unrelated, two-years-younger, healthy adoptive sister who was also born in Colombia and adopted at 3 months of age. The patient was able to walk independently at 1 year and 4 months, he started speaking his first words at the age of 2 years, and became continent at 3 years and 1 month.
During kindergarten years, an obsessive and ritualistic behavioural repertoire became apparent. Since these behaviours form key elements of autism, for this reason, he was referred to a university outpatient orthopedagogic clinic at the age of 5 years, where, apart from intellectual disability, a diagnosis of autism was formally confirmed. Shortly thereafter, a severe myopia of the left eye (−15dpt) was detected resulting in nearly complete visual loss. Until the age of 8 years he visited a medical child care centre, after which he followed special education until his 20th year. During this period, he showed marked problems in speech and language, and behavioural problems, particularly temper tantrums occurred necessitating a structured and predictable environment. Methylphenidate was started but had to be discontinued because of further deterioration of behaviour. Subsequently, symptomatic treatment with pipamperone (40mg twice daily) was initiated, supplemented with a low dose of benzodiazepine if necessary. Over the years, pipamperone dose ranged between 80 and 180mg. Severity of behavioural problems was contingent upon the intensity of (socioemotional) overestimation.
From 20 year onwards, he frequented a day care facility where he was able to perform unskilled labour. His somatic antecedents during these years mention surgical correction of left and right inguinal hernia, at the age of 24 and 39 years, respectively. One year later, he suffered from perforated appendicitis. Although at age 24 and 36, he moved with his parents to another city, he still succeeded in working at a day care facility. However, due to overestimation, regularly, escalation of obsessive-compulsive and aggressive behaviour occurred, and he escaped from the facility. Shortly thereafter, he returned for a period of several months to his parents' home where his behaviour stabilizes gradually with a structured daily program. During this period, a consulted psychiatrist prescribes symptomatically several psychotropics without, however, any effect.
Finally, at the age of 39 years, he was institutionalized. Discontinuity in nursing staff, however, as well as several internal changes in department led to a general decline in daily life functioning, a marked increase in compulsive and disinhibited behaviours, and autistic withdrawal. Because of persisting aggressive and autistic behaviours, the Dutch Centre for Consultation and Expertise was asked for advice.
Investigations
Somatic and neurological examinations revealed no abnormalities. Weight, height, and head circumference were 79kg, 175cm, and 57cm, respectively. No facial dysmorphisms were present. Psychiatric investigations disclosed a clear autistic behavioural repertoire with prominent stereotyped, hyperactive, obsessive features and compulsively arranging objects. According to the nursing staff, these behaviours intensified and became more frequently contingent upon his level of stress from environmental factors. Lack of information about his family history and the previously noticed but not investigated intellectual disability necessitated extensive genetic and pharmacogenetic investigations as well as detailed neuropsychological assessments.
Relevant standard hematological and biochemical parameters were all within the normal range. Pharmacogenetically, normal genotypes for the various cytochrome isoenzymes were found. Whole Exome Sequencing analysis as described by Neveling and coworkers showed a hemizygotic, likely pathogenic variant (class 5) in the Epsilon-Trimethyllysine Hydroxylase (TMHLE) gene [ChrX(GRCh37):g.154774922_154774924dup NM_018196.4:c.14_16dup p.(Leu6*)].6 No other (likely) pathogenic single nucleotide variants (SNVs) or copy number variants (CNVs) were detected in the WES data. Pathogenic variants in the TMHLE gene have been reported earlier as causal for X-linked autism, type 6 (OMIM #300872). The present variant, a duplication of three nucleotides, causes a premature stop codon in the first exon of the TMHLE gene and has never been described before, nor has it been reported in the Genome Aggregation Database (GnomADv4.1.9; https://gnomad.broadinstitute.org). Subsequent biochemical laboratory diagnostics to confirm the pathogenicity of this DNA variant, using Next-Generation Metabolic Screening (NGMS) essentially as described in 2018 by Coene and coworkers, revealed an increased relative concentration of TML and decreased value of BB.7 This resulted in a severely decreased BB/TML ratio that confirms the pathogenicity of the novel THMLE variant in the present patient (Figure 1). In addition, we evaluated the carnitine status and measured a normal free carnitine concentration (40.8 µmol/l [reference values: 20–55 µmol/l]) and a normal total carnitine (46.8 µmol/l [reference values: 25–65 µmol/l]).
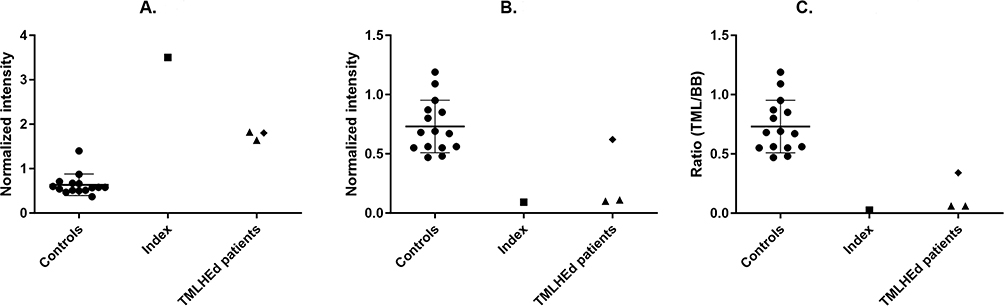 |
Figure 1 Normalized plasma intensities of trimethyllysine (panel A, TML) and gamma-butyrobetaine (panel B, BB), and their ratio (panel C, TML/BB), metabolites measured as [M+H]+ ions in ESI+ positive ionization mode. Data include controls (●), our index patient (■), and three samples of two other patients (▲, ♦) with confirmed N-trimethyllysine hydroxylase (TMLHE) deficiency. Error bars in control plots represent the mean ± standard deviation. All measurements in the index patient significantly differed from controls. |
At neuropsychological examination, moderate intellectual disability was found (IQ =< 55; SON-R) was found.8 Also, severe attentional and inhibitory dysfunctions were found, in addition to heightened sensitivity to external stimuli leading to enhanced distractibility, strongly restricted self-regulation capacities, and constant repetitive behaviours with varying intensity depending on his social interactional context. Adaptive functioning as measured with the Vineland Adaptive Behaviour Scale (Vineland-II) was moderately to severely limited with respect to communication and daily skills, but profoundly low as to socialization skills.9 Social-emotional development as measured by the Dutch scale for emotional development in people with intellectual disability (ESSEON-R) corresponded with a developmental age of 1.5 years (social developmental and emotional developmental ages of 0.5 years and 2.5 years, respectively).10 Finally, with the revised scale for autism and related disorders (AVZ-R) a total score of 18 was established (scores between 10 and 19 are indicative for autism spectrum disorder).11
Outcome and Follow-Up
After extensive multidisciplinary discussion of the etiological diagnosis and because of his normal carnitine status in blood that provides no indication for carnitine suppletion, it was decided to focus on individual daily guidance as much as possible to the autistic behavioural repertoire of the patient. In addition, he moved to another sheltered home facility of Sherpa, Institute for People with Intellectual Disabilities, with much less turmoil around him. After this, aggressive outbursts rarely occurred and prescription of a symptomatic psychotropic compound was not necessary. However, eight months later at follow-up, neuropsychiatric examination demonstrated symptoms from the catatonic spectrum. As assessed with the Bush-Francis Catatonia Rating Scale (BFCRS), the most pronounced scores were established on the items ambitendency and impulsivity and to lesser extent on stereotypy, mannerisms, and inability/stupor (BFCRS Total score = 25).12 Consequently, the patient was treated with lorazepam in an increasing dose to 2mg twice daily, which gradually led to improvement of speech and interaction, decrease in compulsivity, and generally to more behavioural flexibility and relaxation.
Discussion
The 46-years-old male non-dysmorphic male patient with moderate intellectual disability and autism, described here, had a hemizygous pathogenic variant in the TMLHE gene that has never been reported and which resulted in a deficiency of 6-N-trimethyllysine hydroxylase and subsequent deficiency of endogenous carnitine synthesis. This corroborates the diagnosis of X-linked autism and fits the hypothesis that this X-linked inborn error of carnitine biosynthesis is a risk factor for the development of autism spectrum disorder.3,13
So far, only a very limited number of patients have been described with this monogenetic neurodevelopmental disorder, of which variable levels of intellectual disability and persisting autistic behaviours form the core features.4,14 While carnitine is also endogenously synthesized in the human body, generally, a normal carnitine plasma status is usually guaranteed by a standard healthy diet containing carnitine. This may also limit the detection of this disease in regular biochemical screening (which does not evaluate TML and BB levels). A normal carnitine status may not be the case in carnitine auxotrofic people (ie, fully dependent on the external intake of carnitine), particularly when they follow a plant-based (non-animal) diet. Because of the aforementioned role of carnitine in the development of the central nervous system, in young children with autism, detection of carnitine deficiency is crucial because in early stages, autistic regressive episodes – that are often triggered by stressful events like infections – can be improved or even reversed by means of immediate carnitine suppletion.5 It follows that etiological investigation by means of whole exome sequencing and extensive metabolic screening is mandatory when delayed developmental milestones and symptoms from the autism spectrum coincide, even in the absence of any dysmorphisms.15–17
Conclusion
The phenotype of X-linked autism type 6, here etiologically related to the novel pathogenic variant in the TMLHE gene described above, is characterized by moderate intellectual disability and symptoms from the autism spectrum in the absence of any dysmorphisms. This and comparable patterns in people, referred for the differential diagnosis of autism and/or challenging behaviour, point at the increasing importance of genetic and biochemical evaluation by means of next-generation metabolic screening and next-generation sequencing techniques, particularly when symptoms occur already in early childhood.
Acknowledgments
The authors are indebted to the staff members of the Sherpa Institute for People with Intellectual Disabilities, Baarn, The Netherlands, for their careful observations of the behavioural status of the patient. Dr Bregje van Bon, clinical geneticist and associate professor at Radboudumc, is greatly acknowledged for her detailed examination of the patient. Thanks are extended to Mr Hugo Bijsterbosch, clinical neuropsychologist/CCE-consultant, for his assessment of the adaptive capacities of the patient, as well as to the non-biological sister and Mrs Anne Bos, psychologist at Sherpa Institute for People with Intellectual Disabilities, for their detailed analysis of his developmental history.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval for the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
None of the authors report any conflicts of interest for this work.
References
1. Vaz FM, Wanders RJA. Carnitine biosynthesis in mammals. Biochem J. 2002;361(3):417–429. doi:10.1042/0264-6021:3610417
2. Celestino-Soper PBS, Shaw CA, Sanders SF, et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet. 2011;20:4360–4370. doi:10.1093/hmg/ddr363
3. Celestino-Soper PBS, Violante S, Crawford EL, et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc Natl Acad Sci USA. 2012;109:7974–7981. doi:10.1073/pnas.1120210109
4. Nava C, Lamari F, Héron D, et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl Psychiatry. 2012:23:e179. doi:10.1038/tp.2012.102
5. Ziats MN, Comeaux MS, Yang Y, et al. Improvement of regressive autism symptoms in a child with TMLHE deficiency following carnitine suppletion. Am J Med Genet. 2015;167A:2162–2167. doi:10.1002/ajmg.a.3644
6. Neveling K, Feenstra I, Gilessen C, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34:1721–1726. doi:10.1002/humu.22450
7. Coene KLM, Kluijtmans LAJ, van der Heeft E, et al. Next-generation metabolic screening: targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J Inherit Metab Dis. 2018;41:337–353. doi:10.1007/s10545-017-0131-6
8. Tellegen PJ, Laros JA. Snijders-Oomen Non-Verbal Intelligence Test. [Snijders-Oomen Niet-Verbale Intelligentietest: Verantwoording]. Göttingen, Duitsland: Hogrefe; 2011. Dutch.
9. Sparrow SS, Cicchetti DV, Balla DA. Vineland Adaptive Behavior Scales.
10. Hoekman J, Miedema A, Otten B, Gielen J. ESSEON-R Scale for the Assessment of Social-Emotional Development. [ESSEON-R Schaal voor het Sociaal-Emotioneel Ontwikkelingsniveau]. Amsterdam: Hogrefe; 2014. Dutch.
11. Kraaijer DW. Scale for Autism and Related Disorders [AVZ-R: Autisme- En Verwante Stoornissenschaal-Z – Revised]. Amsterdam: Swets & Zeitlinger; 1999. Dutch.
12. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129–136. doi:10.1111/j.1600-0447.1996.tb09814.x
13. Beaudet AL. Hypothesis: brain carnitine deficiency causes nonsyndromic autism with an extreme male bias. Bioessays. 2017;39. doi:10.1002/bies.201700012
14. Kepka A, Ochocińska A, Chojnowska S, et al. Potential role of L-carnitine in autism spectrum disorder. J Clin Med. 2021;10:1202. doi:10.3390/jcm10061202
15. Sánchez-Luqyez KY, Carpena MX, Karam SM, Tovo-Rodrigues L. The contribution of whole exome sequencing to intellectual disability diagnosis and knowledge of underlying molecular mechanisms: a systematic review and meta-analysis. Mutat Res Rev Mutat Res. 2022;
16. Willemsen MH, Kleefstra T. Making headway with genetic diagnostics of intellectual disabilities. Clin Genet. 2014;85:101–110. doi:10.1111/cge.12244
17. Wortmann SB, Koolen DA, Smeitking JA, et al. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis. 2015;38(3):437–443. doi:10.1007/s10545-015-9823-y
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.