Back to Journals » The Application of Clinical Genetics » Volume 7
Wiskott–Aldrich syndrome: diagnosis, current management, and emerging treatments
Authors Buchbinder D ![]() , Nugent D, Fillipovich A
, Nugent D, Fillipovich A
Received 28 November 2013
Accepted for publication 31 December 2013
Published 3 April 2014 Volume 2014:7 Pages 55—66
DOI https://doi.org/10.2147/TACG.S58444
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
David Buchbinder,1 Diane J Nugent,1 Alexandra H Fillipovich2
1Division of Hematology, Children's Hospital of Orange County, Orange, CA, USA; 2Division of Immunology, Cincinnati Children's Hospital, Cincinnati, OH, USA
Abstract: Wiskott–Aldrich syndrome (WAS) is a rare X-linked primary immunodeficiency disorder characterized by the triad of eczema, thrombocytopenia, and severe and often recurrent infections. Despite the rarity of this disorder, our understanding of the molecular and cellular pathogenesis of WAS has continued to increase. Advances in the use of diagnostic tools, the provision of supportive care, and improvements in allogeneic hematopoietic stem cell transplantation have significantly reduced the morbidity and mortality associated with this disorder. Exciting advancements in the care of patients with WAS have also occurred, including the successful application of autologous gene-modified hematopoietic stem cell transplantation.
Keywords: primary immunodeficiency, Wiskott–Aldrich syndrome, hematopoietic stem cell transplantation, gene therapy
Introduction
Wiskott–Aldrich syndrome (WAS) is an X-linked primary immunodeficiency disorder that is characterized by the classic triad of severe immunodeficiency, microthrombocytopenia, and eczema.1 The incidence of this rare X-linked primary immunodeficiency disorder is approximately one to four cases per 1,000,000 live male births, with an average age at diagnosis of 24 months in families without a previously affected family member.2 The estimated prevalence of WAS in the US is 1.2% of patients with identified primary immune defects.3
The gene responsible for WAS is located on the short arm of the X chromosome at Xp11.22–p11.23.1 The WAS gene encodes the WAS protein (WASp), which is a 501-amino acid protein expressed within the cytoplasm of nonerythroid hematopoietic cells.1 WASp is involved in actin polymerization and associated coupling of receptor engagement, signaling events, and cytoskeletal rearrangement.1 Heterogeneous mutations spanning the entire WAS gene have been described.1 These mutations in WAS alter the function and/or expression of the intracellular protein, WASp.1 Perturbation in the function and/or expression of WASp is correlated with the spectrum of clinical findings of “classic” WAS and its associated phenotypic variations, such as X-linked thrombocytopenia (XLT) and X-linked neutropenia (XLN).1
The purpose of this review is to provide clinicians with a comprehensive review of the tools used in the clinical diagnosis and treatment of WAS and its associated complications. We will not focus on the molecular basis of WAS, including the structure and function of WASp. Recent developments that have occurred with respect to the pathogenesis and treatment of the complications that occur in association with WAS will be reviewed. Curative approaches, including the use of allogeneic hematopoietic stem cell transplantation (HSCT), will be discussed. Emerging therapeutic approaches, such as autologous gene-modified HSCT, will also be reviewed.
Diagnosis
WAS is an X-linked disorder that is manifest in males, with an absence of clinical symptomatology in obligate female carriers. Rare cases of females with WAS have been described involving a deleterious mutation of the paternally derived X chromosome and nonrandom inactivation of the maternally derived X chromosome.4 The diagnosis of WAS should be considered in any male presenting with a clinical history, physical exam findings, and laboratory data suggestive of the disorder (see Figure 1).2 Clinical findings consistent with WAS may be present or absent at various times across the trajectory of illness. Reassessment is necessary due to the evolution of clinical, physical, and laboratory findings over time.
 | Figure 1 Clues to the diagnosis of Wiskott–Aldrich syndrome. |
Despite the wide clinical spectrum seen in WAS, a strong genotype and phenotype correlation exists.5 Mutations resulting in absent WASp expression are associated with the “classic” syndrome. Mutations resulting in residual WASp expression are associated with the milder XLT,1 and gain-of-function mutations in the WAS gene result in XLN.1 The phenotype of the patient may also be altered by genetic reversion events that may endow a selective advantage upon hematopoietic cells with normal WASp expression.6 In addition to revertant mosaicism, mutations resulting in the restoration of a wild-type sequence of compensatory alterations have been described. These states can be suggested by a milder phenotype than would be predicted by the genotype.
WASp detection and genetic analysis
The use of flow-cytometry analysis of WASp expression as a screening tool in all cases of suspected WAS is recommended. The quantification of intracytoplasmic WASp expression serves as a rapid and inexpensive screening test for WAS.7 Abnormal WASp expression may indicate a variety of disease states, including the presence of the disease, carrier state, revertant states, or mixed chimerism following transplantation.8,9 If the flow-cytometry analysis of WASp expression is normal, further testing should be dictated by the clinical suspicion of the diagnosis.
For those cases in which the diagnosis is still under consideration, sequencing of the WAS gene is required. This typically includes sequencing of the coding regions as well as the intron and exon boundaries of the WAS gene. This approach will identify the majority of mutations in males; however, mutations impacting upstream regulatory elements or untranslated areas may be missed. Furthermore, WAS occurring in the setting of large deletions may also be missed. For those families in which the WAS mutation is known, targeted analysis of the WAS gene is recommended.10 These approaches have been utilized in the setting of prenatal diagnosis in at-risk couples. More recently, they have also been utilized in the setting of preimplantation genetic testing for at-risk couples seeking to avoid the birth of a child with WAS11 or to ensure the birth of an unaffected child who may be utilized as a human leukocyte antigen (HLA)-matched donor in the setting of allogeneic HSCT.12
Clinical scoring
Once a diagnosis of WAS is made, a clinical score may be assigned.13,14 The clinical scoring system is derived from a variety of clinical parameters, including the presence of thrombocytopenia, eczema, immunodeficiency, autoimmunity, and malignancy. The WAS scoring system ranges between 0 and 5. A WAS score greater than or equal to 3 is considered “classic” WAS. These patients have presence of thrombocytopenia, eczema, immunodeficiency, and infectious sequelae. The presence of autoimmunity and/or malignancy is consistent with a WAS score of 5. Transient eczema, immunodeficiency, and mild infectious complications may also occur and may not portend a bad prognosis. These patients may still be classified as XLT with a WAS score of 2. A WAS score of 0 is reserved for those patients with gain-of-function mutations associated with XLN and/or myelodysplasia. Patients may transition from a lower WAS score to a higher WAS score. The mechanisms and risk factors underlying this transition are unknown; however, data suggest that this transition may occur secondarily to epigenetic changes.15 Patients with a higher WAS score (ie, 5) at a younger age (ie, during the first 2 years of life) may represent a group at a high risk of morbidity and mortality.16 The most common first clinical manifestation in these patients was severe refractory thrombocytopenia with a platelet count less than 10,000/μL.16
Immunodeficiency
Abnormalities in immune system function (ie, cell-mediated, humoral, and innate immunity) among patients with WAS result in susceptibility to a wide variety of infectious pathogens.2 Despite this, infectious complications as a sole first manifestation of WAS are uncommon (<5% of cases).2 Patients with WAS are susceptible to opportunistic infections with organisms such as Pneumocystis jirovecii.2 Patients with WAS may develop severe and disseminated forms of viral infections, with herpes simplex virus I or II (6% of cases) and varicella (3% of cases) as the most common pathogens.2 Invasive yeast and fungal infections (10% of cases) have also been described.2 Sinopulmonary infections are the most common infectious complications prior to diagnosis, including otitis media (64% of cases) and pneumonia (25% of cases).2 Other severe infectious complications may occur, such as sepsis (7% of cases) and meningitis (4% of cases).2
The immunodeficiency in WAS involves T cells, and is associated with both quantitative and qualitative defects in T cells.2 T-cell lymphopenia is often present in patients with WAS, and may occur due to increased apoptosis.17 Patients with WAS typically have an absolute lymphocyte count >1,000/μL, demonstrating the absence of the profound lymphopenia seen in other primary immunodeficiency disorders. An absolute lymphocyte count of <1,000/μL was noted in only 22% of patients with WAS.2 Functional abnormalities in T cells may be present, as demonstrated by abnormal proliferative responses to mitogens; however, 46% of patients have normal responses.2 T-cell chemotaxis, activation, and cytokine secretion are also impaired in patients with WAS.18,19
Humoral immune responses are abnormal in patients with WAS.2 Serum levels of immunoglobulin (Ig)-G, IgM, and IgA are often low and IgE levels often high in patients with WAS.2 Functional antibody responses may be abnormal as demonstrated by abnormal isohemagglutinin titers (84% of cases) and diminished vaccine responses to protein (eg, 62% with abnormal responses to tetanus vaccine), polysaccharide (eg, 69% with abnormal responses to pneumococcal vaccine), and conjugate vaccines (eg, 66% with abnormal responses to Hib (haemophilus influenzae type b) conjugate vaccine).2 Perturbations in T-cell function mentioned earlier may impair the maturation and differentiation of B cells into antibody-producing cells and memory cells; however, intrinsic defects in B cells are also notable in patients with WAS.20
Abnormalities in elements of the innate immune response may also be present. Natural killer (NK) cells and numbers may be normal or increased in patients with WAS; however, NK cell function is often abnormal, including formation of the immunological synapse, mobilization of secretory granules, and subsequent cytolytic activity.21 Despite the presence of normal numbers of neutrophils, monocytes, and other phagocytes, functional abnormalities may also be present in patients with WAS.2 Chemotaxis, the initiation of degranulation, the formation of a functional respiratory burst, and antibody-mediated phagocytosis may be impaired in patients with WAS.22,23
Autoimmunity
Autoimmunity is a frequent occurrence in patients with WAS. Estimates suggest 40% of patients with WAS develop autoimmunity, with many patients developing several manifestations of autoimmune disease.2 As an example, Sullivan et al reported that two-thirds of patients with autoimmune manifestations of WAS developed multiple autoimmune disorders.2 The most common manifestations of autoimmunity in the setting of WAS include autoimmune hemolytic anemia (14%), vasculitis (13%), renal disease (12%), and chronic arthritis (10%).2 Autoimmunity in the setting of WAS may be due to the formation of autoantibodies or the presence of autoreactive T-cell clones. The development of autoimmunity as a complication of WAS may also be related to perturbations in regulatory T-cell24 and B-cell20 homeostasis.
Malignancy
The development of malignancy is a frequent occurrence in the setting of WAS. Sullivan et al noted that 13% of patients with WAS developed malignancy at a mean age of 9.5 years.2 Lymphomas were the most commonly observed malignancy in the setting of WAS.2 Moreover, there is a preponderance of non-Hodgkin’s lymphoma (NHL) in patients with WAS when compared to Hodgkin’s lymphoma.25 The most common histologic variants of pediatric NHL, such as Burkitt’s lymphoma and lymphoblastic lymphoma, are poorly represented among patients with WAS. Other forms of NHL, including large-cell lymphoma variants, are more commonly described. Other malignancies, including myelodysplasia, leukemia, and myeloproliferative disorders, have also been described.26 The aggressive nature of malignancy in the setting of WAS portends a poor prognosis, as demonstrated by historical data suggesting >95% mortality among those affected.2 Genetic susceptibility conferred by abnormal WASp function, associated abnormalities in tumor-surveillance mechanisms (eg, impairment in NK cell cytotoxicity), and environmental factors (eg, Epstein–Barr virus [EBV]) are examples of factors that increase the risk of malignancy in patients with WAS.27
Thrombocytopenia/bleeding
Thrombocytopenia is nearly universal in patients with WAS; however, the severity is variable. Approximately half of patients with WAS will have severe thrombocytopenia with a platelet count <20,000/μL.2 Moreover, the severity is correlated with the risk of bleeding.28 Bleeding complications occur in >80% of WAS patients.2 These bleeding complications may be non-life-threatening (eg, epistaxis, ecchymoses, petechiae, etc) or life-threatening (eg, gastrointestinal hemorrhage, intracranial hemorrhage).2 Sullivan et al found that life-threatening bleeding complications occurred in 30% of patients; however, intracranial hemorrhage occurred in only 2% of patients prior to diagnosis.2 Megakaryocyte numbers are typically normal in patients with WAS; however, platelet formation is abnormal.29 Moreover, platelet destruction in association with antiplatelet antibodies may be a contributing factor in the maintenance of thrombocytopenia.30 Perturbations in platelet function are also notable, and may result in increased risk of bleeding and diminished platelet survival.29 These defects in platelet production and function relate to the role WASp plays in the process of platelet formation, activation, and associated cytoskeletal remodeling.31
X-linked thrombocytopenia and X-linked neutropenia
XLT patients have excellent overall survival in contrast to patients with WAS; however, event-free survival demonstrates decrements over time.32 Data from the XLT registry suggest an overall survival of 97% at 15 years, with only minor decrements over the life span. Event-free survival is similarly high at 15 years (74%); however, there is a sharper decrement over the life span. For example, by 30 years, event-free survival drops to 56%. Serious bleeding episodes occurred in 13.9%, life-threatening infections in 6.9%, autoimmunity in 12.1%, and malignancy in 5.2% of XLT patients at median ages of 4.9 years, 24.8 years, 12.2 years, and 34.0 years, respectively.32 XLN is a form of severe chronic neutropenia associated with variable degrees of neutropenia and associated infectious complications. In contrast to WAS, infectious complications suggestive of T-cell immunodeficiency are absent. Perturbations in immune function are similar to those described for WAS; however, low numbers of NK cells are a consistent finding in XLN. Mild reductions in platelet counts have also been described. The potential risk of myelodysplastic syndrome and myeloid leukemia exists, which requires regular surveillance.
Prognosis and outcome
Once a diagnosis is confirmed, the patient should be evaluated in a timely manner at a center with expertise in the management of WAS. Without appropriate care and intervention, morbidity and mortality are frequent. Sullivan et al found that 36% of patients with WAS experienced non-HSCT-associated deaths at a mean age of 8 years.2 These deaths were attributed to infection (44%), bleeding (23%), and malignancy (26%). Many centers that provide care for patients with WAS and other primary immunodeficiency disorders have a multidisciplinary approach to the care of patients and affected family members. This includes genetic counseling, psychosocial support services, and subspecialist support, such as transplant immunology, hematology/oncology, infectious disease, and critical care. With appropriate care and timely intervention, patients with WAS have an excellent prognosis. As an example, long-term survival following the use of allogeneic HSCT is >80%.1
Current management
Treatment – infection
The prevention of infectious complications is required. Intravenous Ig (IVIG) is an important adjunct in the treatment of WAS patients.33 Patients with recurrent infectious complications, abnormal quantitative Igs, or abnormal antibody responses should be provided with IVIG at physiologic replacements doses. Alternatively, subcutaneous administration may also be considered. Patients with recurrent bacterial sinopulmonary infections may benefit from prophylactic antibiotic use.33 All patients who have undergone splenectomy, regardless of vaccination status, should be placed on lifelong prophylactic penicillin or an equivalent agent if they are allergic to penicillin or intolerant. Patients should be immunized with conjugated and unconjugated vaccines; however, the avoidance of live viral and attenuated viral vaccines is necessary. All patients with a diagnosis of WAS should be placed on Bactrim or an equivalent agent to prevent P. jirovecii pneumonia. For patients who have had significant viral infections, consideration can be given to the use of prophylactic antiviral agents.
For those patients with clinical signs or symptoms of infection, prompt evaluation and treatment is necessary. The initiation of empiric parenteral antibiotic treatment is necessary in the majority of cases. The evaluation should be exhaustive until the source of the infection is uncovered. This may include invasive assessments, as cultures and isolation of the offending organism should be sought in order to guide therapy. In those cases where HSCT is being considered, attention to the prevention and treatment of infectious complications is necessary to limit pretransplant morbidity.
Treatment – autoimmunity
For severe manifestations of autoimmunity, immunomodulatory therapy including IVIG may improve symptoms. A variety of immunosuppressive agents can also be utilized. Corticosteroids are widely utilized; however, the toxicity (eg, increased risk of fungal infections) associated with the use of these agents is often large.16 Limited data exist on the use of other immunosuppressive agents and immunomodulatory therapies, such as cyclosporine, azathioprine, cyclophosphamide, and plasma exchange.16 Targeted immunosuppressive therapy utilizing monoclonal antibodies (eg, anti-CD20) may be a useful adjunct in cases of EBV or where a definitive pathophysiological role for autoantibodies is suspected.16 For some types of autoimmunity, it is important to note that these can be secondary to infectious pathogens. As an example, arthritis can be secondary to chronic EBV, cytomegalovirus, mycoplasma, or other pathogens. In those cases where HSCT is being considered, attention to the treatment of autoimmunity is also necessary to limit pretransplant morbidity. Moreover, pre-HSCT autoimmunity may be associated with an increased risk of post-HSCT autoimmunity.16,34,35
Treatment – thrombocytopenia
Early studies in WAS patients provided an assessment of agents that have been useful in the treatment of other forms of thrombocytopenia (eg, immune thrombocytopenic purpura).36 IVIG did not have any appreciable impact on platelet counts. Splenectomy is associated with improvement and/or normalization in the platelet count, although important limitations exist.33 If the use of HSCT is under consideration, splenectomy should be avoided. Splenectomy is associated with increased morbidity and mortality following HSCT.34,35 A recent report of long-term HSCT recipients with WAS documented that 21% of those who underwent splenectomy developed significant infectious complications.34 No infectious complications were noted in those who did not have a history of splenectomy. Other agents, such as antifibrinolytic agents, are excellent adjuncts in the prevention and treatment of mucosal bleeding, with the exception of genitourinary bleeding, where they should be avoided. Platelet transfusions using irradiated and cytomegalovirus-negative products may be necessary for patients with clinically severe bleeding (eg, intracranial hemorrhage) or for those patients at high risk for severe bleeding (eg, perioperative); however, the risks of alloimmunization remain. Newer agents, including thrombopoietin mimetic drugs, have also been used in the setting of WAS; however, these approaches did not yield consistent improvements in thrombocytopenia or bleeding.37
Treatment – transplantation
Transplantation is the current accepted curative approach for patients with WAS (see Table 1).16,34,35,38–55 HLA typing of all potential family donors should be completed. If a family donor is not identified, an unrelated donor search should be performed, regardless of the WAS score. This approach allows for the identification of potential donors and also highlights those patients who lack donors, for whom alternative therapeutic approaches may be needed. For patients with WAS and an HLA-identical sibling donor or a matched related or unrelated donor, HSCT should be considered. For those patients with a high WAS score (≥3) lacking a sibling or matched related or unrelated donor, alternative donor HSCT should be considered. Younger patients (<2 years of age) with severe refractory thrombocytopenia (<10,000/μL) may also benefit from early intervention with HSCT.16 For XLT patients lacking a sibling donor, supportive care and clinical monitoring may be considered. HSCT should be avoided in patients with isolated thrombocytopenia, unless there is an HLA-identical sibling donor.32
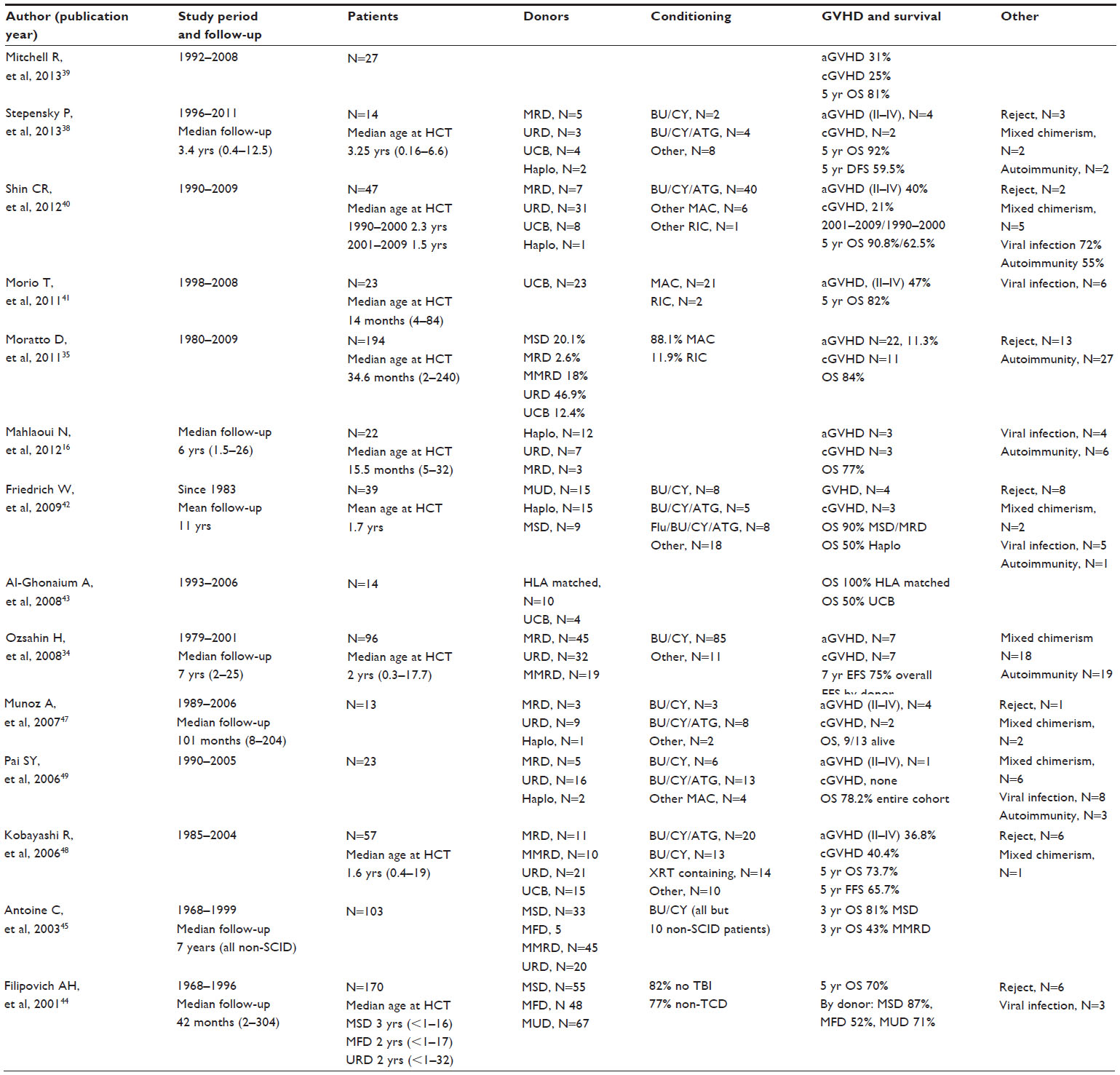 | Table 1 Results of allogeneic transplantation in the treatment of Wiskott–Aldrich syndrome published 2000–2013 |
Transplant considerations
All patients should be considered for HSCT at a young age. In the setting of WAS, age at the time of HSCT is an important consideration.16,35,40,44 Patients with WAS who undergo unrelated donor HSCT at younger ages (ie, less than 5 years of age) have outcomes that are comparable to those associated with the use of matched-sibling donor HSCT. Recommendations regarding the timing of HSCT when considering very young children (ie, neonates and infants) cannot be made, due to a lack of data focusing on the pharmacokinetics, pharmacodynamics, and toxicity of conditioning agents in neonates and young infants.
Donor type is an important consideration in the utilization of HSCT for patients with WAS.35,43–45 In general, HSCT outcomes have improved over time for all donor types.34,35,40 HSCT utilizing HLA-identical sibling donors is associated with excellent outcomes, as demonstrated by an overall survival over 80%.16,34,35,38–55 Matched unrelated donor HSCT has also demonstrated success in the setting of WAS, with the expectation of outcomes that are similar to the HLA-identical sibling donor experience.16,34,35,38–55 For those patients with a high WAS score lacking an HLA-identical sibling donor or a well-matched related or unrelated donor, alternative donor HSCT may be considered. In general, the data are poor for haploidentical HSCT, due to the high morbidity and mortality associated with graft-versus-host disease (GVHD) and rejection. Survival is approximately 50% in the majority of the larger published cohorts.35,42,44,45,48
Graft source is an additional consideration in the utilization of HSCT for patients with WAS.34–43 The majority of the clinical experience in HSCT for patients with WAS is associated with the use of bone marrow as a graft source. A matched unrelated donor marrow that is 10/10 (considering HLA-A, -B, -C, -DRB1, -DQ) using high-resolution typing provides excellent outcomes. Recent data also demonstrate that cord blood as a graft source has excellent outcomes as well.35,41,48 A cord blood unit that is 5/6 or 6/6 (considering HLA-A and -B using low-resolution typing and HLA-DRB using high-resolution typing) with an adequate cell dose is acceptable, and is associated with good outcomes. Cord blood units with a 4/6 match may also be considered if an adequate cell dose is available. Other donor cell sources, such as peripheral blood stem cells, may also be considered, although there is a higher risk of chronic GVHD.
The choice of preparatory therapy is an important consideration in the care of a patient with WAS who will be undergoing HSCT.38,48 Particularly for WAS patients, mixed chimerism has been associated with post-HSCT complications. A myeloablative preparative approach with the goal of obtaining complete and durable engraftment is ideal. Matched related or unrelated donor HSCT using a myeloablative preparatory combination, such as busulfan, cyclophosphamide, and antithymocyte globulin, is a proven approach with a marrow donor or suitable cord blood unit.38–48 The use of myeloablative preparatory regimens associated with reduced toxicity (eg, reduced-dose busulfan-, treosulfan-based regimens) are also being adopted in the setting of primary immunodeficiency disorders, including WAS.51,52 These approaches may be considered in patients without comorbidities, in order to limit early and late morbidity and mortality associated with HSCT. If the patient is unable to tolerate a myeloablative preparatory regimen due to underlying infections or other comorbidities, a reduced-intensity approach to conditioning is recommended. Various groups have demonstrated the ability to achieve complete and durable engraftment using a reduced-intensity approach with matched related or unrelated marrow donors or with cord blood.53,54 Additional data are needed; however, promising data in high-risk WAS patients are available. Nonmyeloablative preparatory regimens followed by HLA-matched related or unrelated bone marrow have also been utilized successfully; however, the data using these approaches are limited.55
Posttransplant complications
Mixed chimerism
Mixed chimerism posttransplant is an undesirable outcome following HSCT for WAS, secondary to the association of mixed chimerism with a variety of post-HSCT complications, including lymphopenia, autoimmunity, and thrombocytopenia.34,35,40 Recent data focusing on long-term outcomes and lineage-specific chimerism in 194 HSCT recipients with WAS demonstrated that mixed chimerism frequently impacts the myeloid compartment (16.5% of cases), followed by the B-cell compartment (7.4% of cases), and uncommonly the T-cell compartment (3.2% of cases).35 With current approaches, the majority of patients with WAS will achieve full and stable donor chimerism (72.1% of cases).35 Those patients who undergo a reduced-intensity approach or nonmyeloablative approach have an increased risk of mixed chimerism post-HSCT. All HSCT recipients with a diagnosis of WAS should undergo an assessment of lineage-specific chimerism posttransplant. This analysis can be achieved using a variety of techniques, such as variable nucleotide tandem-repeat analysis on purified CD3, CD19, CD16/56, and CD14 (or CD15) cells. The quantification of lineage-specific WASp expression by flow cytometry may be even more informative, and would also provide data on specific T-lymphocyte subsets, such as CD4+ and CD8+ T cells. For those HSCT recipients who develop mixed chimerism, the use of cellular therapy should be considered, including the use of donor lymphocyte infusions. As an example, Shin et al noted that four of five HSCT recipients with WAS and mixed chimerism experienced an increase in donor chimerism following cellular therapy.40
Autoimmunity, infections, malignancy
Post-HSCT patients with WAS are at risk for specific complications, including post-HSCT autoimmunity, infections, and malignancy.34,35,40,41,44 Estimates of post-HSCT autoimmunity are variable (13.9%–20%).34,35 The development of post-HSCT autoimmunity typically occurs early (eg, the first 2 years post-HSCT); however, the development of these complications may be delayed and the course may be protracted.34,35,40 The median time to development of post-HSCT autoimmunity is 1.5 years post-HSCT (range 4 months to 10 years), with a median duration of 4 years.34 Autoimmune cytopenias are the most common manifestation of autoimmunity post-HSCT and may require treatment. Shin et al noted that 55% of HSCT recipients with post-HSCT autoimmunity developed autoimmune cytopenias.40 Moreover, it is important to note that HSCT recipients who have experienced autoimmunity prior to HSCT appear to have a greater risk of severe and prolonged post-HSCT autoimmunity.
WAS patients who have undergone HSCT may also experience a significant risk of viral infections post-HSCT.34,35,40,41,44 Shin et al noted that 84% of HSCT recipients with WAS experienced viral reactivation.40 This complication may be more pronounced in the setting of HSCT utilizing cord blood as a graft source. Hematopoietic progenitor cells that are contained within cord blood are virally naive. Moreover, the majority of WAS patients are infected with a variety of viruses from an early age. As a result of these issues, it is anticipated that post-HSCT reactivation of viral infections in the host may be a significant challenge. Careful and frequent surveillance (eg, with quantitative polymerase chain reaction), prevention, and treatment of these infectious sequelae are critical in the care of HSCT recipients with WAS.
With regard to primary immunodeficiency disorders, the incidence of malignancies posttransplant are also among the highest in patients with WAS.50 Estimates suggest an incidence of 3.3% in patients with WAS compared to an overall estimate of 2.3% in patients with other primary immunodeficiency disorders. The types of malignancies documented in post-HSCT recipients with a diagnosis of WAS include lymphoproliferative disorders, such as posttransplant lymphoproliferative disorder (non-EBV-associated and EBV-associated), B-cell lymphoma, large-cell lymphoma, myelodysplasia, and myeloid leukemia.34,35,40,41,44 Full donor engraftment and freedom from immunosuppression post-HSCT may limit the risk of malignancy.
Emerging treatments
Autologous gene-modified HSCT has been an emerging therapy in the treatment of primary immunodeficiency, including WAS.56–59 The avoidance of the immunological barriers imposed by graft rejection and GVHD seen in allogeneic HSCT supports the use of autologous gene-modified HSCT. Utilizing these techniques, CD34+-enriched grafts undergo ex vivo gene correction. Gene correction is carried out with the use of a variety of viruses that are able to deliver and integrate the desired gene into the genome. Nonmyeloablative conditioning is used to confer a selective advantage to the gene-modified cells. The manipulated graft is then reinfused, with stable persistence of the corrected gene resulting in clinical improvement.
Results from studies using γ-retroviral vectors have been reported.57,59 A total of ten patients with WAS received autologous gene-modified HSCT using cells that were transduced with retroviral vectors expressing WASp. Stable and durable integration of the corrected gene and expression of WASp was noted.59 Following gene-modified autologous HSCT, improvement in the associated complications of WAS, including infection, bleeding, and autoimmunity, was noted in nine of ten patients.59 Five patients demonstrated insertional oncogenesis with subsequent development of T-cell leukemia, and one patient developed myelodysplasia followed by acute myeloid leukemia.59 Three of the patients with T-cell leukemia and the one patient who developed acute myeloid leukemia required subsequent allogeneic HSCT.59
Efforts at improving the efficacy and safety of autologous gene-modified HSCT have focused on the use of lentiviral vectors.56,57 Three patients with WAS received autologous gene-modified HSCT using cells that were transduced with lentiviral vectors expressing WASp. Stable and durable integration of the corrected gene and expression of WASp was noted. Following gene-modified autologous HSCT, the patients with WAS demonstrated clinical improvement. In these cases, the gene-modified cells were reinfused after a conditioning regimen consisting of anti-CD20 monoclonal antibody, busulfan, and fludarabine. No insertional oncogenesis-associated events have been recorded to date; however, time will be needed to assess long-term safety and efficacy.
In addition to the advances in autologous gene-modified HSCT, newer approaches to the treatment of WAS may hold promise.60 As an example, technology that will support gene correction as opposed to the addition of a new gene using autologous gene-modified HSCT is rapidly advancing. Whether or not these techniques can be successfully applied clinically to WAS patients is not known at this time. Furthermore, our understanding of WAS as it relates to actin polymerization is continuing to unfold. As an example, WASp-interacting protein, a binding partner of WASp that serves to stabilize and prevent degradation of WASp, has been characterized. Efforts aimed at stabilizing WASp through WASp-interacting protein may prove useful and clinically beneficial in patients with WAS.
Conclusion
Despite the rarity of WAS, we have continued to make impressive strides in the strategies used to diagnose WAS. Improvements in the management of WAS have also been large, as demonstrated by the meticulous supportive care that is provided to these patients by experienced centers. Exciting advances in allogeneic HSCT have also ensured curative options and excellent long-term outcomes for the majority of patients with WAS. The immunological barriers of graft rejection and GVHD, which are the key challenges in allogeneic HSCT, have led to the development and successful application of autologous gene-modified HSCT in patients with WAS. The commitment of clinical and basic science resources across a wide array of disciplines, such as immunology, hematology, and transplantation, ensure a bright future for patients impacted by WAS, and also provide an exciting model for the care of other primary immunodeficiency disorders.
Disclosure
The authors report no conflicts of interest in this work.
References
Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. 2013;1285:26–43. | |
Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A mutliinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994; 125(6 Pt 1):876–885. | |
Modell V, Gee B, Lewis DB, et al. Global study of primary immunodeficiency diseases (PI) – diagnosis, treatment, and economic impact: an updated report from the Jeffrey Modell Foundation. Immunol Res. 2011;51(1):61–70. | |
Boonyawat B, Dhanraj S, Al Abbas F, et al. Combined de-novo mutation and non-random X-chromosome inactivation causing Wiskott-Aldrich syndrome in a female with thrombocytopenia. J Clin Immunol. 2013;33(7):1150–1155. | |
Imai K, Morio T, Zhu Y, et al. Clinical course of patients with WASP gene mutations. Blood. 2004;103(2):456–464. | |
Davis BR, Candotti F. Revertant somatic mosaicism in the Wiskott-Aldrich syndrome. Immunol Res. 2009;44(1–3):127–131. | |
Kawai S, Minegishi M, Ohashi Y, et al. Flow cytometric determination of intracytoplasmic Wiskott-Aldrich syndrome protein in peripheral blood lymphocyte subpopulations. J Immunol Methods. 2002;260(1–2):195–205. | |
Yamaguchi K, Ariga T, Yamada M, et al. Mixed chimera status of 12 patients with Wiskott-Aldrich syndrome (WAS) after hematopoietic stem cell transplantation: evaluation by flow cytometric analysis of intracellular WAS protein expression. Blood. 2002;100(4):1208–1214. | |
Yamada M, Ariga T, Kawamura N, et al. Determination of carrier status for the Wiskott-Aldrich syndrome by flow cytometric analysis of Wiskott-Aldrich syndrome protein expression in peripheral blood mononuclear cells. J Immunol. 2000;165(2):1119–1122. | |
Siminovitch KA. Prenatal diagnosis and genetic analysis of Wiskott-Aldrich syndrome. Prenat Diagn. 2003;23(12):1014–1016. | |
Simpson JL, Carson SA, Cisneros P. Preimplantation genetic diagnosis (PGD) for heritable neoplasia. J Natl Cancer Inst Monogr. 2005;(34):87–90. | |
Rechitsky S, Kuliev A, Tur-Kaspa I, Morris R, Verlinsky Y. Preimplantation genetic diagnosis with HLA matching. Reprod Biomed Online. 2004;9(2):210–221. | |
Zhu Q, Watanabe C, Liu T, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression and phenotype. Blood. 1997;90(7):2680–2689. | |
Imai K, Nonoyama S, Ochs HD. WASP (Wiskott-Aldrich syndrome protein) gene mutations and phenotype. Curr Opin Allergy Clin Immunol. 2003;3(6):427–436. | |
Buchbinder D, Nadeau K, Nugent D. Monozygotic twin pair showing discordant phenotype for X-linked thrombocytopenia and Wiskott-Aldrich syndrome: a role for epigenetics? J Clin Immunol. 2011;31(5):773–777. | |
Mahlaoui N, Pellier I, Mignot C, et al. Characteristics and outcome of early-onset, severe forms of Wiskott-Aldrich syndrome. Blood. 2013;121(9):1510–1516. | |
Rawlings SL, Crooks GM, Bockstoce D, Barsky LW, Parkman R, Weinberg KI. Spontaneous apoptosis in lymphocytes from patients with Wiskott-Aldrich syndrome: correlation of accelerated cell death and attenuated bcl-2 expression. Blood. 1999;94(11):3872–3882. | |
Zhang J, Shehabeldin A, da Cruz LA, et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J Exp Med. 1999;190(9):1329–1342. | |
Calvez R, Lafouresse F, De Meester J, Galy A, Valitutti S, Dupré L. The Wiskot-Aldrich syndrome protein permits assembly of a focused immunological synapse enabling sustained T-cell receptor signaling. Haematologica. 2011;96(10):1415–1423. | |
Recher M, Burns SO, de la Fuente MA, et al. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice. Blood. 2012;119(12):2819–2828. | |
Orange JS, Ramesh N, Remold-O’Donnell E, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK-cell activating immunological synapses. Proc Natl Acad Sci U S A. 2002;99(17):11351–11356. | |
Kumar S, Xu J, Perkins C, et al. Cdc42 regulates neutrophil migration via crosstalk between WASp, CD11b, and microtubules. Blood. 2012;120(17):3563–3574. | |
Dart AE, Donnelly SK, Holden DW, Way M, Caron E. Nck and Cdc42 co-operate to recruit N-WASP to promote FcγR-mediated phagocytosis. J Cell Sci. 2012;125(Pt 12):2825–2830. | |
Humblet-Baron S, Sather B, Anover S, et al. Wiskott-Aldrich syndrome protein is required for regulatory T cell hemostasis. J Clin Invest. 2007;117(2):407–418. | |
Cotelingam JD, Witebsky FG, Hsu SM, Blaese RM, Jaffe ES. Malignant lymphoma in patients with the Wiskott-Aldrich syndrome. Cancer Invest. 1985;3(6):515–522. | |
Yohshimi A, Kamachi Y, Imai Y, et al. Wiskott-Aldrich syndrome presenting with a clinical picture mimicking juvenile myelomonocytic leukemia. Pediatr Blood Cancer. 2013;60(5):836–841. | |
Kersey JH, Shapiro RS, Filipovich AH. Relationship of immunodeficiency to lymphoid malignancy. Pediatr Infect Dis J. 1988;7(Suppl 5):S10–S12. | |
Notarangelo LD. In Wiskott-Aldrich syndrome, platelet count matters. Blood. 2013;121(9):1484–1485. | |
Shcherbina A, Cooley J, Lutskiy MI, Benarafa C, Gilbert GE, Remold-O’Donnell E. WASP plays a novel role in regulating platelet responses dependent on alphaIIbbeta3 integrin outside-in signaling. Br J Haematol. 2010;148(3):416–427. | |
Marathe BM, Prislovsky A, Astrakhan A, Rawlings DJ, Wan JY, Strom TS. Antiplatelet antibodies in WASP(-) mice correlate with evidence of increased in vivo platelet consumption. Exp Hematol. 2009;37(11):1353–1363. | |
Lutskiy MI, Shcherbina A, Bachli ET, Cooley J, Remold-O’Donnell E. WASP localizes to the membrane skeleton of platelets. Br J Haemotol. 2007;139(1):98–105. | |
Albert MH, Bittner TC, Nonoyama S, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. 2010;115(16):3231–3238. | |
Litzman J, Jones A, Hann I, Chapel H, Strobel S, Morgan G. Intravenous immunoglobulin, splenectomy, and antibiotic prophylaxis in Wiskott-Aldrich syndrome. Arch Dis Child. 1996;75(5):436–439. | |
Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, et al. Long-term outcome following hematopoietic stem cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and the European Group for Blood and Marrow Transplantation. Blood. 2008;111(1):439–445. | |
Moratto D, Giliani S, Bonfim C, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an international collaborative study. Blood. 2011;118(6):1675–1684. | |
Matthew P, Conley ME. Effect of intravenous gammaglobulin (IVIG) on the platelet count in patients with Wiskott-Aldrich syndrome. Pediatr Allergy Immunol. 1995;6(2):91–94. | |
Bussel JB, Frelinger AL, Mitchell WB, Pinheiro MP, et al. Platelet function and response to thrombopoietin mimetics in Wiskott-Aldrich syndrome/X-linked thrombocytopenia. Poster presented at: ASH Meeting December 4–7, 2010; Orlando FL. Abstract 1429. | |
Stepensky P, Krauss A, Goldstein G, et al. Impact of conditioning on outcome of hematopoietic stem cell transplantation for Wiskott-Aldrich syndrome. J Pediatr Hematol Oncol. 2013;35(6):e234–e238. | |
Mitchell R, Nivison-Smith I, Anazodo A, et al. Outcomes of hematopoietic stem cell transplantation in primary immunodeficiency: a report from the Australian and New Zealand Children’s Haematology Oncology Group and the Australasian Bone Marrow Transplant Recipient Registry. Biol Blood Marrow Transplant. 2013;19(3):338–343. | |
Shin CR, Kim MO, Li D, et al. Outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome. Bone Marrow Transplant. 2012;47(11):1428–1435. | |
Morio T, Atsuta Y, Tomizawa D, et al. Outcome of unrelated umbilical cord blood transplantation in 88 patients with primary immunodeficiency in Japan. Br J Haematol. 2011;154(3):363–372. | |
Friedrich W, Schütz C, Schulz A, Benninghoff U, Hönig M. Results and long-term outcome in 39 patients with Wiskott-Aldrich syndrome transplanted from HLA-matched and mismatched donors. Immunol Res. 2009;44(1–3):18–24. | |
Al-Ghonaium A. Stem cell transplantation for primary immunodeficiencies: King Faisal Specialist Hospital experience from 1993 to 2006. Bone Marrow Transplant. 2008;42 Suppl 1:S53–S56. | |
Filipovich AH, Stone JV, Tomany SC, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood. 2001;97(6):1598–1603. | |
Antonie C, Müller S, Cant A, et al. Long-term survival and transplantation of haematopoietic stem cells for immunodeficiencies: a survey of the European experience 1968–99. Lancet. 2003;361(9357):553–560. | |
Gennery AR, Slatter MA, Grandin L, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010;126(3):602–610. | |
Muñoz A, Olivé T, Martinez A, et al. Allogeneic hematopoietic stem cell transplantation (HSCT) for Wiskott-Aldrich syndrome: a report of the Spanish Working Party for Blood and Marrow Transplantation in Children (GETMON). Pediatr Hematol Oncol. 2007;24(6):393–402. | |
Kobayahsi R, Ariga T, Nonoyama S, et al. Outcome in patients with Wiskott-Aldrich syndrome following stem cell transplantation: an analysis of 57 patients in Japan. Br J Haematol. 2006;135(3):362–366. | |
Pai SY, DeMartiis D, Forino C, et al. Stem cell transplantation for the Wiskott-Aldrich syndrome: a single-center experience confirms efficacy of matched unrelated donor transplantation. Bone Marrow Transplant. 2006;38(10):671–679. | |
Kaymani NR, Kumar S, Hassebroek A, et al. Malignancies after hematopoietic cell transplantation for primary immune deficiencies: a report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2011;17(12):1783–1789. | |
Güngör T, Teira P, Slatter M, et al. Reduced-intensity conditioning and HLA-matched haematopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. Epub October 22, 2013. | |
Slatter MA, Rao K, Amrolia P, et al. Treosulfan-based conditioning regimens for hematopoietic stem cell transplantation in children with primary immunodeficiency: United Kingdom experience. Blood. 2011;117(16):4367–4375. | |
Barnes YJ, Hayashi RJ, Witty S, Shenoy S. Allogeneic transplantation for Wiskott-Aldrich syndrome using a reduced intensity conditioning regimen. Biol Blood Marrow Transplant. 2007;13(Suppl 2):58. | |
Gocho Y, Koh K, Kato M, et al. Reduced-intensity stem cell transplantation is a promising therapeutic strategy for pediatric patients with Wiskott-Aldrich syndrome: a case series of 7 patients with a uniform reduced-intensity conditioning. Poster presented at: ASH Meeting December 10–13, 2011; San Diego, CA Abstract 1950. | |
Burroughs LM, Storb R, Leisenring WM, et al. Intensive postgrafting immune suppression combined with nonmyeloablative conditioning for transplantation of HLA-identical hematopoietic cell grafts: results of a pilot study for treatment of primary immunodeficiency disorders. Bone Marrow Transplant. 2007;40(7):633–642. | |
Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M. Gene therapy of primary T cell immunodeficiencies. Gene. 2013;525(2):170–173. | |
Auiti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341(6148):1233151. | |
Boztug K, Schmidt M, Schwarzer A, et al. Stem cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010;363(2):1918–1927. | |
Witzel MG, Braun CJ, Boztug K, et al. Hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Blood. 2013;122(21):718. | |
Massaad MJ, Ramesh N, Le Bras S, et al. A peptide derived from the Wiskott-Aldrich syndrome (WAS) protein-interacting protein (WIP) restores WAS protein level and actin cytoskeleton reorganization in lymphocytes from patients with WAS mutations that disrupt WIP binding. J Allergy Clin Immunol. 2011;127(4):998–1005. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.