Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Whole-Exome Sequencing in Patients Affected by Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis Reveals New Variants Potentially Contributing to the Phenotype
Authors Fonseca DJ ![]() , Morel A
, Morel A ![]() , Llinás-Caballero K
, Llinás-Caballero K ![]() , Bolívar-Salazar D, Laissue P
, Bolívar-Salazar D, Laissue P
Received 1 November 2020
Accepted for publication 26 December 2020
Published 1 March 2021 Volume 2021:14 Pages 287—299
DOI https://doi.org/10.2147/PGPM.S289869
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Dora Janeth Fonseca,1,* Adrien Morel,1,* Kevin Llinás-Caballero,1 David Bolívar-Salazar,1 Paul Laissue1,2
1Center for Research in Genetics and Genomics-CIGGUR, GENIUROS Research Group, School of Medicine and Health Sciences, Universidad Del Rosario, Bogotá, Colombia; 2BIOPAS Laboratoires, Orphan Diseases Unit, BIOPAS GROUP, Bogotá, Colombia
*These authors contributed equally to this work
Correspondence: Paul Laissue
BIOPAS Laboratoires, BIOPAS Group, Calle 127A #53ª-45, Bogotá, CP, 11001, Colombia
Tel +57 3212010179
Email [email protected]
Background: Adverse drug reactions (ADRs) are frequent occurring events that can essentially be defined as harmful or unpleasant symptoms secondary to the use of a medicinal product. ADRs involve a wide spectrum of clinical manifestations ranging from minor itching and rash to life-threatening reactions. Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are rare ADRs. SJS-TEN may be considered a polygenic pathology due to additive/epistatic effects caused by sequence variants in numerous genes. Next-generation sequencing (NGS) represents a potentially interesting exploration tool in such scenario as it facilitates the simultaneous analysis of large genomic regions and genes at affordable cost.
Methods: The present study has involved using whole-exome sequencing (WES) for the first time on SJS-TEN patients. It involved robust and innovative multistep bioinformatics analysis focusing on 313 candidate genes potentially participating in the disease’s aetiology, specific drugs’ metabolism and gene regulation.
Results: We identified combinations of frequently occurring and rare variants that may contribute to the disease’s pathogenesis. Depending on the specific drug being taken, different variants (and alleles) in NAT2, CYP2D8, CYP2B6, ABCC2, UGT2B7 and TCF3 were identified as coherent candidates representing potential future markers for SJS-TEN.
Conclusion: The present study proposed and has described (for the first time) a large-scale genomic analysis of patients affected by SJS-TEN. The genes and variants identified represent relevant candidates potentially participating in the disease’s pathogenesis. Corroborating that proposed by others, we found that complex combinations of frequently occurring and rare variants participating in particular drug metabolism molecular cascades could be associated with the phenotype. TCF3 TF may be considered a coherent candidate for SJS-TEN that should be analysed in new cohorts of patients having ADRs.
Keywords: whole-exome sequencing, Stevens-Johnson syndrome, toxic epidermal necrolysis, molecular aetiology
Introduction
Adverse drug reactions (ADRs) are frequent occurring events that can essentially be defined as harmful or unpleasant symptoms secondary to the use of a medicinal product.1,2 ADRs involve a wide spectrum of clinical manifestations ranging from minor itching and rash to life-threatening reactions. Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are rare ADRs (affecting ~1.7 individuals per million inhabitants worldwide, per year). They are characterised by erosion of the skin and mucous membranes, blistering, skin detachment, purpura, confluent erythema, renal impairment, lymphopenia, transitory neutropenia and eye lesions.3 Clinically, these diseases are variations of the same systemic disorder and their classification depends on the total body surface area compromised by lesions: SJS <10%, SJS/TEN 10–30% and TEN >30%. It is thus considered that SJS and TEN involve common pathophysiological processes, molecular cascades and potentially aetiological genetic/epigenetic factors. Biochemical and genetic prognostic markers are necessary for detecting individuals having an increased risk of contracting the disease due to the high mortality rates related to SJS and TEN.
Research into the genetic basis of SJS and TEN to date has been focused on the human leukocyte antigen (HLA) system which binds to specific receptors (eg, T-cell receptor; TCR), cytotoxic proteins and immunocyte subsets during the disease’s pathogenesis.4 It has been proposed that specific HLA alleles in particular ethnical populations/subpopulations contribute to the phenotype and increase the risk of suffering SJS and TEN. For instance, it has been established that Asian and South-Eastern populations carrying the HLA-B*15:02 allele have an increased risk of suffering SJS-TEN caused by antiepileptic drug intake, especially carbamazepine (CBZ).5–9 Other HLA-B subtypes have also been linked to SJS-TEN, such as HLA-B*14:02, 38:01, 51:01 and 58:01.9,10 However, these findings have not been explored and/or replicated in populations having different ethnic origins. Other proteins and gene variants have been proposed as potential biomarkers of the disease, such as FasL, numerous cytokines, perforin, granulysin and EPHX1, CYP2C9, CYP2B6 and IKZFI polymorphisms.11–15
Recently, the c.11G>A (p.Trp4Ter) mutation located in the GNLY gene (encoding a main cytotoxic protein involved in SJS-TEN) has been functionally related to the disease’s aetiology.16,17 Interestingly, a full-length GNLY mutant protein having subcellular mislocalisation was synthesised despite this mutation having been predicted as generating a very early premature stop codon.17
Unfortunately, such efforts have not enabled markers for screening individuals having a high risk of developing SJS-TEN to be used. Relative ineffectiveness in identifying new SJS-TEN molecular determinants may partly have been because this complex phenotype results from the dysfunction of several genes belonging to different molecular pathways. In fact, SJS-TEN may be considered a polygenic pathology due to additive/epistatic effects caused by sequence variants in numerous genes. Next-generation sequencing (NGS) represents a potentially interesting exploration tool in such scenario as it facilitates the simultaneous analysis of large genomic regions and genes at affordable cost.
The present study has involved using whole-exome sequencing (WES) for the first time on SJS-TEN patients. It involved robust and innovative multistep bioinformatics analysis focusing on 313 candidate genes potentially participating in the disease’s aetiology, specific drugs’ metabolism and gene regulation. This led to identifying that combinations of frequently occurring and rare variants that may contribute to the disease’s pathogenesis. Depending on the specific drug being taken, different variants (and alleles) in combinations of frequently occurring and rare variants that may contribute to the disease’s pathogenesis. Depending on the specific drug being taken, different variants (and alleles) in NAT2, CYP2D8, CYP2B6, ABCC2, UGT2B7 and TCF3 were identified as coherent candidates representing potential future markers for SJS-TEN.
Patients and Methods
Patients
Eleven SJS-TEN patients (previously described by Fonseca, et al) were included in the present study (patient identifiers: SJS-2, SJS-4, SJS-5, SJS-6, SJS-12, SJS-13, SJS-14, SJS-16, SJS-17, SJS-18 and SJS-19)17 (Supplementary Table 1). Their enrolment took Roujeau’s criteria into account for clinical classification,18 SJS-5 and SJS-19 were affected by SJS while SJS-4, SJS-6, SJS-13, SJS-14, SJS-16, SJS-17 and SJS-18 had TEN. SJS-2 and SJS-12 had SJS/TEN. All patients lacked GNLY pathogenic variants.17 The Universidad del Rosario (Code: DVG-098) and Fundación Valle de Lili’s (Code: P-515) Ethical Committees approved the study. The clinical investigation followed Helsinki Declaration guidelines (1975, as revised in 1996). All individuals had signed informed consent forms.
Whole-Exome and Sanger Sequencing
The DNA was obtained by conventional procedures from all patients. Three micrograms of DNA were sent to an external platform for 6Gb NGS experiments. Briefly, after verifying DNA quality and concentration, 1µg was used for library preparation. An Agilent SureSelect Human All Exon kit (Agilent Technologies, CA, USA) was used for sequencing the libraries, following the manufacturer’s recommendations. A hydrodynamic shearing system (Covaris, Massachusetts, USA) was used for generating 180–280 bp fragments. Adapter oligonucleotides were added after adenylating DNA fragment 3ʹ ends. After PCR reaction, the library was captured with magnetic beads complexed with streptomycin/biotin labelled probes. After PCR had been used for adding index tags, the amplicons were purified using the AMPure XP system (Beckman Coulter, Beverly, USA) and quantified by Agilent high sensitivity DNA assay using the Agilent Bioanalyzer 2100 system. The library quality was checked prior to Illumina sequencing.
The 150 pb paired ends were sequenced on the NovaSeq 6000 platform. The raw data was obtained in FASTQ format. Raw reads were trimmed and filtered to remove adapter sequences and low-quality reads. Quality control was performed according to the following procedure: a) discard a read pair if either one read contains adapter contamination, b) discard a read pair if more than 10% of bases are uncertain in either one read and, c) discard a read pair if the proportion of low-quality bases is over 50% in either one read. Burrows-Wheeler Aligner (BWA) was used to map the paired-end reads to the human reference genome (hg19). Variants were called using GATK v3.8. Variants were called using the HaplotypeCaller in DISCOVERY mode with a CONSERVATIVE PCR indel_model. SNPs and INDELs were analyzed separately after using the SelectVariants selection and were hard-filtered by removing variants displaying the following parameters: for SNPs: QD (QualByDepth) < 2.0, FS (FisherStrand) > 60.0, MQ (RMSMappingQuality) < 40.0, HaplotypeScore > 13.0 (for legacy reasons), MQRankSum (MappingQualityRankSumTest) < −12.5, and ReadPosRankSum (ReadPosRankSumTest) < −8.0. For INDELs: QD (QualByDepth) < 2.0, FS (FisherStrand) > 200.0, ReadPosRankSum (ReadPosRankSumTest) < −20.0. In average we obtained 6.5Gb of raw data and 21,779,003 reads (18,739,336–32,164,978) per sample. >80% bases had a phred-scaled quality score greater than 30 (>Q30). The average mapping efficiency was 99.88%, and the average sequencing depth on target and the coverage of target region were 63.89 and 99.72%, respectively. The variant annotation was performed using ANNOVAR for location and predicted function. Library preparation and sequencing were carried out by Novogene Inc. (Beijing-China). The VarSeq v.2.1.1 (Golden Helix) software was used for variant filtering. Rare variants were filtered according to minor allele frequencies (MAF) <1% (gnomAD v2.1.1) and potentially damaging effects assessed by SIFT or PolyPhen-2 tools. Variants were verified by Sanger sequencing. The primer sequences and PCR conditions used for generating the amplicons for direct sequencing are available upon request.
Creating Gene Subsets
Three main groups of genes (subsets) were created for the parallel filtering of sequence variants. The aetiopathology (AE) subgroup included 88 candidate genes participating in molecular cascades related to SJS-TEN pathophysiological processes (Supplementary Table 2). Genes playing roles in skin cytolytic activity, proinflammatory processes, immunity and apoptosis were included in that group. The AE gene list was compiled by investigating public databases such as PubMed, Highwire, Geoprofiles and MGI using the following keywords: genetic susceptibility, SJS-TEN, cytotoxic protein, keratinocyte death, severe cutaneous adverse reaction, cytolytic activity, cytokine, chemokine, SJS-TEN pathobiology, SJS-TEN aetiology, SJS-TEN immune molecule, cell apoptosis, skin allergy, pharmacogenomics of cutaneous adverse drug reactions, immune reactions and drug hypersensitivity reactions.
The pharmacogenetic (PH) subset consisted of 91 genes involved in the pharmacological metabolism of drugs taken by our patients: CBZ (n=23 genes), lamotrigine (LTG) (n=39 genes), metoclopramide (n=8 genes), pyrimethamine-sulfadoxine (PYR-SULF, n=5 genes), and trimethoprim-sulfamethoxazole (TS, n=16 genes) (Supplementary Table 1). This information was obtained from the DrugBank database (www.drugbank.ca) which includes detailed information on most relevant molecules involved in frequently administered drugs. It should be stressed that since little is known regarding Urtica dioica´s metabolism (the medication suspected as leading to the SJS-2 phenotype), we did not include PH candidate genes potentially involved in this case. Patient SJS-5 was affected by various SJS episodes triggered by different drugs; we considered trimethoprim-sulfamethoxazole for analysis since the contrast medium metabolic cascade has not been precisely defined.
The transcription factor (TF) subset included 141 TF-encoding genes. This subset was constructed by first selecting the promoter regions (−2000 bp to the first ATG codon) from the 184 most up (n=138) and down-regulated genes (n=46) identified by Chung et al, in SJS-TEN patients’ skin lesions.16 Genomatix software (https://www.genomatix.de/) was used for identifying and quantifying TF binding sites (TFBS) located in these regions. TF families having >5 and <-5 Z-scores were selected. TFs belonging to these families were considered as candidates and used for filtering sequence variants, following a specific pipeline (see below).
Filtering Variants and in silico Analysis
Golden Helix software (VarSeq v.2.1.1) was used for filtering the variants. Only non-synonymous substitutions (missense, nonsense, splice-site and indels) were considered for downstream analysis. The Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org) was used for establishing sequence variants’ MAFs.
Genes having <1% MAF in the AE and TF subsets were filtered for subsequent analysis (Figure 1). All sequence variants in the PH subgroup (<1% MAF and >1% MAF) were selected (Figure 1). It should be noted that PH subset use (and therefore the analysed genes) depended on the specific drug being taken by each patient (CBZ, LTG, metoclopramide, pyrimethamine-sulfadoxine, trimethoprim-sulfamethoxazole). Variants carried by more than one patient having >1% MAF in the PH subset were selected. SIFT and Polyphen prediction tools were used on rare (<1% MAF) missense variants from the AE, PH and TF subsets. Sequence variants having scores compatible with potentially damaging effects in one out of two programmes were considered for further analysis.
 |
Figure 1 Methodological scheme for filtering genomic variants in SJS-TEN patients. |
A more detailed protocol is available at: dx.doi.org/10.17504/protocols.io.bk7vkzn6.
Results
WES experiments gave an average >98% for genomic coverage; >49% of the exome was sequenced in all samples (50X average read depth). The 11 SJS-TEN patients had 3,415,863 variants (synonymous and non-synonymous) (Figure 1); 132,469 were non-synonymous. Forty-three non-synonymous common variants (>1% MAF) were identified in the PH group. Bioinformatics filtering of rare variants led to identifying 11 nucleotide changes in the TF gene subset and one variant in the PH subset; all of these were validated by Sanger sequencing. No rare variants were identified in the AE subgroup for downstream analysis. Table 1 shows the distribution of rare and frequently occurring variants in the PH group. Table 2 displays the distribution of rare variants in the TF subset.
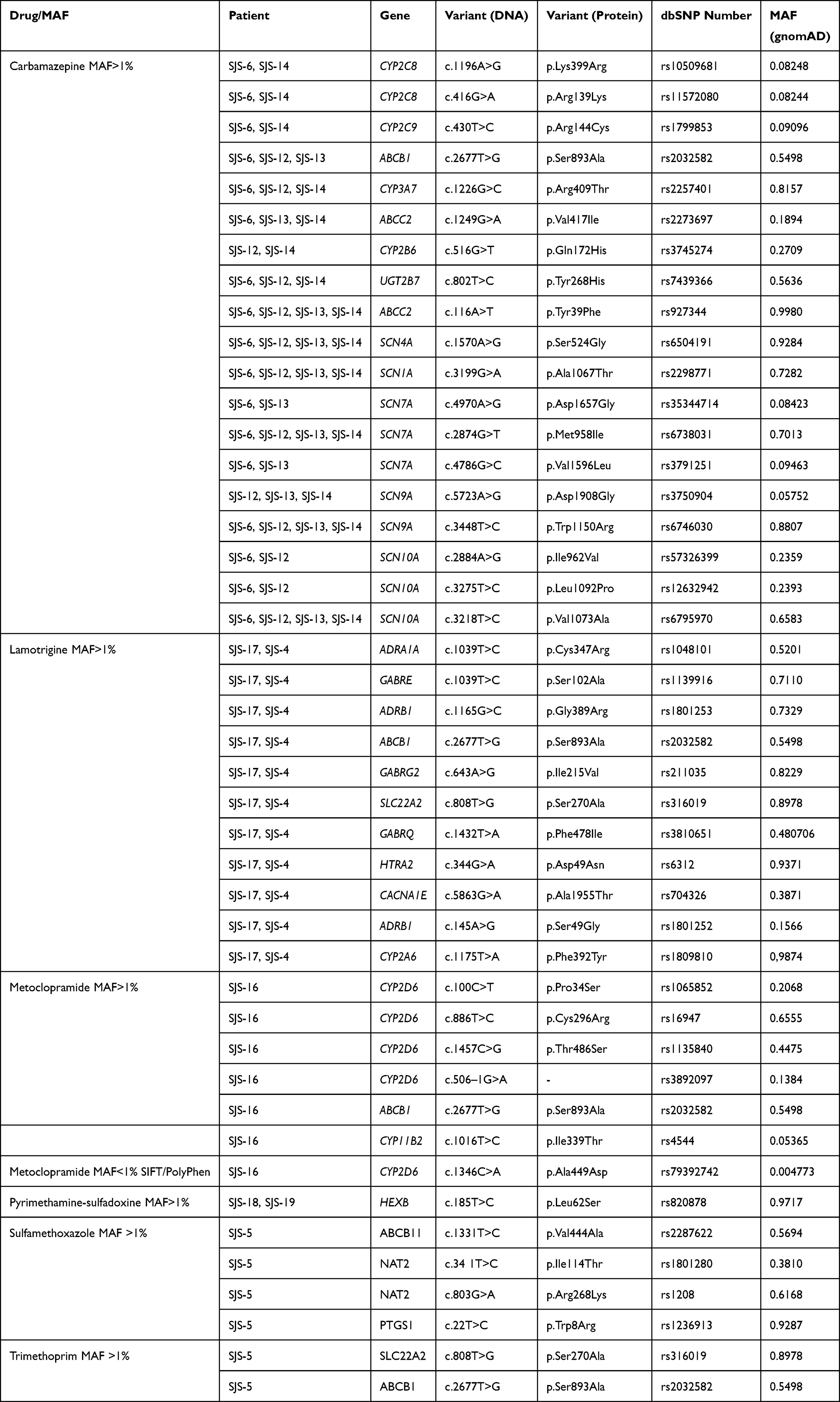 |
Table 1 PH Subset Genetic Variants |
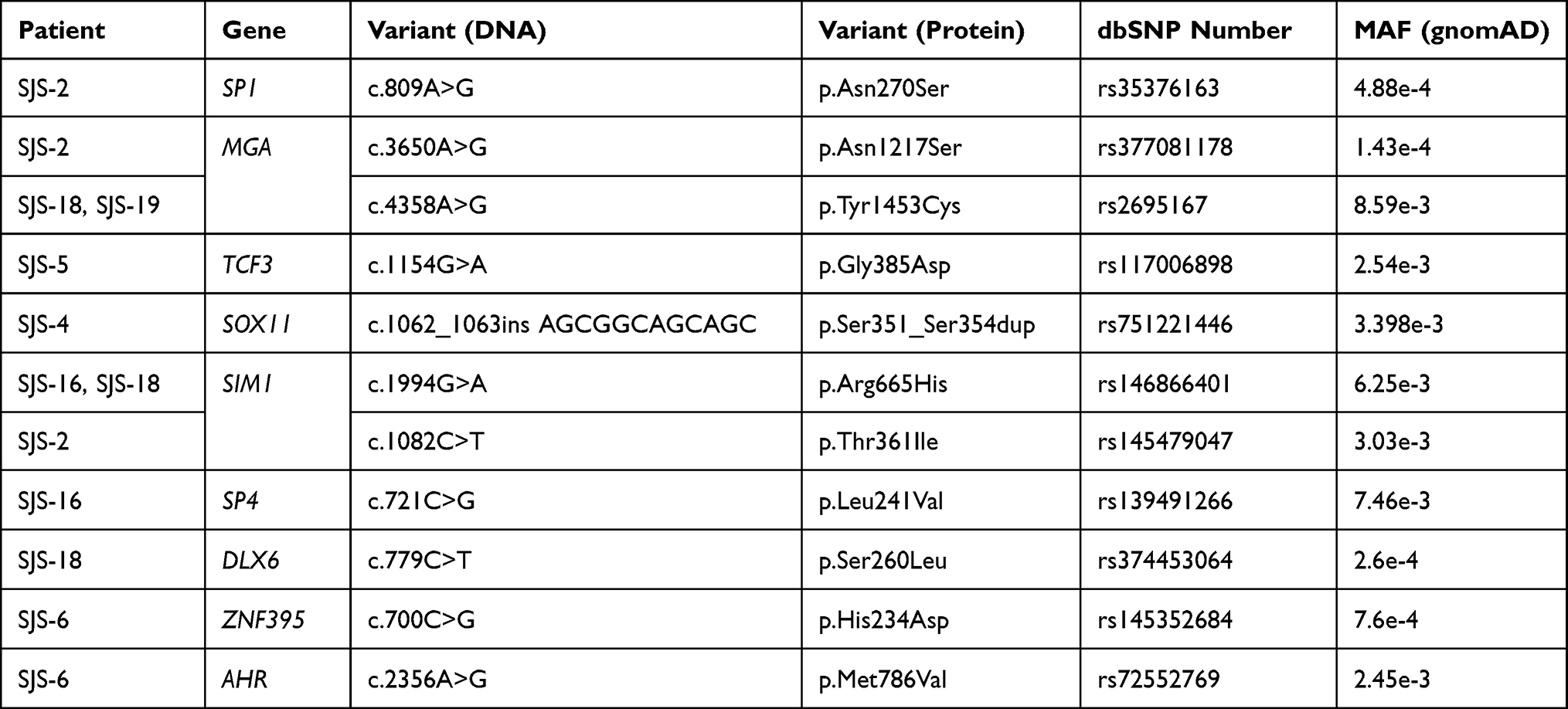 |
Table 2 Variants of the TF-Subset Displaying MAF≤0.01 |
Discussion
WES Gene Subset Analysis: An Innovative Genomic Approach for SJS/TEN
This work has attempted for the first time to identify, via WES, new potential elements participating in SJS-TEN’s genetic architecture. We thus selected 11 previously described SJS-TEN patients in whom the phenotype was triggered by them taking different drugs and who lacked GNLY encoding mutations.17 These patients were selected from Fonseca et al’s study, just depending on their DNA quality/availability to be used in NGS experiments.
Our multilevel bioinformatics analysis took into account ADR pathophysiology complexity (AE gene subset), the specific molecular pathways involved in the metabolism of 5 drugs (CBZ, LTG, metoclopramide, pyrimethamine-sulfadoxine, trimethoprim-sulfamethoxazole) (PH gene subset) and the relevance of the trans-regulation of genes having transcriptomic imbalance during the disease’s cytotoxic process affecting skin/mucosae (TF gene subset). It has been established that creating gene subsets from WES for studying unrelated patients on a large genomic scale is a powerful strategy for identifying new genes and mutations related to complex polygenic phenotypes.19–22
Three subsets (AE, PH and TF) were created in this study, forming the starting point for filtering candidate sequence variants regarding different genomic and pathophysiology hypotheses. We consider that the PH category contains a comprehensive collection of genes that have been well-documented concerning drug metabolism, some of which are available for commercial/diagnostic purposes (eg, ABCB1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP3A4, UGT1A1, SLC47A2, D2 and HTR2A). Due to the limited understanding of the molecular mechanisms underlining SJS-TEN, the AE subset may lack some relevant candidate genes. However, as WES contains data on most encoding genomic regions, the present results can be re-analysed at any time by creating new gene subsets and/or bioinformatics filtering pipelines. This strategy has been shown to be a useful approach for describing new variants having a functional aetiological impact on complex phenotypes.19,23,24
The TF subset was created following the assumption that the promoter regions of massively dysregulated genes in skin and mucosae (during SJS-TEN’s acute phase) are enriched by TFBS for proteins belonging to particular TF families. A previous study using this methodology identified FOXD1 as a major gene involved in the regulation of embryo implantation in mice and various reproductive disorders in humans.20,25
Variant filtering assumed that rare (MAF <1%) non-synonymous variants in AE, PH and/or TF subsets (underlining potential moderate/drastic functional effects) might contribute towards the origin of SJS-TEN. Indeed, it has been demonstrated that using NGS for studying extreme phenotypes facilitates mapping rare sequence variants contributing to complex traits’ origins.26–28 PH subset examination included filtering variants having >1% MAF as a large body of evidence has demonstrated that polymorphism in pharmacogenes contributes to pharmacogenetic phenotypes.29–32 It is undeniable that both rare and common variants in pharmacogenes (which nowadays can be mapped via NGS at affordable costs) have to be considered as relevant determinants for drug responses.32–36
Our approach, based on studying both rare and frequent variants belonging to different molecular cascades and functional mechanisms involved in the metabolism of particular drugs, revealed that patients have different genetic configurations (Figure 2). Some of them (SJS4, SJS5, SJS-6, SJS16, SJS18 and SJS-19) had frequent variants in the PH subset and rare ones in the TF subset. This argues in favour of the disease’s complex polygenic origin. Frequent variants could confer a predisposing condition while rare encoding mutations underlying moderate functional effects act as a stronger determinant for the disease’s onset. Patients SJS-12, SJS-13, SJS-14 and SJS-17 only carried frequently occurring variants in PH genes whilst SJS-2 had exclusively rare nucleotide changes in genes belonging to the TF subset. To note, in this patient the PH subset was not analysed because the Urtica dioica´s metabolism pathway is unknown.
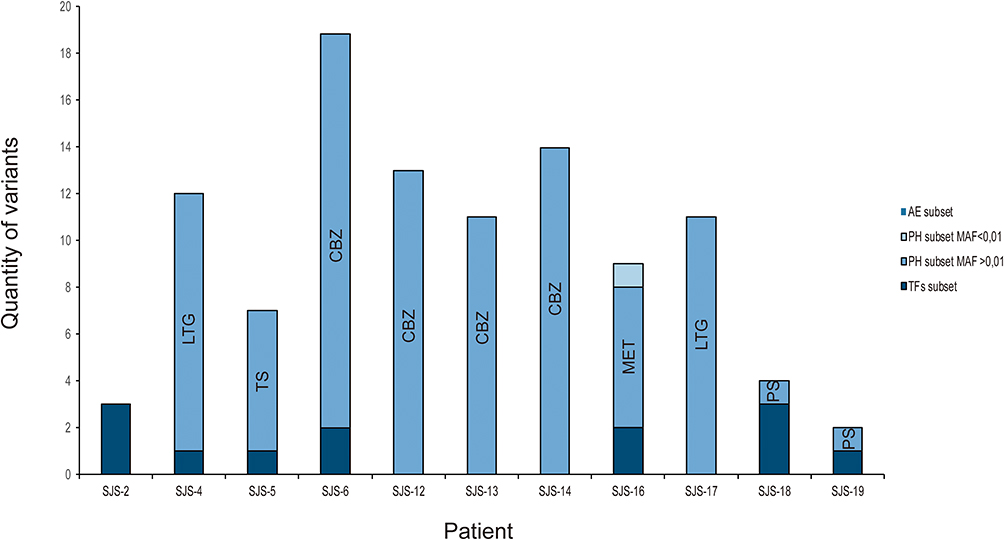 |
Figure 2 Distribution of genomic variants, filtered from specific subsets, found in SJS-TEN patients. |
Taken together, these features indicate that severe ADRs must result from different molecular determinants underlying complex traits’ regulation in which numerous genes (and mutations) interact to produce the phenotype.
PH Gene Subset Analysis
Forty-three frequently occurring variants (>1% MAF) involved in the metabolism of specific drugs were identified in the PH set: CBZ (19 variants in 12 genes), LTG (11 variants in 9 genes) and metoclopramide (6 variants in 3 genes) (Table 1). Only one rare variant (CYP2D6-c.1346C>A, p.Ala449Asp) was identified in one patient (SJS-16) who had taken metoclopramide (Table 1 and see below).
Regarding CBZ metabolism, which was associated in the present study with three cases of TEN and one of SJS, we identified frequently occurring variants in CYP2C8, CYP2C9, CYP2B6, ABCB1, ABCC2 and UGT2B7 genes.
CYP2C8-c.416G>A (p.Arg139Lys) and c.1196A>G (p.Lys399Arg), which were present in two TEN cases (SJS-6 and SJS-14 patients), determine a specific allele (CYP2C8*3) linked with drug-induced toxicity.37 The p.Lys399Arg missense variant (generated by the c.1196A>G transition) was predicted to affect protein folding/stability leading to disturbances in enzymatic activity.38,39 Interestingly, it has been stated that CYP2C8 and CYP3A4 participate in the major CBZ metabolism route through its conversion to CBZ-10.11-epoxide.40 Decreased CYP2C8 activity can theoretically increase this drug’s bioavailability by favouring the activation of metabolic hydroxylation cascades to form 2-OH and 3-OH-CBZ, leading, in turn, to the synthesis of reactive metabolites capable of forming covalent adducts. It has been proposed that CBZ metabolism bioactivating monoxygenated species can occur in keratinocytes and be related to detoxification disturbances leading to cell death and hypersensitivity reactions.2 Interestingly, TEN patients who took CBZ had early peripheral blood T-cell activation with skin homing receptors.41
Patient SJS12 affected by SJS/TEN and SJS-14 by TEN carried the heterozygous CYP2B6*9 (c.516G>T, p.Gln172His) allele.42,43 CYP2B6*9 has been associated with nevirapine-induced SJS/TEN; subjects carrying the heterozygous genotype have a ~two fold risk of suffering the disease compared to its wild type counterpart, thereby arguing in favour of an allele-dose association effect.11
To date, CBZ-induced SJS/TEN has been exclusively related to human leukocyte antigen (HLA) alleles (eg, HLA-B15:02 and HLA-B*31:01).44 We suggest that, similar to phenytoin and nevirapine-induced SJS/TEN, reduced drug-CBZ clearance associated with CYP2B6*9 may have contributed to the phenotype in our SJS/TEN patients. Elevated plasmatic drug-CBZ concentration in SJS/TEN CYP2B6*9 carriers increases direct interaction with specific HLA molecules or induces the formation of reactive metabolites such as CBZ iminoquinone, previously associated with the pathogenesis of CBZ-induced hypersensitivity.45,46
Patient SJS-12 displayed the ABCC2-c.1249 G>A (SJS-6, SJS-13, SJS-14) variant in a gene (ABCC2) related to CBZ transport. Patient SJS-14 was carrier of the UGT2B7 *2 allele (c.802T> C) in a gene (UGT2B7) related to Phase II metabolism. ABCC2-c.1249 G>A reduces CBZ transport across the cell membrane and clearance while UGT2B7*2 c.802T>C could affect its steady-state concentration.47,48 Such CBZ pharmacokinetic variations may potentially modify CBZ plasma levels thereby contributing to SJS/TEN pathogenesis.
Regarding LTG metabolism, 11 genetic variants were identified, some affecting genes (eg, ADRA1A, GABRE, CACNA1E, SCN1A, SCN5A and ADRB1) linked to the drug’s pharmacodynamics (www.drugbank.ca). Similar to that observed for CBZ metabolism, LTG administration may contribute to SJS/TEN aetiology via the synthesis of LTG-N-oxide and the induction of immunogenic haptens triggering T-cell clonal expansion in the skin.10,49
We did not identify sequence variants potentially linked with SJS/TEN pathogenesis in the CYP2A6 nor CYP2D6 enzymes participating in the formation of minor metabolites such as LTG-N-oxide.50 Other genetic determinants in CYP2D6 or CYP2A6, such as copy-number variations (not analysed in this work), could contribute to the phenotype. It is worth noting that our group has recently described a significant distribution (7.7%) of CYP2D6 duplications in a Colombian population, which may perturb the metabolism of drugs, including LTG.51
LTG is extensively metabolised via UGT1A4, leading to the formation of LTG-N-2 and LTG-N-5 glucuronide which may affect LTG levels.52,53 Patient SJS-17, who was affected by TEN, took LTG combined with valproic acid (VPA); the concomitant administration of these drugs has been associated with increased LTG serum concentration. Furthermore, it leads to its decreased clearance due to glucuronidation inhibition.54 LTG can be bioactivated in the absence of N-glucuronidation, contributing to TEN pathogenesis. Interestingly, several reports have supported the clinical evidence stating that a combination of LTG and VPA can increase susceptibility to SJS/TEN.53,55,56
Regarding metoclopramide, we identified one patient (SJS-16) who had developed TEN after taking this drug. To date, few cases of metoclopramide-induced SJS/TEN have been reported.57,58 SJS-16 carried CYP2D6*10-c.100C>T (p.Pro34Ser) and CYP2D6*4-c.506–1G>A alleles which are related to impaired enzymatic activity determining an intermediate metabolizer phenotype. This effect has been linked to ineffective metabolism of CYP2D6 substrates and adverse reactions resulting from increased drug plasma levels.59,60
Patient SJS-5 had suffered from various SJS episodes apparently related to the administration of contrast medium and TS. Recurrent risk for SJS-TEN has been estimated at 7% and it has been linked to potential genetic determinants.61 Genomic analysis of SJS-5 only included genes belonging to TS metabolism, as contrast medium molecular pathways have not yet been precisely described. This patient carries the heterozygous NAT2-c.341T>C (p.Ile114Thr) and NAT2-c.803G>A (p.Arg268Lys) variants defining the NAT2*5C allele and determining the intermediate acetylator phenotype. Interestingly, it has been stated that impaired acetylation capacity leads to sulphonamide hypersensitivity and SJS/TEN.62,63 Interestingly, patient SJS-5 also carried a variant in TCF3, a gene belonging to the TF subset, arguing in favour of the contribution of both frequently occurring and rare variants to the disease’s pathogenesis (see below). Our study did not identify frequent variants in genes related to pyrimethamine-sulfadoxine metabolism which may have been due to the size of the present SJS/TEN cohort and/or to the potential contribution of other yet-to-be discovered genetic determinants.
Regarding rare variants, we identified only one candidate in the PH subset (CYP2D6-c.1346C>A, p.Ala449Asp) (patient SJS-16). At protein level, Ala449 is strictly conserved during the evolution of mammalian species, thereby arguing in favour of its functional role (Supplementary Figure 1). This feature agrees with the results from in silico analysis (SIFT and PolyPhen) predicting a potentially harmful effect. It has been shown recently that this variant confers decreased activity (44.4%) on the CYP2D6 protein due to heme binding perturbation.64 Patient SJS-16 also carried common polymorphisms in CYP2D6 (CYP2D6*10 and CYPY2D6*4) related to decreased activity, compared to the reference CYP2D6.1 wild type protein.65 These findings indicated that rare and common variants in CYP2D6 might contribute to severe metoclopramide-induced ADRs.
TF and AE Gene Subset Analysis
Regarding the TF subset, we filtered 11 different variants located in 9 genes (Table 2). Although all these changes may have represented potential genetic determinants contributing to the phenotype, 5 variants (SP1-c.809A>G, MGA-c.3650A>G, TCF3-c.1154G>A, DLX6-c.779C>T and ZNF395-c.700C>G) were of particular interest, as they were not included in GnomAD databases or in the C population (data not shown). Furthermore, they involved residues which were conserved during mammalian evolution (ie, at protein level), suggesting possible functional impact (Supplementary Figure 1). Theoretically, the effect conferred by these variants should contribute to the onset/maintenance of the SJS-TEN skin/mucosae phenotype, as our computational approach was aimed at identifying TFBS enrichment regarding gene promoters having transcriptional disturbances during the disease’s acute phase.16 This could have been caused by a combination of direct (target promoter transactivation/repression) or indirect dysregulation of target genes belonging to a complex latticework of transcriptional regulation.
Amongst TFs carrying rare sequence variants, TCF3 (also known as TCF7L1, E2A and E47) is a relevant candidate as a future marker for SJS-TEN as it plays a key role during skin development, stem cell homeostasis and malignancy (see below). TCF3 is a member of the E protein family of helix-loop-helix transcription factors, which have a C-terminal bHLH domain involved in E-protein dimerisation and binding to DNA at CANNTG motifs located on target gene promoters.66–68 These factors also have two transcription activation domains located in the protein’s N-terminal (TAD1) and central (TAD2) regions, playing a key role in target gene regulation.69,70 Interestingly, the p.Gly385Asp variant in patient SJS-5 was located in the TAD2 domain, near the highly conserved LDEAI (L397 to I401) sequence which directly binds to the KIX domain of the CBP/p300 transcriptional co-activator.67 The p.Gly385Asp variant implies a significant local change in the protein’s physicochemical properties as glycine is a tiny non-polar positively charged amino acid while aspartic acid is negatively charged. This change may therefore lead to local energetic disturbances between residues perturbing the native protein’s state which might affect macromolecular transcriptional complexes’ structure/function (eg, EA2/CBP300/p300).
Tcf3 is expressed in mouse skin during development in the primordial epithelium and during adult life in the stem cell niche.71–73 Tcf3 overexpression impairs epithelial differentiation whilst Tcf3 and Tcf4 deletion generates skin hyper-proliferation and a long-term inability for self-renewal.74 Moreover, Tcf3 promotes keratinocyte migration thereby enhancing wound healing.75 Interestingly, the CBP/p300 protein is also involved in skin homeostasis in mice since reduced expression contributes towards creating a keratinocyte hyperplastic phenotype via Ras-Erk signalling induction.76
Such features suggested that the TCF3- p.Gly385Asp variant might confer a predisposing transcriptomic environment facilitating the occurrence of severe cutaneous ADRs. Interestingly, patient SJS-5 suffered several ADRs which may underline her increased risk of becoming affected by SJS-TEN. This TCF3 mutant form might also be involved in the severity of skin lesions. Regarding other TFs (eg, SP1, MGA, DLX6 and ZNF395) carrying candidate sequence variants, it is difficult to propose further potential mechanisms related to the phenotype’s aetiology due to the lack of sufficient information regarding their role in skin homeostasis.
Concluding Remarks and Future Directions
The present study proposed and has described (for the first time) a large-scale genomic analysis of patients affected by SJS-TEN. The genes and variants identified represent relevant candidates potentially participating in the disease’s pathogenesis. Corroborating that proposed by others, we found that complex combinations of frequently occurring and rare variants participating in particular drug metabolism molecular cascades could be associated with the phenotype. TCF3 TF may be considered a coherent candidate for SJS-TEN that should be analysed in new cohorts of patients having ADRs.
The small population size and the lack of functional assays for the filtered candidate variants constitute this study’s relative limitations. It has been generally assumed that the use of small sample size in genomic studies may generate weak or unreliable results. This is particularly true for large-scale genomic mapping approaches, such as genome-wide association studies (GWAS). The present approach was slightly different, because we analyzed the coding regions of gene subsets potentially enriched by rare variants (eg, those selected in the TF and AE gene subsets) and performed a downstream astringent bioinformatic variant filtering. As mentioned above, this strategy allows determining variants underlying moderate-to-severe functional effects, which are rare in the general healthy population as they tend to be negatively selected.
Due to the rarity of the disease, we consider that the strategy proposed here provides a relevant description of new potential genetic determinants of SJS-TEN. Although WES involves analysing millions of nucleotides, non-encoding genomic regions may also be relevant and must be analysed in future projects. Epigenetic factors and environmental variables must also be considered as having a relevant effect on the disease’s onset, development and severity. Although the TCF3-c.1154G>A (p.Gly385Asp) variant may contribute to the phenotype we cannot affirm that it is an absolute genetic marker of SJS/TEN. It constitutes an interesting and coherent candidate that deserves further analysis. Functional tests, to validate its association to the phenotype, and the genotyping of largest sets of patients are necessary to establish it as marker with clinical significance. We consider that our results constitute a starting point for future studies aimed at SJS-TEN’s molecular dissection and proposing new biomarkers which can be useful in clinical environments.
Abbreviations
ADR, Adverse drug reaction; AE, Aetiopathology; CBZ, Carbamazepine; DNA, Deoxyribonucleic acid; GNLY, Granulysin; HLA, Human leukocyte antigen; LTG, Lamotrigine; MAF, Minor allelic frequency; MET, Metoclopramide; NGS, Next-generation sequencing; PH, Pharmacogene, PYR-SULF, Pyrimethamine sulfadoxine; SJS, Stevens-Johnson syndrome; TEN, Toxic epidermal necrolysis; TF, Transcription factor; TS, trimethoprim-sulfamethoxazole; WES, Whole exome sequencing.
Ethics Approval and Consent to Participate
The Universidad del Rosario (Code: DVG-098) and Fundación Valle de Lili’s (Code: P 515) Ethical Committees approved the study. The clinical investigation followed Helsinki Declaration guidelines (1975, as revised in 1996). All individuals had signed informed consent forms.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The present study was supported by the Universidad del Rosario (Grant CS/Genetics/ABN062-2019).
Disclosure
PL declares that his present salary is paid by BIOPAS Laboratories for working in projects different to that described in the present manuscript. The authors report no other conflicts of interest in this work.
References
1. Aronson JK, Ferner RE. Clarification of terminology in drug safety. Drug Saf. 2005;28(10):851–870. doi:10.2165/00002018-200528100-00003
2. Yip VL, Alfirevic A, Pirmohamed M. Genetics of immune-mediated adverse drug reactions: a comprehensive and clinical review. Clin Rev Allergy Immunol. 2015;48(2–3):165–175. doi:10.1007/s12016-014-8418-y
3. Duong TA, Valeyrie-Allanore L, Wolkenstein P, Chosidow O. Severe cutaneous adverse reactions to drugs. Lancet. 2017;390(10106):1996–2011. doi:10.1016/s0140-6736(16)30378-6
4. Abe R. Immunological response in Stevens-Johnson syndrome and toxic epidermal necrolysis. J Dermatol. 2015;42(1):42–48. doi:10.1111/1346-8138.12674
5. Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428(6982):486. doi:10.1038/428486a
6. Man CB, Kwan P, Baum L, et al. Association between HLA-B*1502 allele and antiepileptic drug-induced cutaneous reactions in Han Chinese. Epilepsia. 2007;48(5):1015–1018. doi:10.1111/j.1528-1167.2007.01022.x
7. Tassaneeyakul W, Tiamkao S, Jantararoungtong T, et al. Association between HLA-B*1502 and carbamazepine-induced severe cutaneous adverse drug reactions in a Thai population. Epilepsia. 2010;51(5):926–930. doi:10.1111/j.1528-1167.2010.02533.x
8. Fan WL, Shiao MS, Hui RC, et al. HLA association with drug-induced adverse reactions. J Immunol Res. 2017;2017:3186328. doi:10.1155/2017/3186328
9. Mullan KA, Anderson A, Illing PT, Kwan P, Purcell AW, Mifsud NA. HLA-associated antiepileptic drug-induced cutaneous adverse reactions. Hla. 2019;93(6):417–435. doi:10.1111/tan.13530
10. Chen CB, Abe R, Pan RY, et al. An updated review of the molecular mechanisms in drug hypersensitivity. J Immunol Res. 2018;2018:6431694. doi:10.1155/2018/6431694
11. Ciccacci C, Di Fusco D, Marazzi MC, et al. Association between CYP2B6 polymorphisms and Nevirapine-induced SJS/TEN: a pharmacogenetics study. Eur J Clin Pharmacol. 2013;69(11):1909–1916. doi:10.1007/s00228-013-1549-x
12. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part I. Introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69(2):
13. Caruso A, Bellia C, Pivetti A, et al. Effects of EPHX1 and CYP3A4 polymorphisms on carbamazepine metabolism in epileptic patients. Pharmgenomics Pers Med. 2014;7:117–120. doi:10.2147/pgpm.S55548
14. He XJ, Jian LY, He XL, et al. Association of ABCB1, CYP3A4, EPHX1, FAS, SCN1A, MICA, and BAG6 polymorphisms with the risk of carbamazepine-induced Stevens-Johnson syndrome/toxic epidermal necrolysis in Chinese Han patients with epilepsy. Epilepsia. 2014;55(8):1301–1306. doi:10.1111/epi.12655
15. Ueta M, Sawai H, Sotozono C, et al. IKZF1, a new susceptibility gene for cold medicine-related Stevens-Johnson syndrome/toxic epidermal necrolysis with severe mucosal involvement. J Allergy Clin Immunol. 2015;135(6):1538–45.e17. doi:10.1016/j.jaci.2014.12.1916
16. Chung WH, Hung SI, Yang JY, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med. 2008;14(12):1343–1350. doi:10.1038/nm.1884
17. Fonseca DJ, Caro LA, Sierra-Diaz DC, et al. Mutant GNLY is linked to Stevens-Johnson syndrome and toxic epidermal necrolysis. Hum Genet. 2019;138(11–12):1267–1274. doi:10.1007/s00439-019-02066-w
18. Roujeau JC. Stevens-Johnson syndrome and toxic epidermal necrolysis are severity variants of the same disease which differs from erythema multiforme. J Dermatol. 1997;24(11):726–729. doi:10.1111/j.1346-8138.1997.tb02524.x
19. Patino LC, Beau I, Morel A, et al. Functional evidence implicating NOTCH2 missense mutations in primary ovarian insufficiency etiology. Hum Mutat. 2019;40(1):25–30. doi:10.1002/humu.23667
20. Quintero-Ronderos P, Jimenez KM, Esteban-Perez C, et al. FOXD1 mutations are related to repeated implantation failure, intra-uterine growth restriction and preeclampsia. Mol Med. 2019;25(1):37. doi:10.1186/s10020-019-0104-3
21. About F, Bibert S, Jouanguy E, et al. Identification of an endoglin variant associated with HCV-related liver fibrosis progression by next-generation sequencing. Front Genet. 2019;10:1024. doi:10.3389/fgene.2019.01024
22. Rojnueangnit K, Sirichongkolthong B, Wongwandee R, et al. Identification of gene mutations in primary pediatric cardiomyopathy by whole exome sequencing. Pediatr Cardiol. 2020;41(1):165–174. doi:10.1007/s00246-019-02240-x
23. Delcour C, Amazit L, Patino LC, et al. ATG7 and ATG9A loss-of-function variants trigger autophagy impairment and ovarian failure. Genet Med. 2019;21(4):930–938. doi:10.1038/s41436-018-0287-y
24. Machini K, Ceyhan-Birsoy O, Azzariti DR, et al. Analyzing and reanalyzing the genome: findings from the MedSeq project. Am J Hum Genet. 2019;105(1):177–188. doi:10.1016/j.ajhg.2019.05.017
25. Laissue P, Lakhal B, Vatin M, et al. Association of FOXD1 variants with adverse pregnancy outcomes in mice and humans. Open Biol. 2016;6(10):Oct. doi:10.1098/rsob.160109
26. Emond MJ, Louie T, Emerson J, et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012;44(8):886–889. doi:10.1038/ng.2344
27. Li B, Liu DJ, Leal SM. Identifying rare variants associated with complex traits via sequencing. Curr Protoc Hum Genet. 2013;78(1). doi:10.1002/0471142905.hg0126s78
28. Johar AS, Anaya JM, Andrews D, et al. Candidate gene discovery in autoimmunity by using extreme phenotypes, next generation sequencing and whole exome capture. Autoimmun Rev. 2015;14(3):204–209. doi:10.1016/j.autrev.2014.10.021
29. Ciccacci C, Latini A, Politi C, et al. Impact of glutathione transferases genes polymorphisms in nevirapine adverse reactions: a possible role for GSTM1 in SJS/TEN susceptibility. Eur J Clin Pharmacol. 2017;73(10):1253–1259. doi:10.1007/s00228-017-2295-2
30. Salvador-Martin S, Garcia-Gonzalez X, Garcia MI, et al. Clinical utility of ABCB1 genotyping for preventing toxicity in treatment with irinotecan. Pharmacol Res. 2018;136:133–139. doi:10.1016/j.phrs.2018.08.026
31. Galvez JM, Restrepo CM, Contreras NC, et al. Creating and validating a warfarin pharmacogenetic dosing algorithm for Colombian patients. Pharmgenomics Pers Med. 2018;11:169–178. doi:10.2147/pgpm.s170515
32. Margarit C, Ballester P, Inda MD, et al. OPRM1 gene interaction with sleep in chronic pain patients treated with opioids. Pain Physician. 2019;22(1):97–107.
33. Ahn E, Park T. Analysis of population-specific pharmacogenomic variants using next-generation sequencing data. Sci Rep. 2017;7(1):8416. doi:10.1038/s41598-017-08468-y
34. Ingelman-Sundberg M, Mkrtchian S, Zhou Y, Lauschke VM. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum Genomics. 2018;12(1):26. doi:10.1186/s40246-018-0157-3
35. Wright GEB, Carleton B, Hayden MR, Ross CJD. The global spectrum of protein-coding pharmacogenomic diversity. Pharmacogenomics J. 2018;18(1):187–195. doi:10.1038/tpj.2016.77
36. Lauschke VM, Ingelman-Sundberg M. Prediction of drug response and adverse drug reactions: from twin studies to next generation sequencing. Eur J Pharm Sci. 2019;130:65–77. doi:10.1016/j.ejps.2019.01.024
37. Daily EB, Aquilante CL. Cytochrome P450 2C8 pharmacogenetics: a review of clinical studies. Pharmacogenomics. 2009;10(9):1489–1510. doi:10.2217/pgs.09.82
38. Gao Y, Liu D, Wang H, Zhu J, Chen C. Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype-dependent effects on in vitro and in vivo drug-drug interactions. Xenobiotica. 2010;40(7):467–475. doi:10.3109/00498254.2010.487163
39. Backman JT, Filppula AM, Niemi M, Neuvonen PJ. Role of cytochrome P450 2C8 in drug metabolism and interactions. Pharmacol Rev. 2016;68(1):168–241. doi:10.1124/pr.115.011411
40. Pearce RE, Lu W, Wang Y, Uetrecht JP, Correia MA, Leeder JS. Pathways of carbamazepine bioactivation in vitro. III. The role of human cytochrome P450 enzymes in the formation of 2,3-dihydroxycarbamazepine. Drug Metab Dispos. 2008;36(8):1637–1649. doi:10.1124/dmd.107.019562
41. Posadas SJ, Padial A, Torres MJ, et al. Delayed reactions to drugs show levels of perforin, granzyme B, and Fas-L to be related to disease severity. J Allergy Clin Immunol. 2002;109(1):155–161. doi:10.1067/mai.2002.120563
42. Paganotti GM, Russo G, Sobze MS, et al. CYP2B6 poor metaboliser alleles involved in efavirenz and nevirapine metabolism: CYP2B6*9 and CYP2B6*18 distribution in HIV-exposed subjects from Dschang, Western Cameroon. Infect Genet Evol. 2015;35:122–126. doi:10.1016/j.meegid.2015.08.003
43. Hofmann MH, Blievernicht JK, Klein K, et al. Aberrant splicing caused by single nucleotide polymorphism c.516G>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J Pharmacol Exp Ther. 2008;325(1):284–292. doi:10.1124/jpet.107.133306
44. Amstutz U, Shear NH, Rieder MJ, et al. Recommendations for HLA-B*15:02 and HLA-A*31:01 genetic testing to reduce the risk of carbamazepine-induced hypersensitivity reactions. Epilepsia. 2014;55(4):496–506. doi:10.1111/epi.12564
45. Pearce RE, Uetrecht JP, Leeder JS. Pathways of carbamazepine bioactivation in vitro: II. The role of human cytochrome P450 enzymes in the formation of 2-hydroxyiminostilbene. Drug Metab Dispos. 2005;33(12):1819–1826. doi:10.1124/dmd.105.004861
46. Wei CY, Chung WH, Huang HW, Chen YT, Hung SI. Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. J Allergy Clin Immunol. 2012;129(6):1562–9.e5. doi:10.1016/j.jaci.2011.12.990
47. Kim WJ, Lee JH, Yi J, et al. A nonsynonymous variation in MRP2/ABCC2 is associated with neurological adverse drug reactions of carbamazepine in patients with epilepsy. Pharmacogenet Genomics. 2010;20(4):249–256. doi:10.1097/FPC.0b013e328338073a
48. Lu Q, Huang YT, Shu Y, et al. Effects of CYP3A5 and UGT2B7 variants on steady-state carbamazepine concentrations in Chinese epileptic patients. Medicine. 2018;97(30):e11662. doi:10.1097/md.0000000000011662
49. Krauss G. Current understanding of delayed anticonvulsant hypersensitivity reactions. Epilepsy Curr. 2006;6(2):33–37. doi:10.1111/j.1535-7511.2006.00089.x
50. Chen H, Grover S, Yu L, Walker G, Mutlib A. Bioactivation of lamotrigine in vivo in rat and in vitro in human liver microsomes, hepatocytes, and epidermal keratinocytes: characterization of thioether conjugates by liquid chromatography/mass spectrometry and high field nuclear magnetic resonance spectroscopy. Chem Res Toxicol. 2010;23(1):159–170. doi:10.1021/tx9003243
51. Ramirez B, Nino-Orrego MJ, Cardenas D, et al. Copy number variation profiling in pharmacogenetics CYP-450 and GST genes in Colombian population. BMC Med Genomics. 2019;12(1):110. doi:10.1186/s12920-019-0556-x
52. Doig MV, Clare RA. Use of thermospray liquid chromatography-mass spectrometry to aid in the identification of urinary metabolites of a novel antiepileptic drug, Lamotrigine. J Chromatogr. 1991;554(1–2):181–189. doi:10.1016/s0021-9673(01)88448-x
53. Vazquez M, Maldonado C, Guevara N, et al. Lamotrigine-valproic acid interaction leading to Stevens-Johnson syndrome. Case Rep Med. 2018;2018:5371854. doi:10.1155/2018/5371854
54. Kaur S, Dogra A. Toxic epidermal necrolysis due to concomitant use of lamotrigine and valproic Acid. Indian J Dermatol. 2013;58(5):406. doi:10.4103/0019-5154.117319
55. Kocak S, Girisgin SA, Gul M, Cander B, Kaya H, Kaya E. Stevens-Johnson syndrome due to concomitant use of lamotrigine and valproic acid. Am J Clin Dermatol. 2007;8(2):107–111. doi:10.2165/00128071-200708020-00007
56. Maduemem K, Vatca A, O’Neill T, Buckley D. Stevens-Johnson Syndrome induced by combination of lamotrigine and valproic acid in a 9-year-old boy. Ir Med J. 2017;110(6):586.
57. Bautista L, Aleta L, Berba R, Sumpaico M. Toxic epidermal necrolysis from intramuscular metoclopramide in a 52-year old male. Ann Allergy Asthma Immunol. 2008;100:A34–A34.
58. Rajagopalan S, Kaur S, Dogra S, et al. Toxic epidermal necrolysis induced by rarely implicated drugs. Indian J Pharmacol. 2012;44(2):272–273. doi:10.4103/0253-7613.93871
59. Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5(1):6–13. doi:10.1038/sj.tpj.6500285
60. Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19(1):69–76. doi:10.1038/gim.2016.80
61. Finkelstein Y, Macdonald EM, Li P, Hutson JR, Juurlink DN. Recurrence and mortality following severe cutaneous adverse reactions. JAMA. 2014;311(21):2231–2232. doi:10.1001/jama.2014.839
62. Dietrich A, Kawakubo Y, Rzany B, Mockenhaupt M, Simon JC, Schopf SE. Low N-acetylating capacity in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. Exp Dermatol. 1995;4(5):313–316. doi:10.1111/j.1600-0625.1995.tb00211.x
63. Furet Y, Bechtel Y, Le Guellec C, Bechtel PR, Autret-Leca E, Paintaud G. Pertinence clinique du polymorphisme genetique de la N-acetyltransferase de type 2 (NAT2) [Clinical relevance of N-acetyltransferase type 2 (NAT2) genetic polymorphism]. Therapie. 2002;57(5):427–431.
64. Dalton R, Lee SB, Claw KG, et al. Interrogation of CYP2D6 structural variant alleles improves the correlation between CYP2D6 genotype and CYP2D6-mediated metabolic activity. Clin Transl Sci. 2020;13(1):147–156. doi:10.1111/cts.12695
65. Naranjo MG, Rodrigues-Soares F, Penas-Lledo EM, et al. Interethnic variability in CYP2D6, CYP2C9, and CYP2C19 genes and predicted drug metabolism phenotypes among 6060 Ibero- and native Americans: ribef-ceiba consortium report on population pharmacogenomics. Omics. 2018;22(9):575–588. doi:10.1089/omi.2018.0114
66. Henthorn P, Kiledjian M, Kadesch T. Two distinct transcription factors that bind the immunoglobulin enhancer microE5/kappa 2 motif. Science. 1990;247(4941):467–470. doi:10.1126/science.2105528
67. Denis CM, Langelaan DN, Kirlin AC, et al. Functional redundancy between the transcriptional activation domains of E2A is mediated by binding to the KIX domain of CBP/p300. Nucleic Acids Res. 2014;42(11):7370–7382. doi:10.1093/nar/gku206
68. Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20(2):429–440. doi:10.1128/mcb.20.2.429-440.2000
69. Aronheim A, Shiran R, Rosen A, Walker MD. The E2A gene product contains two separable and functionally distinct transcription activation domains. Proc Natl Acad Sci U S A. 1993;90(17):8063–8067. doi:10.1073/pnas.90.17.8063
70. Kee BL. E and ID proteins branch out. Nat Rev Immunol. 2009;9(3):175–184. doi:10.1038/nri2507
71. Merrill BJ, Gat U, DasGupta R, Fuchs E. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 2001;15(13):1688–1705. doi:10.1101/gad.891401
72. DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126(20):4557–4568.
73. Howard JM, Nuguid JM, Ngole D, Nguyen H. Tcf3 expression marks both stem and progenitor cells in multiple epithelia. Development. 2014;141(16):3143–3152. doi:10.1242/dev.106989
74. Nguyen H, Merrill BJ, Polak L, et al. Tcf3 and Tcf4 are essential for long-term homeostasis of skin epithelia. Nat Genet. 2009;41(10):1068–1075. doi:10.1038/ng.431
75. Miao Q, Ku AT, Nishino Y, et al. Tcf3 promotes cell migration and wound repair through regulation of lipocalin 2. Nat Commun. 2014;5(1):4088. doi:10.1038/ncomms5088
76. Ichise T, Yoshida N, Ichise H. CBP/p300 antagonises EGFR-Ras-Erk signalling and suppresses increased Ras-Erk signalling-induced tumour formation in mice. J Pathol. 2019;249(1):39–51. doi:10.1002/path.5279
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.