Back to Journals » International Medical Case Reports Journal » Volume 12
What cardiologists should know about essential thrombocythemia and acute myocardial infarction: report of two cases and advanced heart failure therapies considerations
Authors Soucy-Giguère MC, Turgeon PY ![]() , Sénéchal M
, Sénéchal M ![]()
Received 29 May 2019
Accepted for publication 9 July 2019
Published 8 August 2019 Volume 2019:12 Pages 253—259
DOI https://doi.org/10.2147/IMCRJ.S217568
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Marie-Camille Soucy-Giguère, Pierre Yves Turgeon, Mario Sénéchal
Department of Cardiology, Institut Universitaire de Cardiologie et de Pneumologie de Québec, Québec City, QC, Canada
Abstract: We present the cases of two young male patients aged 22 and 31 without prior medical history nor cardiovascular risk factors, who presented to the hospital with large anterior ST-elevation myocardial infarction (STEMI). Urgent coronary angiography revealed acute thrombotic occlusion of the proximal left anterior descending artery in both patients. Persistent thrombocytosis was noted and subsequent investigations led to the diagnosis of essential thrombocythemia (ET) with positive JAK2-V617F mutation. Myocardial infarction as a first clinical manifestation of ET is rare but must be considered in patients without cardiovascular risk factors who show persistent thrombocytosis. In young patients without risk factors, there may be great delays before the diagnosis of STEMI is made. Longer time to revascularization of extensive STEMI is associated with adverse outcomes and cardiogenic shock which can lead to advanced therapies like heart transplant and left ventricular assist device (LVAD). Considering the favorable long-term prognosis of patients with ET, advanced therapies may be a valuable option in the presence of severe left ventricular dysfunction.
Keywords: thrombocytosis, JAK2 mutation, heart transplant, left ventricular assist device
Introduction
Essential thrombocythemia (ET) is a chronic myeloproliferative neoplasm (MPN) characterized by an excessive clonal platelet production. JAK2-V617F mutation is involved in about 60% of the patients.1,2
Common clinical manifestations of this disease result from thromboembolic events or from the thrombocytosis itself.3 Common vasomotor symptoms include headaches, dizziness, visual disturbances, and burning dysesthesia. Most people with ET experience at least one thrombotic or hemorrhagic event at some point during the course of their disease.4 For instance, patients can present with Budd-Chiari syndrome, multiple first trimester abortions or a hemorrhagic complication secondary to acquired von Willebrand disease associated with extreme thrombocytosis. The incidence of myocardial infarction (MI) in patients with known ET is under 10% and is uncommon as a first clinical manifestation of the disease.5
Case 1
A 31-year-old male patient without medical history, familial history of coronary heart disease, or cardiovascular risk factors was evaluated at our hospital for acute retrosternal pain.
Anterolateral ST-elevation myocardial infarction (STEMI) was diagnosed (Figure 1) and the patient underwent urgent coronary angiography, revealing an acute occlusion by an intramural thrombus of the proximal left anterior descending artery (LAD). There was no other significant lesion. A drug-eluting stent was successfully implanted in the proximal LAD (Figure 2). Echocardiography showed a depressed left ventricular ejection fraction (LVEF) at 15% with extended areas of akinesia in the LAD territory, left ventricular end-diastolic diameter (LVEDD) of 59 mm and left ventricular end-systolic diameter (LVESD) of 53 mm.
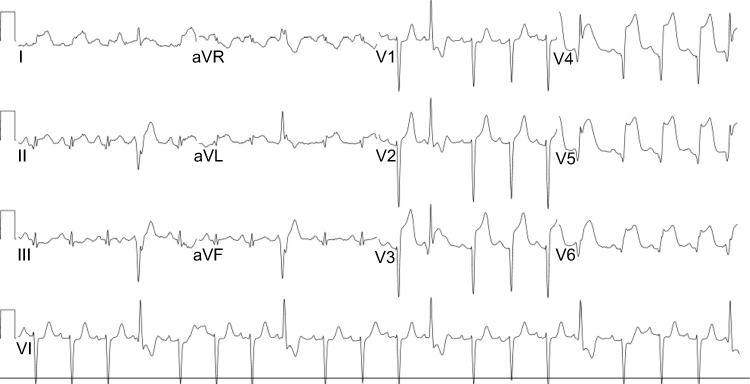 |
Figure 1 ECG, ST waves elevation in anterolateral leads, pathologic Q waves in lateral leads, ventricular ectopic beats. |
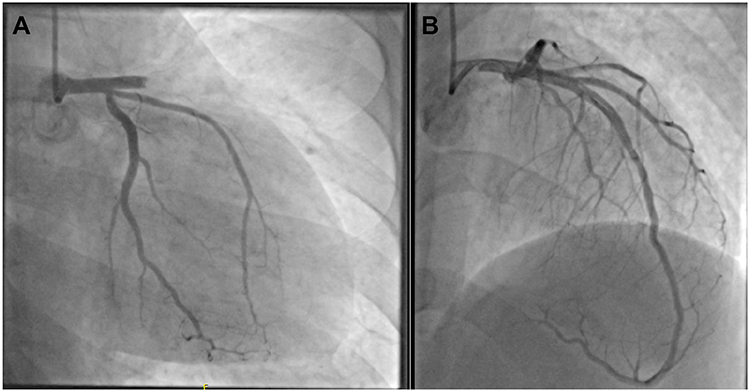 |
Figure 2 (A) RAO 20-caudal 20 view left coronary angiography: proximal LAD thrombus and thrombolysis in myocardial infarction (TIMI) flow grade 0. (B) RAO 10-cranial 40 view: a 4.0×22 mm drug-eluting stent was placed in the proximal LAD to restore normal TIMI flow grade 3. Right coronary angiography (not shown) was normal. |
Blood tests revealed isolated but severe thrombocytosis with 1260×109 platelets/L, which persisted on controls. ET was confirmed by positive JAK2-V617F mutation, negative BCR-ABL1 translocation, and compatible bone marrow biopsy. Hematology consultant started cytoreductive therapy with hydroxyurea, targeting less than 400×109 platelets/L, and recommended anticoagulation for primary prevention of ventricular thrombus given the diffuse area of akinesia and the prothrombotic state induced by ET. The patient underwent a favorable in-hospital evolution and was discharged 4 days later with warfarin for at least 6 months, acetylsalicylic acid 80 mg PO QD, clopidogrel 75 mg PO QD, bisoprolol 2,5 mg PO QD, perindopril 4 mg PO QD, eplerenone 25 mg PO QD, and hydroxyurea 500 mg PO TID. The follow-up complete blood count revealed a steady platelet count of 350x109/L on hydroxyurea.
Despite initial improvement, the patient experienced worsening of symptoms at 6 months and a control echocardiography showed a new severe functional mitral regurgitation and deterioration of LVEF to 20% with LVEDD of 63 mm and LVESD of 54 mm.
At this point, the patient was referred to our specialized heart failure clinic and full pre-transplant assessment was made. Fortunately, the patient benefited from the intensive follow-up which included the substitution of perindopril to sacubitril-valsartan. After up-titration of sacubitril-valsartan to 97–103 mg PO BID, a stable New York Heart Association (NYHA) functional class I was maintained as of now. Last echocardiography showed improvement in LVEF to 25%, stable left ventricular dimensions, and mild mitral regurgitation.
Case 2
A 22-year-old male patient without medical history nor family history of coronary heart disease presented to the emergency department of a regional hospital after 3 days suffering from vague retrosternal pain and nausea.
A recent transmural MI with anterolateral Q waves and persistent ST-elevation was diagnosed on electrocardiogram (Figure 3). Thrombolytics were not administered because of the late presentation. Intravenous furosemide and oxygen supplementation were begun for acute lung edema, and the patient was immediately transferred to our institution. Coronary angiography revealed a complete occlusion of proximal LAD (Figure 4). No coronary angioplasty was performed considering the late presentation with complete transmural cardiomyonecrosis that was confirmed by cardiac magnetic resonance imaging. An intra-aortic balloon pump (IABP) was installed at the end of the procedure for hypotension and signs of organs hypoperfusion. Echocardiography showed a LVEF of 10%, LVEDD of 52 mm, LVESD 40 mm, diffuse akinesia in the LAD territory and severe apical stasis, for which intravenous heparin as primary prevention of thrombus formation was continued.
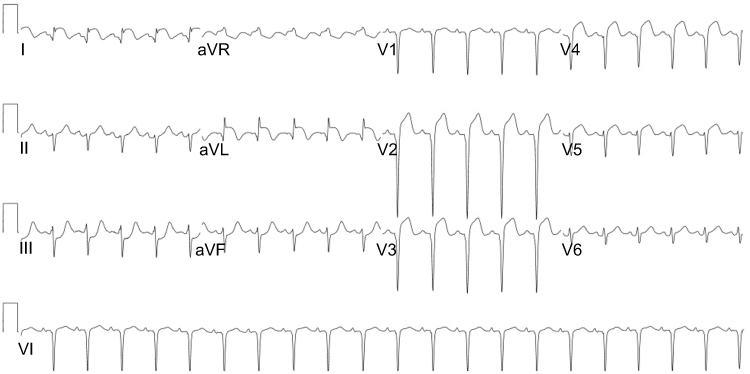 |
Figure 3 ECG, sinus tachycardia with ST waves elevation and pathologic Q waves in the anterolateral leads. |
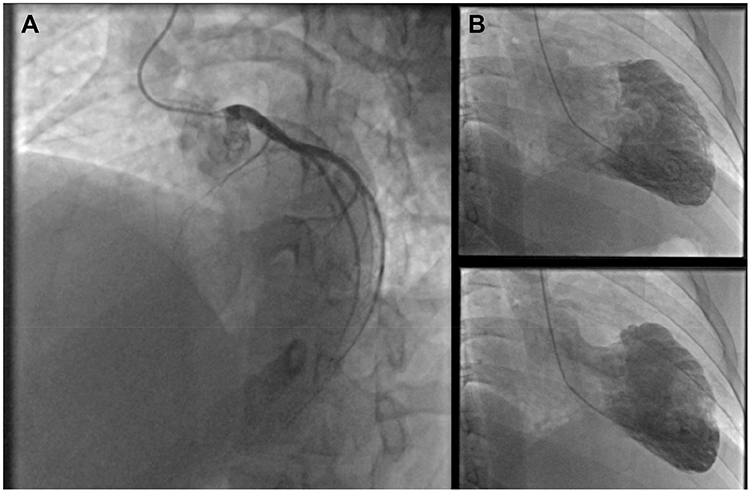 |
Figure 4 (A) LAO 45-cranial 25 view left coronary angiography: complete proximal LAD occlusion. (B) Left ventriculography revealing severe left ventricular systolic dysfunction and antero-apical akinesia. Right coronary angiography (not shown) was normal. |
Blood tests revealed initial and persistent thrombocytosis with platelets level of 700×109/L but otherwise normal complete blood count. ET was eventually confirmed by positive JAK2-V617F mutation, negative BCR-ABL1 translocation, and compatible bone marrow biopsy. Cytoreductive therapy with hydroxyurea was started.
The patient was weaned from the IABP within 48 hrs but the initiation of usual heart failure therapies was limited due to symptomatic hypotension with systolic blood pressure of 85 mmHg and lower. Considering his NYHA class IV heart failure and poor tolerability of medication, the patient underwent a complete pre-transplant assessment.
On day 7, the patient developed an acute right thumb pain with blue discoloration that coincided with a drop in the platelets level to 340×109/L. Considering the high probability of heparin-induced thrombocytopenia (HIT), intravenous heparin perfusion was immediately stopped and argatroban was initiated. The testing for anti-platelet factor 4 antibody was positive, and the diagnosis of HIT was confirmed with positive serotonin release assay. Substitution of argatroban by warfarin was then accomplished.
After 4 weeks of gradual up-titration of medication and slow improvement to NYHA class III, the patient was discharged with warfarin for at least 6 months, hydroxyurea 500 mg PO BID, acetylsalicylic acid 80 mg PO QD, metoprolol 50 mg PO BID, ramipril 5 mg PO BID, and spironolactone 12,5 mg PO QD. Echocardiography on discharge revealed a LVEF of 25%, LVEDD 56 mm, and LVESD 40 mm.
A long-term steady platelet count around 250×109/L was achieved with hydroxyurea and the patient did not experience any other thrombotic event. Despite difficult up-titration of medications due to orthostatic hypotension, patient slowly attained NYHA functional class II and the LVEF reached 35% 8 months after the event. Consequently, the heart transplant was no longer considered.
Discussion
ET is characterized by monoclonal proliferation of hematopoietic stem cells resulting in overproduction of platelets. It is a MPN, like polycythemia vera (PV), primary myelofibrosis, and chronic myeloid leukemia. The JAK2 enzyme mutations (V617F and exon 12 mutations) are identified in almost all patients with PV and in 60% of ET (V617F).2 This enzyme is involved in signal transduction for erythropoietin and thrombopoietin. CALR exon 9 and MPL exon 10 mutations can be identified in 30% of JAK2 negative ET patients, but the JAK2-V617F mutation clearly confers the highest risk for thrombosis, with a hazard ratio of 1.43 and 2.56 for venous and arterial thrombosis, respectively.6 According to the World Health Organization (WHO), the diagnosis of ET needs a persistent platelet count of ≥450×109/L, the identification of typical ET-associated mutations, a negative BCR-ABL1 rearrangement to exclude chronic myeloid leukemia and the histopathological confirmation with bone marrow biopsy.7
MI in patients with ET often results from in situ thrombosis without associated coronary atherosclerosis. Vianello et al,4 demonstrated that ET patients have endothelial dysfunction and lower coronary flow reserve. In two large cohorts of ET patients,5,6 incidence of MI ranged from 2 to 9, 4% and most had other significant cardiovascular risk factors. None of them were younger than 40 years old.5 However, more recent case reports described STEMI as the first thrombotic event that led to the diagnosis of ET in younger patients.8–13 Reactive thrombocytosis is frequent during acute MI but should not be overlooked. Normalization of platelets level must be confirmed with subsequent complete blood count.
Treatment of ET
General treatment of ET includes controlling the usual cardiovascular risk factors and cytoreductive therapy such as hydroxyurea in high-risk patients. Patients are considered at high risk if they fulfill any of the following criteria: age ≥60 years, history of thrombosis or major bleeding and platelets count ≥1500×10(9)/L.14 Additionally, the presence of cardiovascular risk factors or positive JAK2 mutation confers a significant risk of thrombosis. Low dose acetylsalicylic acid is the mainstay for primary and secondary prevention of thrombosis.14
Cytoreductive therapy is to be added in high-risk patient, including all those who have suffered from a thrombotic cardiac event. Hydroxyurea is the first line choice in the majority of patients because of its favorable side effects profile and its effective lowering of thrombotic events.14 However, hydroxyurea has long-term irreversible gonadal toxicity and is contraindicated in the first trimester of pregnancy. Interferon alpha may be the preferred option in young patients with reproductive intention but is associated with more frequent side effects such as hyperthermia, general malaise, and gastrointestinal upset. The objective of treatment is to maintain a platelet level of less than 400×109/L and if concomitant leukocytosis is present, a white blood cell count of less than 11×109/L.
HIT associated with ET
The second case described a challenging diagnosis of HIT in a patient without thrombocytopenia but a relative platelet count fall of more than 50% and a new thrombotic episode. According to few case reports, ET may be associated with a higher prevalence of HIT so that new thrombotic episodes while patient is receiving unfractionated heparin or low molecular weight heparin should not be overlooked.15–21 Clinicians should be aware that ET patients will not present an absolute thrombocytopenia but a faster than expected fall in platelets count with cytoreductive therapy associated with new thrombosis, skin necrosis, or lesions or systemic reaction to heparin bolus. The approach to diagnosis and treatment of HIT in patient with ET is the same as usual.
Prognosis of ET
ET is associated with a better prognosis than other BCR-ABL1 negative MPN and therefore, WHO diagnostic criteria must be strictly respected to estimate long-term evolution. Life expectancy of ET patients is only slightly worse than general population.22,23 Median survival time of overall ET patients is about 20 years and increases to 33 years in patients diagnosed before 60 years.24 Comparatively, median survival time of PV and primary myelofibrosis patients under 60 years is 24 and 15 years, respectively. Most important predictors of poor prognosis are age of ≥60 years, white blood cell count of ≥11,000/microL and history of thrombosis.25 Even for patients considered at high risk, median survival is about 14 years. Among all BCR-ABL negative MPN, ET has the lowest 15 years incidence of blast transformation and myelofibrosis of only 2–3% and 4–10%, respectively.22,23,26,27
Advanced heart failure therapies and ET
Those two cases raised many questions regarding advanced heart failure therapies. There is only one published case of a patient with ET who had a left ventricular assist device (LVAD) implanted for 3 months without complication, and eventually benefited from orthotopic heart transplantation.28 Unfortunately, the authors do not report the long-term evolution of the patient. Even with the improved management and risk categorization of ET patients in the last years, there is a persistent prothrombotic state that confers up to 3.6% annual risk of thrombosis in high-risk patients.29 Therefore, we do not consider ET as an absolute contraindication to LVAD implantation. If long-term LVAD is considered, close follow-up with hematologists and active monitoring for device thrombosis would be a standard of care. In addition to usual LVAD management, twice daily acetylsalicylic acid may be considered owing to the high platelets production.30–32
Considering the favorable long-term prognosis of ET patients, heart transplantation may also be considered if clinically indicated.33 As for medication, hydroxyurea or alternatives do not have prohibitive drug interaction with usual immunosuppressive medication but special attention must be paid to cytopenias caused by increased myelosuppressive effect.
Conclusion
Although close to 10% of the patients with ET will eventually suffer from MI at some point during the course of their disease, MI as the first clinical manifestation leading to the diagnosis of ET is uncommon. Our two cases emphasize the importance of pursuing the investigations for ET in young adults presenting with MI and thrombocytosis. Considering the favorable prognosis of ET, advanced therapies like heart transplantation or LVAD may be considered in the presence of severe left ventricular dysfunction.
Consent
Written informed consent was obtained from the patients for publication of this case series and accompanying images.
Ethical considerations
Considering the non-nominal and descriptive nature of this case series, it was exempted from ethical approval.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29(5):573–582. doi:10.1200/JCO.2010.29.8711
2. Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014;28:1472–1477. doi:10.1038/leu.2014.3
3. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol. 2005;131(2):208–213. doi:10.1111/j.1365-2141.2005.05764.x
4. Vianello F, Cella G, Osto E, et al. Coronary microvascular dysfunction due to essential thrombocythemia and policythemia vera: the missing piece in the puzzle of their increased cardiovascular risk? Am J Hematol. 2015;90:109–113. doi:10.1002/ajh.23881
5. Rossi C, Randi ML, Zerbinati P, Rinaldi V, Girolami A. Acute coronary disease in essential thrombocythemia and polycythemia vera. J Intern Med. 1998;244:49–53.
6. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117:5857–5859. doi:10.1182/blood-2011-02-339002
7. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi:10.1182/blood-2016-03-643544
8. Derks A, Bloemer MH. Essential thrombocythaemia as cause of myocardial infarction. Neth Heart J. 2001;9:383–385.
9. Gangadharan V, Ticku J, Kasi V. Acute myocardial infarction in a 21-year-old male due to essential thrombocytosis. Cath Lab Digest. 2014;22(3).
10. Pande S, Joshi R, Pande R. Essential thrombocythemia in a young man treated for myocardial infarction. BMJ Case Rep. 2010;2010:bcr0920092234.
11. Bildirici U, Celikyurt U, Ural E. Essential thrombocythemia: a case of acute ST-segment elevation myocardial infarction in a young female. Clin Cardiol. 2009;32:104–105. doi:10.1002/clc.20426
12. Daya SK, Gowda RM, Landis WA, Khan IA. Essential thrombocythemia-related acute ST-segment elevation myocardial infarction. A case report and literature review. Angiology. 2004;55:319–323. doi:10.1177/000331970405500312
13. Nanavati A, Patel N, Burke J. Thrombocytosis and coronary occlusion. JACC Cardiovasc Interv. 2012;5:e18–9. doi:10.1016/j.jcin.2011.12.018
14. Rumi E, Cazzola M. How I treat essential thrombocythemia. Blood. 2016;128:2403–2414. doi:10.1182/blood-2016-05-643346
15. Castelli R, Gallipoli P, Schiavon R, Teatini T, Deliliers GL, Bergamaschini L. Heparin-induced thrombocytopenia in essential thrombocytosis. J Stroke Cerebrovasc Dis. 2012;21(8):
16. Risch L, Pihan H, Zeller C, Huber AR. ET gets HIT–thrombocytotic heparin-induced thrombocytopenia (HIT) in a patient with essential thrombocythemia (ET). Blood Coagul Fibrinolysis. 2000;11(7):663–667.
17. Castelli R, Gallipoli P, Schiavon R, Teatini T, Deliliers GL, Bergamaschini L. Increased risk of heparin induced thrombocytopenia and thrombosis in patients with essential thrombocythemia carrying the homozygous JAK2 V617F mutation. J Thromb Thrombolysis. 2019;47(1):155–156. doi:10.1007/s11239-018-1773-4
18. Richard S, Perrin J, Lavandier K, Lacour JC, Ducrocq X. Cerebral venous thrombosis due to essential thrombocythemia and worsened by heparin-induced thrombocytopenia and thrombosis. Platelets. 2011;22(2):157–159. doi:10.3109/09537104.2010.527399
19. Houston DS. Heparin-induced thrombocytopenia without thrombocytopenia in a patient with essential thrombocythemia. Am J Hematol. 2000;65(4):331–332.
20. Reikvam H, Tiu RV. Venous thromboembolism in patients with essential thrombocythemia and polycythemia vera. Leukemia. 2012;26:563–571. doi:10.1038/leu.2011.314
21. Spectre G, Kalish Y, Schliamser L, Varon D. Heparin-induced thrombocytopenia in myeloproliferative disorders: a rare or under-diagnosed complication? Am J Hematol. 2008;83(5):420–423. doi:10.1002/ajh.21128
22. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755–761. doi:10.1016/j.amjmed.2004.06.032
23. Wolanskyj AP, Schwager SM, McClure RF, Larson DR, Tefferi A. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc. 2006;81(2):159–166. doi:10.4065/81.2.159
24. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507–2513. doi:10.1182/blood-2014-05-579136
25. Passamonti F, Thiele J, Girodon F, et al. A prognostic model to predict survival in 867 World Health Organization-defined essential thrombocythemia at diagnosis: a study by the International Working Group on myelofibrosis research and treatment. Blood. 2012;120(6):1197–1201. doi:10.1182/blood-2012-01-403279
26. Chim CS, Kwong YL, Lie AK, et al. Long-term outcome of 231 patients with essential thrombocythemia: prognostic factors for thrombosis, bleeding, myelofibrosis, and leukemia. Arch Intern Med. 2005;165(22):2651–2658. doi:10.1001/archinte.165.22.2651
27. Barbui T, Thiele J, Passamonti F, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011;29(23):3179–3184. doi:10.1200/JCO.2010.34.5298
28. Haddad M, Veinot JP, Masters RG, Hendry PJ. Essential thrombocytosis causing a massive myocardial infarction. Cardiovasc Pathol. 2003;12:216–218.
29. Barbui T, Finazzi G, Carobbio A, et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood. 2012;120(26):
30. Slaughter MS, Pagani FD, Rogers JG, et al. Clinical management of continuous-flow left ventricular assist devices in advanced heart failure. J Heart Lung Transplant. 2010;29(4 Suppl):S1–S39. doi:10.1016/j.healun.2010.01.011
31. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015;5:e369. doi:10.1038/bcj.2015.94
32. Dillinger JG, Sideris G, Henry P, Bal Dit Sollier C, Ronez E, Drouet L. Twice daily aspirin to improve biological aspirin efficacy in patients with essential thrombocythemia. Thromb Res. 2012;129(1):91–94. doi:10.1016/j.thromres.2011.09.017
33. Mehra MR, Canter CE, Hannan MM, et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: a 10-year update. J Heart Lung Transplant. 2016;35(1):1–23. doi:10.1016/j.healun.2015.10.023
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.