Back to Journals » International Medical Case Reports Journal » Volume 7
Warfarin-induced deep vein thrombosis
Authors Binymin K, Nasher M, Patel D
Received 7 February 2014
Accepted for publication 11 April 2014
Published 9 September 2014 Volume 2014:7 Pages 123—125
DOI https://doi.org/10.2147/IMCRJ.S62100
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Khalid A Binymin.
Views: 1148
Khalid A Binymin,1 Magda Nasher,2 Dipti Patel2
1Liverpool University, Medical School, England, UK; 2Southport and Ormskirk NHS Trust, Southport District General Hospital, Kew, UK
Abstract: We are presenting a 72-year-old female who was admitted to hospital with deep vein thrombosis (DVT). She was known to have atrial fibrillation and was initiated on warfarin for stroke prophylaxis 3 days earlier. She was given warfarin therapy without low molecular weight heparin cover as per “slow-start regimen” protocol. The warfarin dose was increased after 3 days to achieve rapid anticoagulation, resulting in DVT in the left leg. We propose that the higher unopposed warfarin dose utilized in this case resulted in DVT. Warfarin loading doses may paradoxically result in a hypercoagulable state and potential clot formation because of significant reductions in protein C and protein S levels.
Keywords: warfarin, deep vein thrombosis, slow-start regimen, protein C, protein S, warfarin-induced skin necrosis, stroke prophylaxis
Case report
A 72-year-old female was admitted to hospital with deep vein thrombosis (DVT). The patient was recently diagnosed with persistent nonvalvular atrial fibrillation, and it was decided that she would benefit from a warfarin “slow-start regimen” for stroke prophylaxis. Warfarin was prescribed at 2 mg daily as a slow loading dose. On day 3, the international normalized ratio (INR) was at 0.9 (range =0.88–1.12). The primary care physician increased the dose of warfarin to a 5 mg daily dose. After receiving the second 5 mg dose, the patient woke up with pain and swelling in the left leg and upper thigh. The whole leg was swollen and blue in color (Figure 1). The patient was admitted to hospital. A duplex Doppler ultrasound was positive for left femoral DVT. The patient’s past medical and family history was noncontributory for a history of thrombophilia or other major thrombotic risk factors.
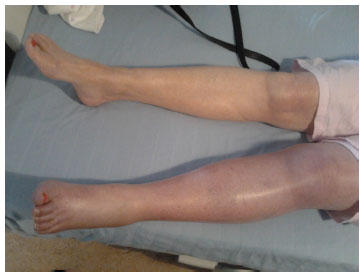 | Figure 1 Left leg deep vein thrombosis. |
Laboratory levels were as follows: protein C activity =13% (range =70%–140%); protein S activity =28% (range =70%–140%), INR =1 (range =0.88–1.12); prothrombin time =10.5 seconds (range =9.5–12.0 seconds); and activated partial thromboplastin time =30.5 seconds (range =23.4–35.4 seconds). These values indicated that the patient was found to be reactive to initial anticoagulant therapy with warfarin.
Laboratory tests including factor V Leiden, lupus screen, antinuclear antibody, and anticardiolipin, were negative for an inherited or acquired prothrombotic state. Ultrasound of the abdomen and pelvis was within the normal limits. The chest radiograph was reported as normal. A plan to rule out protein C and protein S deficiencies and inadequate antithrombin 3 was scheduled to take place after the cessation of warfarin therapy in 3 months’ time (prove to be negative).
The patient was anticoagulated simultaneously with low molecular weight heparin and warfarin sulfate.
Anticoagulation was achieved on day 6 of the patient’s hospitalization. She was discharged on 3 mg of warfarin per day, with instructions to continue the use of compression stockings.
Discussion
The use of unopposed warfarin as monotherapy (without an initial heparin overlap) as advocated by the “slow-start regimen” is often regarded as safe and achieves therapeutic anticoagulation in the majority of patients within 3–4 weeks.1,2 This regimen is suitable for use in both the secondary and primary care setting, and it allows for the induction of anticoagulation therapy requiring only weekly monitoring.2 This appears to avoid over-anticoagulation and bleeding associated with rapid loading.
Oates et al1 developed an algorithm for safe and efficient prophylactic anticoagulation that can be used in outpatient settings. In their study, the INR range of 2-3 was maintained in half of the patients tested during the observation period. No significant over-anticoagulation was reported. No age limits were set for entry into the study, and there were no exclusion criteria at entry.1 Tait and Sefcick2 studied 200 patients who were started on 3 mg of warfarin daily for 1 week, and subsequent doses were determined by weekly INR measurement. Similar results to the Oates et al study were obtained.
Current guidelines advise treating doctors to start warfarin at 2 mg/day for 1 week, then to repeat the INR. If the INR is <2, then the patient should continue with warfarin at 2 mg/day, and the INR should be repeated at the end of the second week. At that stage, the treating doctors can adjust the warfarin dose, as per nomogram, with the aim of achieving a target INR of 2.5.
Warfarin loading doses may paradoxically result in a hypercoagulable state and potential clot formation because of significant reductions in protein C and protein S.3,4 A precipitous reduction in the concentration of protein C and protein S (with an approximate half-life of 8 hours) occurs during the first 36 hours of warfarin therapy, which is when warfarin has a very limited effect on prothrombin (which has an approximate half-life of 50 hours).5 Consequently, because of the potential risk of thromboembolism during the initiation of warfarin therapy, the use of concurrent heparin is extremely important.6
Warfarin-induced skin necrosis is a recognized adverse event during the loading of warfarin therapy. Similarly, it begins on day 3–6 after initiating warfarin.7 It is also thought that the precipitous reduction in the concentration of protein C and protein S creates a hypercoagulable state, which cause thrombotic occlusions of the microvasculature with resulting necrosis of the subcutaneous fat.7,8
The use of a small loading dose of warfarin (2 mg/day) in the “slow-start regimen” is basically designed to combat the precipitous reduction in protein C and protein S, and then to obviate the need to use concurrent heparin therapy.1 In an attempt to achieve rapid anticoagulation, doctors might depart from the standard protocol of the “slow-start regimen” and use a larger unopposed loading dose of warfarin, which could result in the development of DVT with potentially life-threatening complications.5
With the growing popularity of using the slow-start unopposed warfarin regimen in outpatient and community settings, there is an increased risk of warfarin-related adverse events when treating doctors fail to adhere to protocols. The desire of some doctors to achieve rapid anticoagulation by initiating or escalating warfarin monotherapy could be catastrophic. The incidence of warfarin unopposed therapy causing DVT is expected to rise with increase adoption of the “slow-start regimen”.
Although the development of DVT after giving a higher loading dose in our patient could be a pure coincidence, we felt that the temporal relationship between the onset of the DVT timed with the increase in the warfarin loading dose is compelling. Also, the paucity of obvious clinical risk factors for a hypercoagulable state increased our suspicion. The Naranjo score was at +2, which renders the development of DVT as a possible adverse drug reaction to warfarin.9
This case study illustrates the importance of considering DVT as a possible complication of unopposed warfarin therapy. Warfarin-induced skin necrosis is a well-established adverse event of a warfarin loading dose.7 The hypercoagulable state of the blood caused by warfarin is regarded as the main cause for this adverse event. This case is the first ever reported case of a possible iatrogenic warfarin-induced DVT. It highlighted the importance of adhering to the lower loading doses specified in protocols. Understanding the pharmacokinetics and pharmacodynamics of warfarin would help ensure safe and effective prescription of the drug. A high index of suspicion may allow for the rapid reversal of the warfarin effect with therapeutic heparinization before the processes of adverse procoagulation begin.
We propose the acronym “WIDVT” for the adverse event of warfarin-induced DVT in order to help physicians become more alert to and aware of the consequences of initiating an unopposed warfarin loading dose.
Disclosure
The authors report no conflicts of interest in this work.
References
Oates A, Jackson PR, Austin CA, Channer KS. A new regimen for starting warfarin therapy in out-patients. Br J Clin Pharmacol. 1998;46(2):157–161. | |
Tait RC, Sefcick A. A warfarin induction regimen for out-patient anticoagulation in patients with atrial fibrillation. Br J Haematol. 1998;101(3):450–454. | |
Fuster V, Rydén LE, Asinger RW, et al; American College of Cardiology; American Heart Association; European Society of Cardiology; North American Society of Pacing and Electrophysiology. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines and Policy Conferences (Committee to develop guidelines for the management of patients with atrial fibrillation) developed in collaboration with the North American Society of Pacing and Electrophysiology. Eur Heart J. 2001;22(20):1852–1923. | |
Guyatt GH, Akl EA, Crowther M, Gutterman DD, Schuünemann HJ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: Antithrombotic Therapy and Prevention of Thrombosis. 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(Suppl 2):7S–47S. | |
Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 1995;108(Suppl 4):231S–246S. | |
Keeling D, Baglin T, Tait C, et al; British Committee for Standards in Haemotology. Guidelines on oral anticoagulation with warfarin – fourth edition. Br J Haematol. 2011;154(3):311–324. | |
Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg. 2000;87(3):266–272. | |
Ng T, Tillyer ML. Warfarin-induced skin necrosis associated with Factor V Leiden and protein S deficiency. Clin Lab Haematol. 2001;23(4):261–264. | |
Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239–245. |
 © 2014 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.
© 2014 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.