Back to Journals » Journal of Blood Medicine » Volume 12
Waldenström’s Macroglobulinemia: An Exploration into the Pathology and Diagnosis of a Complex B-Cell Malignancy
Authors Askari E, Rodriguez S, Garcia-Sanz R ![]()
Received 14 April 2021
Accepted for publication 19 July 2021
Published 30 August 2021 Volume 2021:12 Pages 795—807
DOI https://doi.org/10.2147/JBM.S267938
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Elham Askari,1 Sara Rodriguez,2 Ramon Garcia-Sanz3
1Hematology Department, Fundación Jiménez Díaz, Centro de Investigación Biomédica en Red-Cáncer (CIBERONC) CB16/12/00369, Madrid, Spain; 2Clinica Universidad de Navarra, Centro de Investigación Medica Aplicada (CIMA), Instituto de Investigación Sanitaria de Navarra (IDISNA), Accelerator project, Centro de Investigación Biomédica en Red-Cáncer (CIBERONC) CB16/12/00369, Pamplona, Spain; 3Haematology Department, University Hospital of Salamanca, Research Biomedical Institute of Salamanca (IBSAL), Accelerator project, Centro de Investigación Biomédica en Red-Cáncer (CIBERONC) CB16/12/00369 and Center for Cancer Research-IBMCC (USAL-CSIC), Salamanca, Spain
Correspondence: Ramon Garcia-Sanz
Department of Hematology, University Hospital of Salamanca, Paseo de San Vicente, 58-182, Salamanca, 37007, Spain
Tel +34 923291100
Fax +34 923294624
Email [email protected]
Abstract: After 77 years since the initial description, Waldenström macroglobulinemia (WM) remains as a bone marrow neoplastic disorder with lymphoplasmacytic differentiation oversecreting a monoclonal immunoglobulin M (IgM). However, many biological and genetic aspects of this entity have been unraveled and it is now easy to correctly diagnose patients with this illness. The diagnosis requires the presence of a monoclonal IgM component and bone marrow lymphoid infiltration must be demonstrated. In addition, other small B-cell lymphoid neoplasms with plasma cell differentiation must be discarded. Although the clinical picture is highly heterogeneous, the diagnosis is much easier today compared to the past, since now we can demonstrate the presence of somatic mutations, especially the L265P mutation in the MYD88 gene, highly characteristic of WM (> 90% of the patients), followed by the WHIM-like mutations in the CXCR4 gene (∼ 35%). The identification of these mutations is very important, because they can modulate the response to new treatments with Bruton’s tyrosine kinase (BTK) inhibitors. Thus, the conventional prognostic factors that predict the outcome of these patients (anemia, thrombopenia, high M component, high B2M, and advanced age), must be complemented with the genetic evaluation of the patient, that can help us in the prediction of the risk of transformation from asymptomatic to symptomatic forms (Del6q) and/or from indolent forms of the disease to aggressive lymphomas (CD79b mutations).
Keywords: Waldenström’s macroglobulinemia, IgM-MGUS, pathology, biology, diagnosis, prognosis
Introduction
Waldenström’s macroglobulinemia (WM) is a B-cell lymphoproliferative disorder which is defined by bone marrow (BM) infiltration by small lymphocytes, lymphoplasmacytoid cells and plasma cells together with the presence of a detectable monoclonal immunoglobulin M (IgM).1,2 According to the 2008 World Health Organization (WHO) classification system of lymphoid neoplasms, the pathological disorder underlying WM is a lymphoplasmacytic lymphoma (LPL).3 By the end of 2011, a remarkable fact that has changed the view of WM was presented at this year annual meeting of the American Society of Hematology, as it was the presence of the MYD88L265P mutation in most cases of this disease,4 which has been highlighted in the last 2016 WHO classification of lymphoid malignancies.5 Other recent advances in the genomic profiling of patients with Waldenström macroglobulinemia (WM) have enhanced our understanding of its pathogenesis.6,7 Thus, the highly recurring somatic mutation in the MYD88L265P is now recognized in >95% of cases, while CXCR4 mutations are present in 30–40% of WM patients.6–8 Such studies have also shown that the genomic status can impact not only the diagnosis but also the clinical presentation, treatment outcome and overall survival of patients.6,7
Classification and Epidemiology
WM patients may have a long asymptomatic course, called Smoldering WM, also known as asymptomatic WM (AWM). Although some authors have defined AWM as a WM with a serum monoclonal IgM ≥30 g/l and/or ≥10% BM lymphoplasmacytic infiltration without end‐organ damage,9 the International Waldenström’s Macroglobulinemia Group (IWMG)1,2 and the WHO1,2 consider that it would be inappropriate to suggest disease definitions based on arbitrary values for laboratory parameters, such as IgM concentration and percentage of BM lymphocytes. Thus, patients with an IgM monoclonal protein and unequivocal evidence of BM infiltration by lymphoplasmacytic lymphoma should be considered to have WM irrespective of the IgM concentration. Then, when we are in front of an asymptomatic patient with an IgM monoclonal component in the absence of unequivocal BM or other tissue infiltration, an IgM Monoclonal Gammopathy of Uncertain Significance (IgM-MGUS) must be diagnosed. We still can have a small group of patients with an IgM monoclonal component, with no demonstrable tissue infiltration but pathological consequences of the IgM protein, such as peripheral neuropathy, amyloidosis, or skin lesions among others; in these cases, we will be in front of a so-called IgM related disorder. Finally, we also can see some isolated patients with IgM monoclonal component and some tissue infiltration by LPL but in the absence of BM infiltration; such patients should be considered as ‘pure’ LPL, together with those rare cases in whom the M-component is an IgG and/or IgA. These definitions can be found in Table 1.
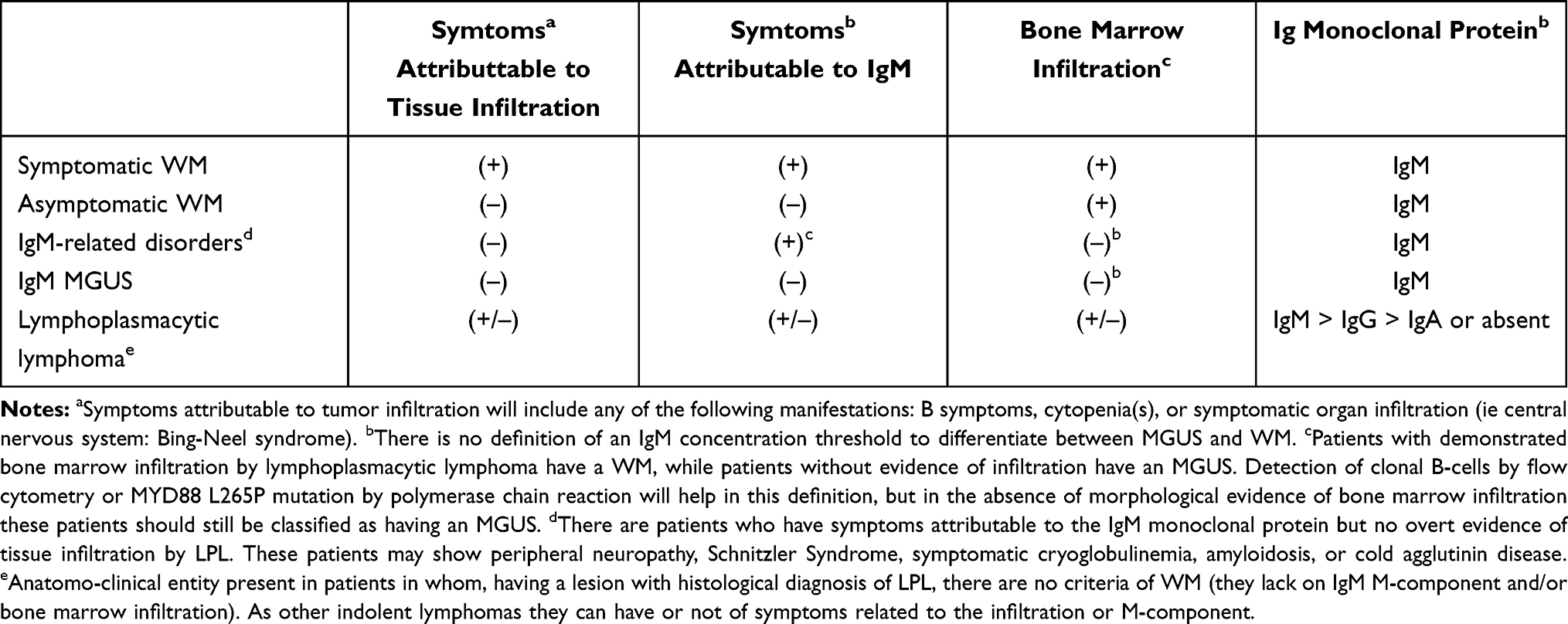 |
Table 1 Classification of Immunoproliferative Disorders with Monoclonal IgM |
WM represents approximately 1–2% of all hematologic malignancies with 150–200 new cases per year in Spain.10 WM mostly occurs in adult Caucasians with a median age in the seventh decade of life. The annual incidence of WM is 3–4 cases per million persons per year and increase with the age or familial predisposition that is observed in approximately 20% of patients.11,12 Virus C Hepatitis is also associated with WM.13 IgM monoclonal gammopathy of undetermined significance (IgM-MGUS) and AWM are defined as clinical precursor states to symptomatic WM.14 Thus, patients with IgM-MGUS have a 10.8 higher relative risk of developing WM.15
Clinical Features
The clinical presentation of WM is variable (Tables 2 and 3). Symptoms can be related to tumor infiltration, either in the BM or as extramedullary infiltration, or they can be related to specific immunological or chemical properties of the monoclonal component.
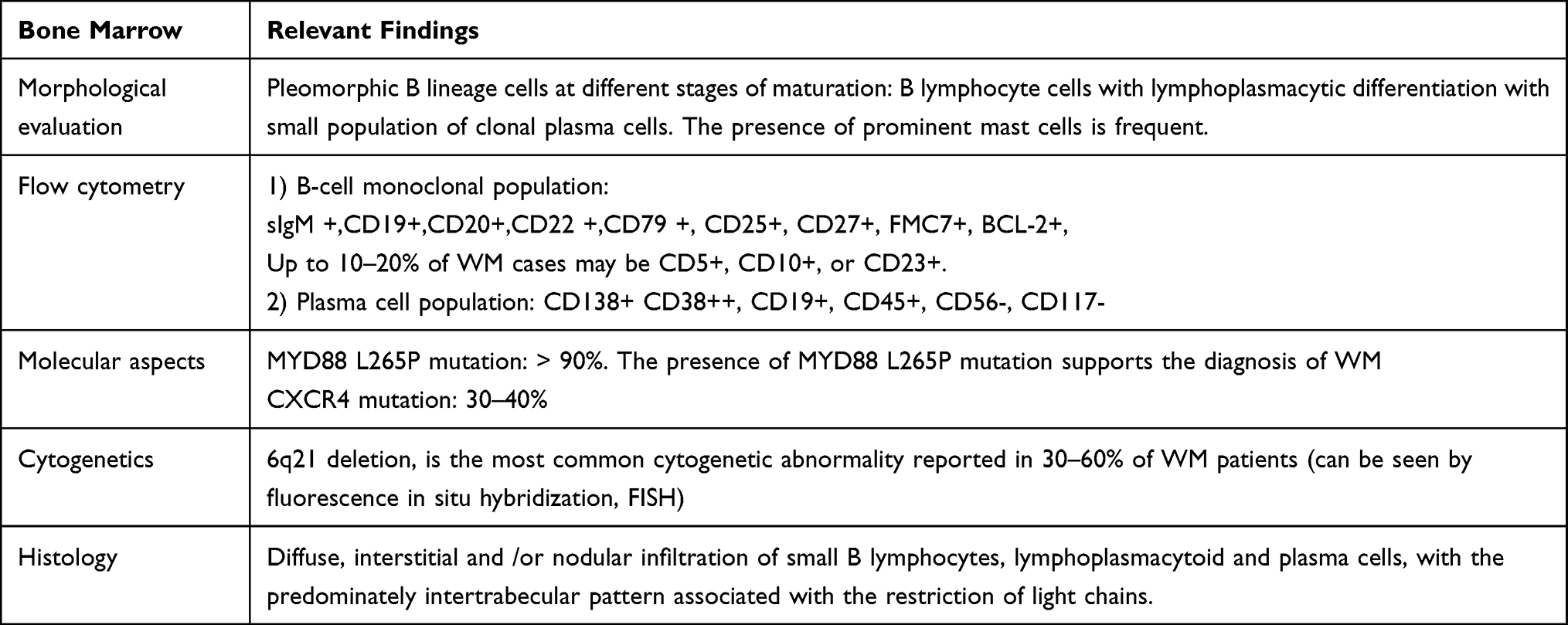 |
Table 2 Inside the Bone Marrow: Histology, Immunophenotype and Molecular Features |
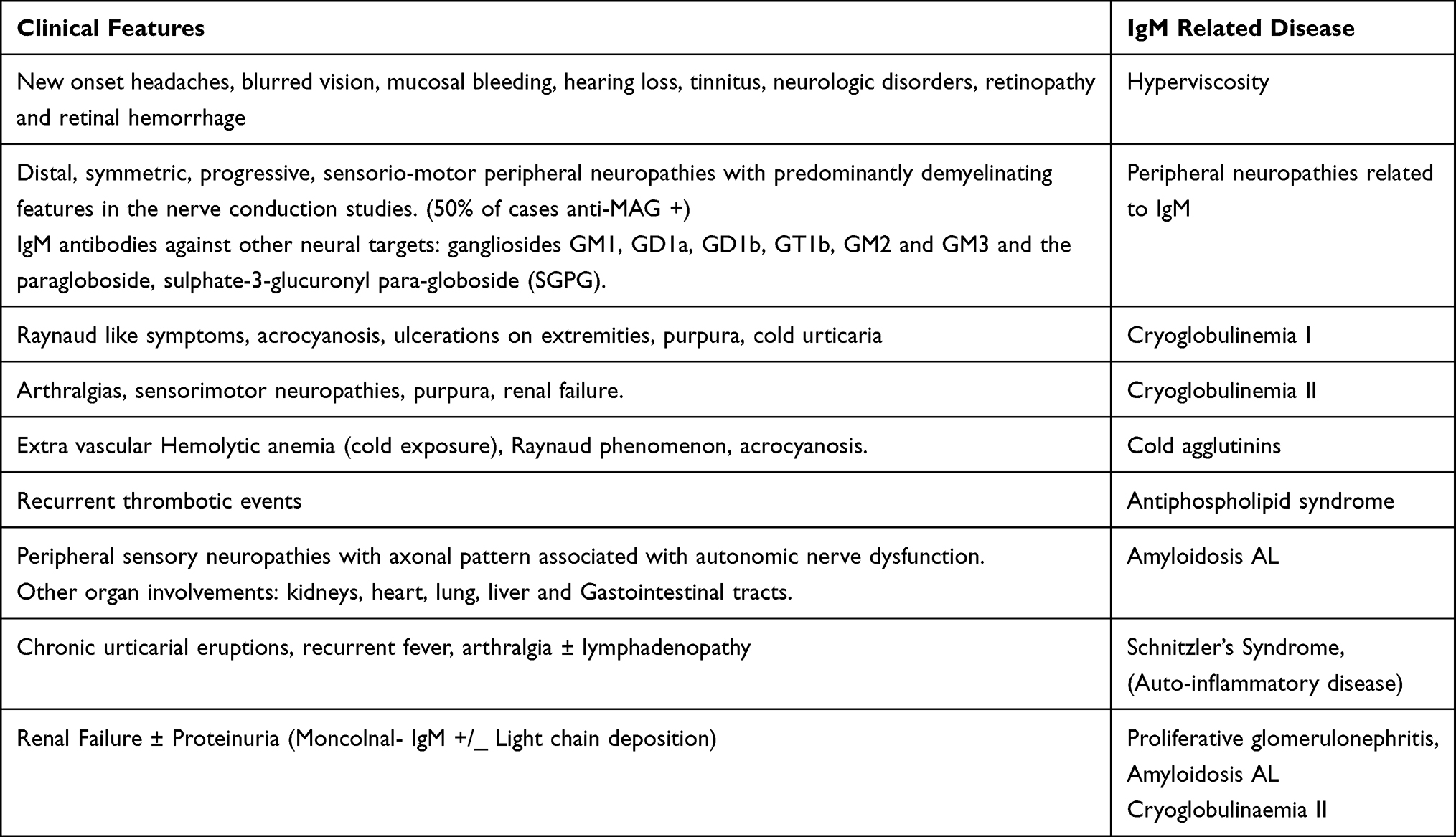 |
Table 3 Outside the Bone Marrow: IgM Related Symptoms |
Symptoms and Signs Derived from Lymphoma Infiltration
B Symptoms
In symptomatic WM patients, the presence of B symptoms including fever, night sweats, and weight loss is not uncommon.
Bone Marrow Failure
Diagnosis of WM requires the presence of lymphoplasmacytic lymphoma in the BM (Table 2) with or without other histological infiltration. A BM aspiration and biopsy, together with immunophenotyping and genetics will establish the diagnosis of WM and will help to differentiate it from IgM MGUS, IgM multiple myeloma (MM) and other IgM-secreting lymphoproliferative disorders, such as marginal zone lymphoma (MZL) and chronic lymphocytic leukemia (CLL).16–18 A typical effect of BM infiltration is anemia that is the most common symptom of WM and the usual cause to start therapy. Anemia is due to BM infiltration, but iron metabolism dysfunction and hemolysis can play a role. Iron deficiency is very common in WM because hepcidin, a negative regulator of iron absorption, is elevated in the serum of WM patients, probably related with the MYD88 alterations.19,20 Other cytopenias, as leukopenia and thrombocytopenia can also be present in around 15% of patients.
Extra-Medullary Disease (EMD)
At diagnosis, 10–15% of WM patients have EMD: lymphadenopathy, hepatosplenomegaly, pleural or abdominal effusions, among others (Table 3). At relapse, nearly 60% of patients can present adenopathy. The IWMG recommends an initial assessment of EMD by imaging in all new diagnosed patients, and at the time of relapse.21 In a retrospective single center study in Dana-Farber Cancer Institute, among 985 patients with WM that were evaluated at diagnosis, only 4.4% of patients have extranodal/extramedullary disease; 21% of them presented EMD at diagnosis and 79% developed EMD during the outcome. Most frequent EMD sites involved were lungs (30%), soft tissue (21%), cerebrospinal fluid (23%), kidneys (8%), and bones (9%).22
Bing-Neel Syndrome (BNS) is a rare but interesting presentation of WM, observed in about 1% of patients. BNS should be suspected in patients with WM who develop central neurological symptoms. Such symptoms include motor deficits, balance disorder, gait abnormalities, cranial nerve deficits, seizures, headaches, and atypical peripheral neuropathy. The gold standard for BNS diagnosis is the demonstration of WM cells on cerebro spinal fluid (CSF) examination or brain biopsy. The evaluation of these patients should include brain and whole spine magnetic resonance imaging with gadolinium enhancement, and CSF sampling. CSF should be evaluated by morphology, flow cytometry and molecular studies.23 Flow cytometry of CSF is the most sensitive technique to detect tumor cells in the central nervous system,24 but PCR assays to detect IGH gene rearrangements and mutated MYD88L265Pon CSF can also help to support the diagnosis of BNS. In patients with focal brain lesions but no CSF involvement, a stereotaxic biopsy should be programmed.23
Symptoms Related to IgM
IgM paraprotein can cause specific complications due to its physical‐chemical properties, autoantibody activity, tissue deposition and non‐specific interactions with other proteins.
Hyperviscosity Syndrome (HVS)
Hyperviscosity syndrome (HVS) related to high IgM levels is a hallmark of symptomatic WM. It has been described in 10–15% of cases with variable manifestations that include headache, blurred vision, confusion, and mucosal bleeding.25 We should identify patients at high risk of symptomatic hyperviscosity that might support the decision to treat asymptomatic patients before irreversible damage occurs. Although the size of the monoclonal component is not exactly related to symptoms, they are rarely observed with a serum IgM level below 3 g/dL, while they are frequent in patients with a serum IgM ≥6 g/dL (median time to symptomatic hyperviscosity of ~3 months).26 The funduscopic examination is very reliable to detect clinically significant hyperviscosity by seeing changes in the retinal vessels. Plasmapheresis should be carried out as an emergency procedure in high-risk HVS patients. The panel of the 8th International workshop on WM recommended that patients with serum IgM levels >3g/dL should undergo funduscopic evaluation by an experienced ophthalmologist to identify vessel tortuosity/retinal hemorrhages.21 These findings would suggest the need for immediate therapy.
Cryoglobulins
Cryoglobulinemia refers to the presence of serum proteins (immunoglobulins) with heterogeneous etiopathogenetic and immunochemical properties that precipitate at the temperatures below 37°C [98.6 F] and redissolve at 37°C. Cryoglobulins can deposit in medium and large-sized blood vessels, leading to a systemic inflammatory syndrome characterized by fatigue, arthralgia, purpura, neuropathy, glomerulonephritis, endothelial injury and end-organ damage. Brouet criteria classify cryoglobulinemia into three (I, II & III) subgroups based on their immunoglobulin (Ig) composition.27 Type I cryoglobulinemia, which develops in the setting of protein-secreting monoclonal gammopathies, is the one usually associated to WM. It is characterized by Raynaud phenomenon, acrocyanosis ulcers, purpura and cold urticaria. In contrast, in type II or mixed cryoglobulinemia, the cryoglobulins are composed of a mix of monoclonal IgM with rheumatoid factor (RF) activity and polyclonal IgG, usually associates Hepatitis C virus infection, and leads to purpura, renal failure, arthralgias and sensorimotor neuropathies.28 Type III is characterized by polyclonal IgM with RF activity and polyclonal IgG, and it is not associated to WM.
Accordingly, in WM patients suspected of having cryoglobulins, serum samples should be obtained in a warm bath to avoid cryoprecipitation. Plasmapheresis will be a choice of therapy in WM patients with severe cryoglobulinemia. A blood warmer will be necessary to prevent cryoglobulin precipitation during plasmapheresis.27
Cold Agglutinin Syndrome (CAD)
Another cause of Anemia is hemolysis in WM patients. A hemolytic panel including reticulocyte counts, lactate dehydrogenase, haptoglobin, direct Coombs test, and cold agglutinins should be performed, in all patients with anemia and WM. We should think about of CAD, if the WM patient presents hemoglobinuria after cold exposure.29 CAD is produced by IgM with immunological activity: IgM acts as an autoantibody against red blood cell antigens, producing Hemolytic anemia, and sometimes Raynaud phenomenon, acrocyanosis, and livedo reticularis. In patients with severe cold agglutininemia, plasmapheresis should be started promptly to remove cold agglutinins.
Polyneuropathy
The prevalence of peripheral neuropathy (PNP) in WM at diagnosis is around 30%, but 50% of them can be affected at some time during the course of their disease.30 Although PNP may be related to lymphoplasmacytic infiltration nerve fibers, it most commonly is due to IgM deposition, presence of autoantibodies, cryoglobulinemia or amyloidosis.
When a WM patient is suspected to be affected by PNP, the first point to evaluate is whether or not the monoclonal gammopathy is the cause of PNP. The second point will be to distinguish if the PNP can be associated to specific plasma cell disorders, such as primary amyloidosis, that could have a specific approach and treatment.31
Usually, IgM monoclonal gammopathy associated PNP presents as a distal, acquired, demyelinating, and symmetric neuropathy with M protein (DADS-M).31 Nerve conduction tests should be performed in all WM patients, and different patterns of nerve damage can appear: demyelinating, axonal or mixture pattern. All these patients should be tested for myelin-associated globulin antibodies (anti-MAG antibodies). A clinically significant result [>70.000 Bühlmann Titer Units (BTU)] strongly suggests a PNP caused by the M-component. Up to 50% of patients with IgM-associated demyelinating PNP have anti-MAG antibodies in the setting of IgM MGUS or WM.31 If anti-MAG are negative, patients should be tested for antibodies against other neural targets, including the gangliosides GM1, GD1a, GD1b, GT1b, GM2 and GM3 and the paragloboside, sulphate-3-glucuronyl para-globoside (SGPG). GM1 antibodies may be causally associated with a multifocal motor neuropathy, as well as IgM GD1b antibodies. IgM disialosyl antibodies could be associated with CANOMAD: Chronic Ataxic Neuropathy with Ophthalmoplegia, M-protein, cold Agglutinins and Disialosyl ganglioside (IgM Anti-GD1b/ GT1b/GQ1b) antibodies.32 In CANOMAD, nerve conduction tests show a mixed pattern of axonal loss and demyelinating features. 30–40% of IgM-related demyelinating neuropathies have no identifiable antibodies.30 In these cases, clinical judgment is the only possibility to connect IgM and PNP. It is important to identify them, because treatment could be needed to avoid a prolonged demyelination, that can induce irreversible axonal damage. In addition, other causes of PNP should be always ruled out: diabetes, cobalamin deficiency, thyroid dysfunction, etc.33
Amyloidosis
Systemic immunoglobulin light-chain (AL) amyloidosis is a rare complication of WM caused by the aggregation of misfolded proteins that deposit as fibrils in several organs, including kidneys, heart, peripheral nerves, liver, and gastrointestinal tract. Monoclonal IgM-related light chain AL accounts for 6% of all cases of AL.34 Several studies have shown that the pattern of organ involvement is different from non-IgM Amyloidosis, with higher frequencies of lung, soft tissue, and peripheral nervous system involvement, and lower frequencies of heart.34,35 In these cases, PNP is usually due to amyloid deposition in the nerves. The neuropathy in AL amyloidosis often has an axonal pattern whereas IgM monoclonal gammopathy associated PNP without amyloidosis often has a demyelinating nature. Autonomic function also tends to be impaired in neuropathy associated with AL amyloidosis, which is not frequent in non-amyloidotic WM neuropathy. The prognosis of patients with IgM-related AL amyloidosis might be more favorable than those with non-IgM AL amyloidosis.36
Diagnosis
The diagnosis of WM is clinicopathological, requiring the histologic evidence of BM infiltration by LPL as well as the presence of an IgM monoclonal gammopathy detected, at least, by immunofixation.1,21,37 A summary of the tests that are essential or highly recommended to be performed during the diagnostic work-up of patients with WM is shown in Table 4.
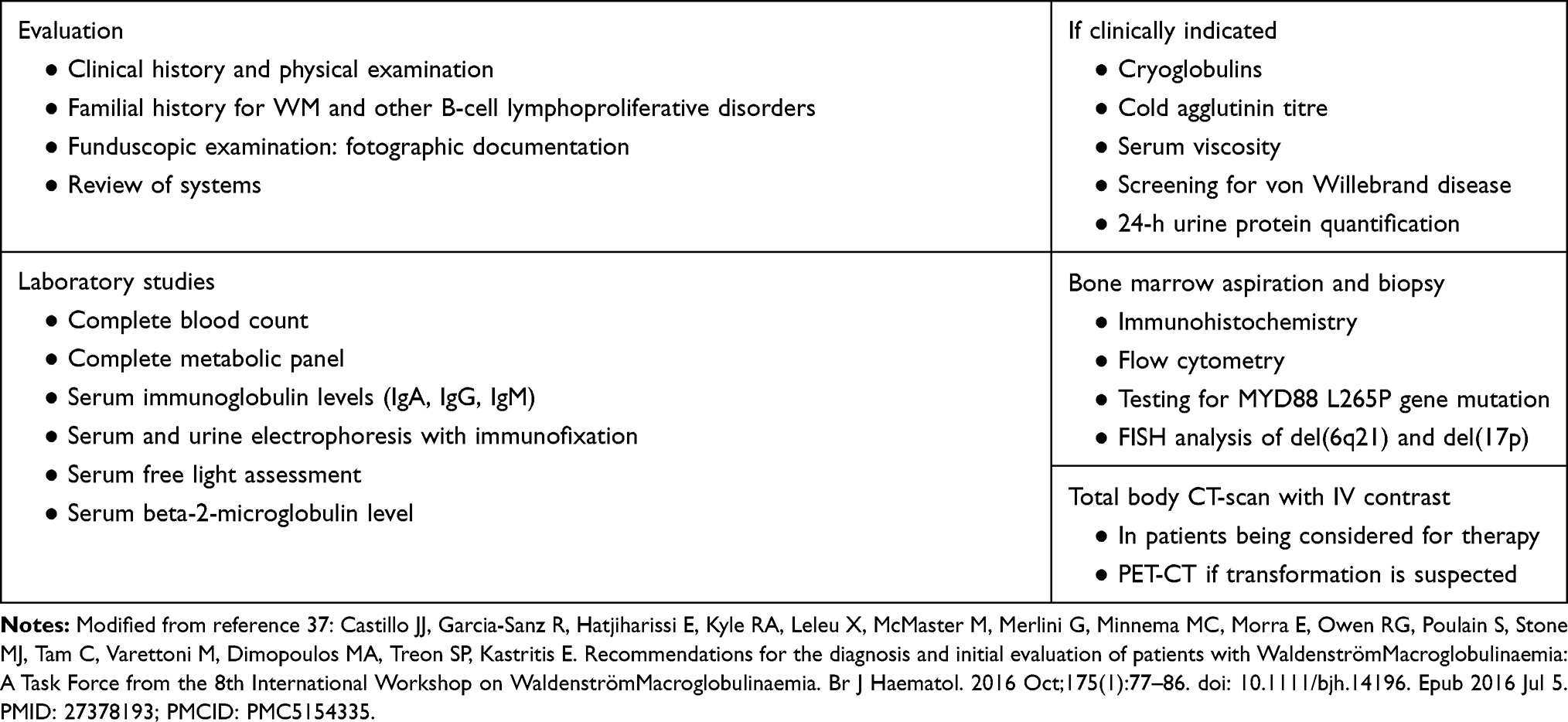 |
Table 4 Essential Evaluation of Patients with Waldenström Macroglobulinemia (WM) |
Laboratory Findings
Hemogram
50–70% of patients have normocytic and central anemia, although a peripheral component can also be found (blood loss, cold agglutinins, hemodilution).21 White blood cells are usually normal, but lymphocytosis (>4×109/L) is present in one-fourth of patients, usually monoclonal, and neutropenia is possible. Thrombopenia only appears in <20% of cases. Blood smear shows stacks or aggregations of red blood cells (rouleaux phenomenon) due to paraproteinemia, and sed rate (SR) is increased.
Biochemistry
The biochemistry of patients with WM is usually normal. Renal damage can occur in patients with WM, but they guide more towards the diagnosis of IgM myeloma. The biochemical profile can also provide data of hemolysis and/or other organ lesions, related or not to WM.21
Viscosity is usually increased in WM because in serum and plasma it is mainly determined by proteins. Among them, IgM has one of the highest molecular weight (950 kDa), with a high length/width ratio and the possibility of pentamerize.25 This provides a very high intrinsic viscosity to IgM, being responsible of plasma viscosity in a higher way than any other immunoglobulin. Thus, while 5000 mg/L of IgM easily promote HVS, an IgA component would require a serum level higher than 7000 mg/dL.
Serum and Urinary Proteinogram
Total proteins are increased by IgM paraprotein. Electrophoresis reveals the existence of a monoclonal IgM band that must be identified by immunofixation. Unlike MM, the levels of polyclonal Ig (IgG and IgA) are usually normal (immunoparesis is rare in WM),1 but this could change with novel tests, such as free Heavy/Light chain assessment.38 Finally, 30–50% of WM patients present with Bence Jones proteinuria, generally low (<2 g/24 hours).
The determination of serum-free light chains (sFLC) is a useful study in MW, since it avoids interpretation problems derived from the polymerization of the IgM chains. sFLC assessment has a prognostic value, because patients with >60 mg/L of the involved light chain have less hemoglobin (Hb) and more Beta2microglobulin (B2M),39 and patients with >80 mg/L of involved light chain had more progressive disease and shorter treatment-free survival.38,40 The Heavy/Light chain (HLC) test could help to identify cases with special risk of progression.38
Detection of Tumor Traits in Peripheral Blood
Flow cytometry has demonstrated that WM cells can be detected in PB, but the efficacy is lower than that we can observe in BM.41 The MYD88L265Pcan also be detected in PB, but we need to use advanced techniques to increase the detection capacity, for instance by using CD19+ selected cells, cell free DNA (liquid biopsy)42 or droplet digital PCR.43 Probably, the most efficient approach could be a combination of cfDNA and ddPCR, although we cannot forget that the presence of the mutation is not sufficient for WM diagnosis, while the absence of MYD88L265P does not completely excludes it.
Histology
The 2008 World Health Organization (WHO) classification defines LPL as a B-cell lymphoid neoplasm composed of small B lymphocytes, lymphoplasmacytoid cells and plasma cells, usually involving BM and sometimes lymph nodes and spleen, which does not fulfill the criteria for any of the other small B-cell lymphoid neoplasms.2 Thus, LPL remains an exclusionary diagnosis because there are no unique and uniform clinically applicable features that characterize the small lymphoid cells or the plasma cells.44 LPL is characterized by BM infiltration of small B lymphocytes with a predominately intertrabecular pattern. In addition, an interstitial, nodular or diffuse pattern can be observed. A paratrabecular pattern of infiltration is unusual, which help to distinguish LPL from other subsets of lymphoma, such as MZL that exhibits this paratrabecular pattern much more frequently.21 For the diagnosis of WM, variable numbers of plasmacytoid lymphocytes and plasma cells, often with positive periodic acid–Schiff intranuclear pseudoinclusions (Dutcher bodies), must also be present.44
BM biopsy (Table 2) shows the presence of light chain restricted lymphocytes, lymphoplasmacytoid and plasma cells, in which monoclonality is easy to be demonstrated by molecular or flow cytometric techniques in simple BM aspirates.45 A typical feature of LPL is the presence of prominent mast cells that are often recognized in the spicules of aspirate smears with Giemsa stain or in the histologic sections with tryptase immunohistochemical stain. Finally, LPL may also be associated with immunoglobulin deposition, amyloid, or crystal storing histiocytosis.2
Outside the BM, the presence of LPL is not frequently seen. Lymphoid tissue infiltration is often difficult to be assessed, especially when differential diagnosis includes nodal MZL, splenic MZL, or other small B-cell neoplasms with plasmacytic differentiation. Three patterns of lymph node involvement by LPL have been described, the most classic being lymph nodes with intact sinuses, a relatively monotonous lymphoplasmacytic proliferation, small residual follicles without large germinal centers or prominent follicular colonization.44 In the spleen, LPL typically involves both the splenic white and red pulp with diffuse pattern. Periarteriolar aggregates of plasmacytoid cells, small lymphocytes, immunoblasts are consistent in LPL. Increased mast cells, and hemosiderin can also be seen. When BM is not involved and monoclonal IgM is not present (IgG/IgA/absent M-component), MYD88L265P mutation is less frequently seen.46,47
Large cells are uncommon in LPL-IgM unless histological transformation occurs. If transformation is suspected, a lymph node biopsy is recommended,48 especially after a PET-CT scan to select one of the most actives adenopathies from the metabolic point of view.49
Immunophenotypic Features
Immunophenotypic evaluation is of great value in the differential diagnosis of B-cell lymphoproliferative disorders and must be performed on BM samples (Table 2). The immunophenotypic profile of lymphoplasmacytic cells in WM expresses pan B-cell antigens CD19, CD20, CD22, and CD79, as well as CD25+, CD27+ and light chain-restricted surface IgM.50,51 It is also typically characterized by the lack of expression of CD5, CD10, CD11c, CD23 and CD103, which is in contrast with most other mature lymphoid malignancies, except MZL. However, several studies have observed that in 10–20% of WM cases tumor cells can express CD5, CD23 or CD10.52–54
Multiparametric flow cytometric immunophenotyping also demonstrates the presence of monoclonal plasma cells (PC) with the same restricted light-chain expression as the lymphoplasmacytic population.50 The antigenic profile of these plasma cells shows a phenotypic profile similar to normal PC and clearly different from that of myeloma patients: CD138+++, CD19+, CD45+, CD38+, CD56–, with high proportion of PC with the same light-chain restriction as the IgM monoclonal component seen in the serum.50 This means that the genetic program of the final B-cell differentiation is altered in WM cells, which can explain why Robert et al observed the abnormal co-expression of CD138 and PAX5 in 23% of plasma cells in samples from a series of LPL patients. This finding contrasts with MZL, where PAX5 cells (B lymphocytes) completely lack of CD138, while CD138+ cells (plasma cells) have switch off PAX5 expression.50
Cytogenetic Features
Conventional karyotypic analysis is not mandatory for the routine diagnostic assessment of WM patients as it is very difficult to obtain tumor metaphases in vitro. 6q deletion (usually from 6q21 to 6q23), that can be seen by fluorescence in situ hybridization (FISH), is the most common cytogenetic abnormality reported in 30–60% of WM patients.55–58 Such deletions are associated with more aggressive IgM gammopathies and a high probability of symptomatic transformation.58 Chromosome 6q deletions involve genes that modulate Nuclear Factor-κB (NF-KB), BCL2, Bruton Tyrosine Kinase (BTK), apoptosis and differentiation, which could help to explain why this deletion is associated with poor clinical features and a higher risk of symptomatic evolution. Other genes whose deletion could justify such relationship are genes with important regulatory functions, such as IBTK, HIVEP2, and FOXO3.8
FISH studies may be also be useful to detect some abnormalities that could help in the differential diagnosis, such as the detection of the t(11;14), very frequent in IgM myeloma60,61 and virtually absent in WM.46 In addition, it can detect several other abnormalities that are in common to other B-cell lymphoproliferative disorders: del(13q14) in 3–16% of cases, del(17p23) in 7–15%, del(11q22) in 8%, +8 in 11%, and +4 in 8%.57,59–61
Molecular Genetics
MYD88 Mutations
Whole genome sequencing (WGS) in WM patients has identified several somatic mutations in WM.8 However, a mutation in the myeloid differentiation primary response 88 (MYD88) gene, more specifically, the MYD88L265P mutation, is now considered the hallmark of WM (and LPL), since it is present in more than 90% of the patients.6 MYD88 is a protein adapter that activates the IL-1 receptor signaling pathway via interleukin-1 promoting BTK constitutive activation. BTK is a kinase with a critical role in B-cell receptor (BCR) signaling that regulates immune response, cell proliferation and cell death and seems to be directly related to B-cell lymphoproliferative disorders as WM. Although rarely, MYD88L265P is also present in other B cells disorders, so it is not completely specific of WM, by now. Some WM patients in whom MYD88L265P mutation is negative can have a MYD88 variant but placed in a different point respect the L265 position.62 Thus, some series reach a 100%41 of incidence of WM patients and 87% of IgM-MGUS patients.46 These frequencies, together with the differential response to first generation BTK inhibitors hast prompted to some authors to consider that wild type MYD88 (MYD88WT) WM could be a different disease.63 In addition, MYD88WT WM patients have a different genomic landscape that shows other NF-κB activating mutations, impart epigenomic dysregulation, or impair DNA damage repair (DDR). These patients show a shorter survival, especially if they have DDR mutations and a higher incidence diffuse large B-cell lymphoma transformation when compared with WM patients who carry the conventional MYD88 mutation.64
The MYD88L265P mutation is consider pathogenic in WM, since it leads to an amino-acid change in the MYD88 protein and is present in most cells and cases. It is an activating change and triggers interleukin-1 receptor–associated kinase (IRAK), Bruton’s tyrosine kinase (BTK), and hematopoietic cell kinase (HCK) growth and survival signaling, that in turn activate NF-KB-p65 dependent nuclear translocation and malignant cell survival.64 The presence of MYD88 mutation in IgM-MGUS reveals its role as a potential early oncogenic factor, but most of these IgM-MGUS patients never evolve into WM or other lymphoproliferative disorders, so this mutation cannot be considered as a unique pathogenic factor in WM.
The method and DNA source for the MYD88 mutation detection can affect the result.65 There is no standardized method for MYD88L265P mutation detection and several methods can be used with various approaches and detection limits. This justify why the detection rate can range between 40% and 87% in IgM MGUS,46,66 and from 71% to 100% in WM.6,46,65 Currently, most authors recommend allele-specific polymerase chain reaction (AS-PCR)67 which usually provides sensitivities beyond 1% and are sufficient for most diagnostic purposes,68 although droplet digital PCR are becoming more and more popular.43
CXCR4 Mutations
In 30–40% of patients with WM, tumor cells have somatic activating mutations in the C-terminal domain of C-X-Chemokine receptor type 4 (CXCR4) gene, similar to nonsense (NS) and frameshift (FS) germline mutations found in the “warts, hypogammaglobulinemia, infections and myelokathexis (WHIM) syndrome.8,69 CXCR4 is a classical G protein coupled receptor (GPCRs) located on chromosome 2 and acts as a conventional chemokine receptor, the normal ligand of CXCR4 is CXCL2 who coupled to CXCR4 initiate intracellular signaling cascades that control chemotaxis, migration, proliferation and stemness.70 CXCR4 mutations in WM were the first ever reported in human cancer with a wide range of possibilities, which makes difficult to develop of a PCR-based diagnostic test. The most frequent (50%) mutated region is the amino acid S338X at position 1013 with nucleotide changes C > G, and C > A, both resulting in a stop codon. The second one is a S338 frameshift mutation in 21% of the cases, but there are more than 40 different mutations described.71 Most of such mutations introduce a premature stop codon or a frameshift that cuts the end of the CXCR4 protein avoiding its metabolization, thus prolonging its effects.
This over function of CXCR4 explains why patients with such mutations have a higher BM disease burden, higher serum immunoglobulin M levels and more cases of symptomatic hyperviscosity.7 Asymptomatic patients also present mutations in CXCR4, but this fact seems to increase the risk of progression into symptomatic disease. In WM patients treated with a BTK-inhibitor the presence of CXCR4 mutations reduce the therapeutic effectiveness, and in vitro CXCR4 mutant cells treated with BTK-inhibitor are rescued by CXCL12 from apoptosis.71 Accordingly, CXCR4 mutation emerges as a relevant molecular abnormality to be taken into account in WM not only for diagnosis, but also as a potential actionable target.
Other Somatic Mutations
Near 50% of WM harbor recurrent mutations in other genes.72–74 Somatic mutations in ARID1A are present in 17% of patients with WM, including nonsense and frameshift variants. Although this frequency has not been reproduced by others,74 patients with ARID1A mutations are presumed to have more advanced disease which concurs with the fact that it is located at chromosome 6q, as its homolog ARID1B. In addition, ARID1A could modulate TP53 and is thought to act as an epigenetic tumor suppressor in ovarian cancer.67 TP53 mutations are rare in WM (<5%), but they are associated with poor survival.67
Mutations in CD79A and CD79B can be found in 8–12% of patients with WM.67,73,74 Both are components of the BCR pathway and can form heterodimers with each other, so activating mutations of these components could contribute to the chronic BCR signaling observed in WM cells.67 In one study, mutations in CD79A and CD79B were nearly exclusive of CXCR4 mutations, suggesting that CD79A/B mutations may also have an independent role in facilitating mutated MYD88-directed progression in WM.75 In addition, CD79B mutations have been associated with disease transformation in a some WM patients.76 The number of detectable genetic abnormalities in IgM monoclonal gammopathies is increased as the aggressiveness of the disease is increased. In a recent study evaluating the 12 most frequently mutated genes in WM, the percentage of patients with alterations was increasing as the monoclonal gammopathy progressed: 21% in IgM MGUS, 35% in AWM and 50% in symptomatic WM.74
Risk Assessment & Prognosis
According to the type of IgM monoclonal gammopathy, we have different classifications of risk assessment that are specific for the different stages.
Risk Assessment in IgM-MGUS
When we are in front of a patient with a MGUS, the first thing that we have to consider is that, among the different heavy chain isotypes of the heavy chain that can exist, IgM is the one with the highest risk of malignant transformation.15 These patients can evolve to non-Hodgkin lymphoma, WM, AL amyloidosis or CLL with a risk 11-fold higher than a matched control population. Probability of progression is higher for patients with an abnormal serum-free light-chain ratio (outside normal limits) and a high serum monoclonal protein level (≥1.5 g/dL), especially if both are associated. Thus, the 20 year probability of malignant progression is 55% when both risk factors are present, 41% if there is one, and 19% there is none of them.15
Risk Assessment in Asymptomatic WM
When a patient fits with the AWM criteria (Table 1), the risk assessment to score the probability of transformation into a symptomatic disease can be done through several systems, but the most recent one is based on four risk factors: Immunoglobulin M ≥4500 mg/L, BM lymphoplasmacytic infiltration ≥70%, β2microglobulin ≥4.0 mg/dL, and albumin ≤3.5 g/dL.77 With these risk factors, the current progression risk-classification identifies three groups of asymptomatic WM with a different median time to develop symptomatic disease: patients whose risk scores were below the first quartile (low risk, have a median time from diagnosis to transformation of 9.3 years), patients whose risk scores were in the interquartile range (intermediate risk, 4.8 years) and those whose risk scores were above the third quartile (high risk, 18 years).77 This new classification, whose access is facilitated through a ready-to-use webpage tool (http://www.awmrisk.com) could be of help to identify patients with high-risk AWM who may need closer follow-up or benefit from early intervention.
Risk Assessment in Symptomatic WM
Finally, if our patient fits the criteria of symptomatic WM, we should use the International Prognostic Scoring System (IPSS)78 that includes five variables associated with poor prognosis:
Anemia: Hb ≤115 g/L,
Thrombopenia: ≤100×109/L,
Monoclonal component: >70 g/L,
Age: >65 years
β2-microglobulin: >3 mg/L.
Using these risk factors we can construct three risk-groups:
- Low risk: when there is no one risk-factor or only one, excluding the age. The median survival is 14y
- Intermediate risk: if the patient is older than 65 or there are 2 risk-factors. The median survival is 8y
- High risk: if there are more than two risk factors. The median survival is 4y
Since this IPSS is based on patients treated with old-fashioned therapies, more recently Kastritis et al developed a revised score (rIPSSWM) that found four parameters associated with poor prognosis: age (66–75 and ≥76 years), β2microglobulin ≥4 mg/L, serum albumin <3.5 gr/dL, and LDH ≥250 IU/L (ULN < 225). Accordingly, five different prognostic groups were constructed with a 3-year WM-related death rate of 0, 10, 14, 38, and 48% (p < 0.001) and 10-year survival rate of 84%, 59%, 37%, 19%, and 9%. This system includes two extremes: very-low risk and very-high risk groups. It is conceivable to recommend the modification of current strategies a better management approach in them.5 However, we should take into account that IPSSWM and rIPSSWM, are merely based on clinical and biochemical parameters, lacking on molecular and genetic characteristics that could be crucial for the future risk assessment in WM.18
Summary
Clinical-biological work-up in WM must include a deep pathological evaluation that help in the correct diagnostic assignment of the patient in order to correctly prescribe the patient treatment. Diagnosis should be precise and identify what specific IgM monoclonal gammopathy is present in our patient, including as much as immunophenotypic and genetic information is possible to be obtained, including bone biopsy and BM cell data. In addition, a prognostic risk group assignment must be provided. Both, genomic and prognostic information will be used for a correct therapeutic indication. According to the 10th IWWM updated consensus, first line of therapy for newly diagnosed symptomatic patients WM could include alkylating drugs (Cyclophosphamide and Bendamustine) plus Rituximab, proteasome inhibitors (Bortezomib, Carfilzomib) plus Rituximab, or BTK inhibitors (Ibrutinib) with or without Rituximab. Second and further therapeutic lines will depend on the former response duration and toxicities, making possible the reuse of previous regimens or changing into others. The treatment initiation should be strictly guided by a correct diagnosis (especially differentiating between symptomatic and asymptomatic forms) knowing that that responses to novel BTK inhibitors could be influenced by the molecular characteristics of the disease.79 This reinforces the necessity of an exquisite evaluating approach for these patients as we have pointed out in this review.
Acknowledgments
This work was partially supported by the Instituto de Salud Carlos III (ISCIII), Spanish Ministry of Economy and Competitiveness PI18/01866, CIBERONC-CB16/12/00233, FUCALHH 2015, Cancer Research UK [C355/A26819], FC AECC and AIRC under the “Accelerator Award Program”, and “Una manera de hacer Europa” (Innocampus; CEI-2010-1-0010). The authors thank C. Jiménez, M.E. Sarasquete, M. Alcoceba & M.C. Chillón for their support.
Disclosure
The authors declare that they have no potential conflicts of interest.
References
1. Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of waldenstrom’s macroglobulinemia: consensus panel recommendations from the second international workshop on waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30(2):110–115. doi:10.1053/sonc.2003.50082
2. Swerdlow S, Campo E, Pileri S, et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi:10.1182/blood-2016-01-643569
3. Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. doi:10.1182/blood-2011-01-293050
4. Hunter Z, Xu L, Zhou Y, et al. Whole-genome sequencing results from 30 patients with waldenstrom’s macroglobulinemia. Blood. 2011;118(21):434. doi:10.1182/blood.V118.21.434.434
5. Lynch RC, Gratzinger D, Advani RH. Clinical impact of the 2016 update to the WHO lymphoma classification. Curr Treat Options Oncol. 2017;18(7):45. doi:10.1007/s11864-017-0483-z
6. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367(9):826–833. doi:10.1056/NEJMoa1200710
7. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123(18):2791–2796. doi:10.1182/blood-2014-01-550905
8. Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637–1646. doi:10.1182/blood-2013-09-525808
9. Pophali PA, Bartley A, Kapoor P, et al. Prevalence and survival of smouldering Waldenström macroglobulinaemia in the United States. Br J Haematol. 2019;184(6):1014–1017. doi:10.1111/bjh.15201
10. García-Sanz R, Montoto S, Torrequebrada A, et al. Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases. Br J Haematol. 2001;115(3):575–582. doi:10.1046/j.1365-2141.2001.03144.x
11. Kristinsson SY, Koshiol J, Goldin LR, et al. Genetics- and immune-related factors in the pathogenesis of lymphoplasmacytic lymphoma/ Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma. 2009;9(1):23–26. doi:10.3816/CLM.2009.n.004
12. Treon SP, Tripsas C, Hanzis C, et al. Familial disease predisposition impacts treatment outcome in patients with Waldenström macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2012;12(6):433–437. doi:10.1016/j.clml.2012.08.006
13. Giordano TP, Henderson L, Landgren O, et al. Risk of non-Hodgkin lymphoma and lymphoproliferative precursor diseases in US veterans with hepatitis C virus. JAMA. 2007;297(18):2010–2017. doi:10.1001/jama.297.18.2010
14. Mailankody S, Landgren O. Monoclonal gammopathy of undetermined significance and Waldenström’s macroglobulinemia. Best Pract Res Clin Haematol. 2016;29(2):187–193. doi:10.1016/j.beha.2016.08.015
15. Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378(3):241–249. doi:10.1056/NEJMoa1709974
16. Swerdlow SH, Kuzu I, Dogan A, et al. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Arch. 2016;468(3):259–275. doi:10.1007/s00428-015-1858-9
17. Sabattini E, Bacci F, Sagramoso C, Pileri SA. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica. 2010;102(3):83–87.
18. Dimopoulos MA, Kastritis E. How I treat Waldenström macroglobulinemia. Blood. 2019;134(23):2022–2035. doi:10.1182/blood.2019000725
19. Ciccarelli BT, Patterson CJ, Hunter ZR, et al. Hepcidin is produced by lymphoplasmacytic cells and is associated with anemia in Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011;11(1):160–163. doi:10.3816/CLML.2011.n.038
20. Samba-Mondonga M, Calvé A, Mallette FA, Santos MM. MyD88 regulates the expression of SMAD4 and the iron regulatory hormone hepcidin. Front Cell Dev Biol. 2018;6:105. doi:10.3389/fcell.2018.00105
21. Castillo JJ, Garcia‐Sanz R, Hatjiharissi E. Recommendations for the diagnosis and initial evaluation of patients with waldenström macroglobulinaemia: a task force from the 8th international workshop on waldenström macroglobulinaemia. Br J Haematol. 2016;175:77.
22. Banwait R, Aljawai Y, Cappuccio J, et al. Extramedullary Waldenström macroglobulinemia. Am J Hematol. 2015;90(2):100–104. doi:10.1002/ajh.23880
23. Castillo JJ, Treon SP. How we manage Bing-Neel syndrome. Br J Haematol. 2019;187(3):277–285. doi:10.1111/bjh.16167
24. Muñiz C, Martín-Martín L, López A, et al. Contribution of cerebrospinal fluid sCD19 levels to the detection of CNS lymphoma and its impact on disease outcome. Blood. 2014;123(12):1864–1869. doi:10.1182/blood-2013-11-537993
25. Gertz MA. Acute hyperviscosity: syndromes and management. Blood. 2018;132(13):1379–1385. doi:10.1182/blood-2018-06-846816
26. Gustine JN, Meid K, Dubeau T, et al. Serum IgM level as predictor of symptomatic hyperviscosity in patients with Waldenström macroglobulinaemia. Br J Haematol. 2017;177(5):717–725. doi:10.1111/bjh.14743
27. Muchtar E, Magen H, Gertz MA. How I treat cryoglobulinemia. Blood. 2017;129(3):289–298. doi:10.1182/blood-2016-09-719773
28. Saadoun D, Sellam J, Ghillani-Dalbin P, Crecel R, Piette JC, Cacoub P. Increased risks of lymphoma and death among patients with non-hepatitis C virus-related mixed cryoglobulinemia. Arch Intern Med. 2006;166(19):2101–2108. doi:10.1001/archinte.166.19.2101
29. Berentsen S. New insights in the pathogenesis and therapy of cold agglutinin-mediated autoimmune hemolytic anemia. Front Immunol. 2020;11:590. doi:10.3389/fimmu.2020.00590
30. D’Sa S, Kersten MJ, Castillo JJ, et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol. 2017;176(5):728–742. doi:10.1111/bjh.14492
31. Chaudhry HM, Mauermann ML, Rajkumar SV. Monoclonal gammopathy-associated peripheral neuropathy: diagnosis and management. Mayo Clin Proc. 2017;92(5):838–850. doi:10.1016/j.mayocp.2017.02.003
32. Le Cann M, Bouhour F, Viala K, et al. CANOMAD: a neurological monoclonal gammopathy of clinical significance that benefits from B-cell-targeted therapies. Blood. 2020;136(21):2428–2436. doi:10.1182/blood.2020007092
33. Castillo JJ, Garcia-Sanz R, Hatjiharissi E, et al. Recommendations for the diagnosis and initial evaluation of patients with waldenström macroglobulinaemia: a task force from the 8th international workshop on waldenström macroglobulinaemia. Br J Haematol. 2016;175(1):77–86. doi:10.1111/bjh.14196
34. Sidana S, Larson DP, Greipp PT, et al. IgM AL amyloidosis: delineating disease biology and outcomes with clinical, genomic and bone marrow morphological features. Leukemia. 2020;34(5):1373–1382. doi:10.1038/s41375-019-0667-6
35. Milani P, Merlini G. Monoclonal IgM-related AL amyloidosis. Best Pract Res Clin Haematol. 2016;29(2):241–248. doi:10.1016/j.beha.2016.08.013
36. Sachchithanantham S, Roussel M, Palladini G, et al. European collaborative study defining clinical profile outcomes and novel prognostic criteria in monoclonal immunoglobulin m-related light chain amyloidosis. J Clin Oncol. 2016;34(17):2037–2045. doi:10.1200/JCO.2015.63.3123
37. Kastritis E, Leblond V, Dimopoulos MA, et al. Waldenström’s macroglobulinaemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(5):860–862. doi:10.1093/annonc/mdy466
38. Andrade-Campos M, Murillo-Flórez I, García-Sanz R, Giraldo P. Immunoparesis in IgM gammopathies as a useful biomarker to predict disease progression. Clin Chem Lab Med. 2017;55(10):1598–1604. doi:10.1515/cclm-2016-0748
39. Leleu X, Moreau AS, Weller E, et al. Serum immunoglobulin free light chain correlates with tumor burden markers in Waldenstrom macroglobulinemia. Leuk Lymphoma. 2008;49(6):1104–1107. doi:10.1080/10428190802074619
40. Itzykson R, Le Garff-tavernier M, Katsahian S, Diemert MC, Musset L, Leblond V. Serum-free light chain elevation is associated with a shorter time to treatment in Waldenstrom’s macroglobulinemia. Haematologica. 2008;93(5):793–794. doi:10.3324/haematol.12107
41. Xu L, Hunter ZR, Yang G, et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121(11):2051–2058. doi:10.1182/blood-2012-09-454355
42. Bagratuni T, Ntanasis-Stathopoulos I, Gavriatopoulou M, et al. Detection of MYD88 and CXCR4 mutations in cell-free DNA of patients with IgM monoclonal gammopathies. Leukemia. 2018;32(12):2617–2625. doi:10.1038/s41375-018-0197-7
43. Drandi D, Genuardi E, Dogliotti I, et al. Highly sensitive mutation detection by droplet digital polymerase chain reaction in Waldenström macroglobulinemia. Haematologica. 2018;103(6):1029–1037. doi:10.3324/haematol.2017.186528
44. Lin P, Molina TJ, Cook JR, Swerdlow SH. Lymphoplasmacytic lymphoma and other non-marginal zone lymphomas with plasmacytic differentiation. Am J Clin Pathol. 2011;136(2):195–210. doi:10.1309/AJCP8FOIVTB6LBER
45. Paiva B, Montes MC, García-Sanz R, et al. Multiparameter flow cytometry for the identification of the Waldenström’s clone in IgM-MGUS and Waldenström’s Macroglobulinemia: new criteria for differential diagnosis and risk stratification. Leukemia. 2014;28(1):166–173. doi:10.1038/leu.2013.124
46. Jiménez C, Sebastián E, Chillón MC, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenström’s macroglobulinemia. Leukemia. 2013;27(8):1722–1728. doi:10.1038/leu.2013.62
47. Hamadeh F, MacNamara SP, Aguilera NS, Swerdlow SH, Cook JR. MYD88 L265P mutation analysis helps define nodal lymphoplasmacytic lymphoma. Mod Pathol. 2015;28(4):564–574. doi:10.1038/modpathol.2014.120
48. Durot E, Kanagaratnam L, Zanwar S, et al. A prognostic index predicting survival in transformed Waldenström macroglobulinemia. Haematologica. 2020. doi:10.3324/haematol.2020.262899
49. Durot E, Tomowiak C, Michallet AS, et al. Transformed Waldenström macroglobulinaemia: clinical presentation and outcome. A multi-institutional retrospective study of 77 cases from the French innovative leukemia organization (FILO). Br J Haematol. 2017;179(3):439–448. doi:10.1111/bjh.14881
50. García-Sanz R, Jiménez C, Puig N, et al. Origin of Waldenstrom’s macroglobulinaemia. Best Pract Res Clin Haematol. 2016;29(2):136–147. doi:10.1016/j.beha.2016.08.024
51. Paiva B, Corchete LA, Vidriales MB, et al. The cellular origin and malignant transformation of Waldenström macroglobulinemia. Blood. 2015;125(15):2370–2380. doi:10.1182/blood-2014-09-602565
52. Morice WG, Chen D, Kurtin PJ, Hanson CA, McPhail ED. Novel immunophenotypic features of marrow lymphoplasmacytic lymphoma and correlation with Waldenström’s macroglobulinemia. Mod Pathol. 2009;22(6):807–816. doi:10.1038/modpathol.2009.34
53. Konoplev S, Medeiros LJ, Bueso-Ramos CE, Jorgensen JL, Lin P. Immunophenotypic profile of lymphoplasmacytic lymphoma/Waldenström macroglobulinemia. Am J Clin Pathol. 2005;124(3):414–420. doi:10.1309/3G1XDX0DVHBNVKB4
54. San Miguel JF, Vidriales MB, Ocio E, et al. Immunophenotypic analysis of Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30(2):187–195. doi:10.1053/sonc.2003.50074
55. Ocio EM, Schop RF, Gonzalez B, et al. 6q deletion in Waldenström macroglobulinemia is associated with features of adverse prognosis. Br J Haematol. 2007;136(1):80–86. doi:10.1111/j.1365-2141.2006.06389.x
56. Braggio E, Fonseca R. Genomic abnormalities of Waldenström macroglobulinemia and related low-grade B-cell lymphomas. Clin Lymphoma Myeloma Leuk. 2013;13(2):198–201. doi:10.1016/j.clml.2013.02.015
57. Schop RF, Fonseca R. Genetics and cytogenetics of Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30(2):142–145. doi:10.1053/sonc.2003.50075
58. García-Sanz R, Dogliotti I, Zaccaria GM, et al. 6q deletion in Waldenström macroglobulinaemia negatively affects time to transformation and survival. Br J Haematol. 2020;192:843.
59. Ocio EM, Hernandez JM, Mateo G, et al. Immunophenotypic and cytogenetic comparison of Waldenstrom’s macroglobulinemia with splenic marginal zone lymphoma. Clin Lymphoma. 2005;5(4):241–245. doi:10.3816/CLM.2005.n.007
60. Chang H, Samiee S, Li D, et al. Analysis of IgH translocations, chromosome 13q14 and 17p13.1(p53) deletions by fluorescence in situ hybridization in Waldenstrom’s macroglobulinemia: a single center study of 22 cases. Leukemia. 2004;18(6):1160–1162. doi:10.1038/sj.leu.2403369
61. Nguyen-Khac F, Lambert J, Chapiro E, et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenström’s macroglobulinemia. Haematologica. 2013;98(4):649–654. doi:10.3324/haematol.2012.070458
62. Treon SP, Xu L, Hunter Z. MYD88 Mutations and Response to Ibrutinib in Waldenström’s Macroglobulinemia. N Engl J Med. 2015;373(6):584–586. doi:10.1056/NEJMc1506192
63. Gracia-sanz R. WM, MYD88, and CXCR4: following the thread. J Am Society Hematol. 2016;128:746.
64. Hunter ZR, Xu L, Tsakmaklis N, et al. Insights into the genomic landscape of MYD88 wild-type Waldenström macroglobulinemia. Blood Adv. 2018;2(21):2937–2946. doi:10.1182/bloodadvances.2018022962
65. Varettoni M, Arcaini L, Zibellini S, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom’s macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121(13):2522–2528. doi:10.1182/blood-2012-09-457101
66. Poulain S, Roumier C, Decambron A, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121(22):4504–4511. doi:10.1182/blood-2012-06-436329
67. Treon SP, Xu L, Guerrera ML, et al. Genomic landscape of waldenström macroglobulinemia and its impact on treatment strategies. J Clin Oncol. 2020;38(11):1198–1208. doi:10.1200/JCO.19.02314
68. Jiménez C, Chillón M, Balanzategui A, et al. Detection of MYD88 L265P mutation by real-time allele-specific oligonucleotide polymerase chain reaction. Appl Immunohistochem Mol Morphol. 2014;22(10):768–773. doi:10.1097/PAI.0000000000000020
69. Schmidt J, Federmann B, Schindler N, et al. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol. 2015;169(6):795–803. doi:10.1111/bjh.13361
70. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16(11):2927–2931. doi:10.1158/1078-0432.CCR-09-2329
71. Kaiser LM, Hunter ZR, Treon SP, Buske C. CXCR4 in Waldenström’s Macroglobulinema: chances and challenges. Leukemia. 2021;35(2):333–345. doi:10.1038/s41375-020-01102-3
72. Hunter ZR, Yang G, Xu L, Liu X, Castillo JJ, Treon SP. Genomics, signaling, and treatment of waldenström macroglobulinemia. J Clin Oncol. 2017;35(9):994–1001. doi:10.1200/JCO.2016.71.0814
73. Varettoni M, Boveri E, Zibellini S, et al. Clinical and molecular characteristics of lymphoplasmacytic lymphoma not associated with an IgM monoclonal protein: a multicentric study of the Rete Ematologica Lombarda (REL) network. Am J Hematol. 2019;94(11):1193–1199. doi:10.1002/ajh.25600
74. Jiménez C, Prieto-Conde MI, García-álvarez M, et al. Unraveling the heterogeneity of IgM monoclonal gammopathies: a gene mutational and gene expression study. Ann Hematol. 2018;97(3):475–484.
75. Poulain S, Roumier C, Venet-Caillault A, et al. Genomic landscape of CXCR4 mutations in waldenström macroglobulinemia. Clin Cancer Res. 2016;22(6):1480–1488. doi:10.1158/1078-0432.CCR-15-0646
76. Jiménez C, Alonso-álvarez S, Alcoceba M, et al. From Waldenström’s macroglobulinemia to aggressive diffuse large B-cell lymphoma: a whole-exome analysis of abnormalities leading to transformation. Blood Cancer J. 2017;7(8):e591. doi:10.1038/bcj.2017.72
77. Bustoros M, Sklavenitis-Pistofidis R, Kapoor P, et al. Progression risk stratification of asymptomatic waldenström macroglobulinemia. J Clin Oncol. 2019;37(16):1403–1411. doi:10.1200/JCO.19.00394
78. Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood. 2009;113(18):4163–4170. doi:10.1182/blood-2008-08-174961
79. Castillo JJ, Advani RH, Branagan AR, et al. Consensus treatment recommendations from the tenth international workshop for waldenström macroglobulinaemia. Lancet Haematol. 2020;7(11):e827–e837. doi:10.1016/S2352-3026(20)30224-6
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.