Back to Journals » Clinical Interventions in Aging » Volume 13
Von Willebrand Disease in the elderly: clinical perspectives
Authors Chapin J
Received 9 May 2018
Accepted for publication 12 July 2018
Published 31 August 2018 Volume 2018:13 Pages 1531—1541
DOI https://doi.org/10.2147/CIA.S136931
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
John Chapin
Department of Hematology, Shire, Lexington, MA, USA
Abstract: Von Willebrand disease (VWD) is an inherited bleeding disorder that affects up to 1% of the population. In most cases, VWD results from a mutation in the von Willebrand Factor (VWF) gene, which alters the amount and function of VWF, a key glycoprotein in both primary and secondary hemostasis. A comprehensive analysis of patients with VWD should include VWF activity, antigen levels, platelet function, and a careful bleeding history. Treatment options include antifibrinolytics, desmopressin, and VWF replacement therapy. VWF levels fluctuate due to age, stress, environmental exposures, and pharmacologic treatment. Treatment guidelines exist to treat and prevent bleeding for patients undergoing surgery and medical procedures, but often these must be reevaluated in the setting of age-related comorbidities including cardiovascular events, venous thrombosis, and malignancy. In addition, many age-related complications are associated with a secondary acquired von Willebrand syndrome (AVWS), including malignancies, hypothyroidism, cardiovascular diseases, and cardiac replacement devices. The current literature is limited by a lack of older patients in clinical trials. Larger studies are needed to determine if age-related comorbidities affect VWD patients at different frequencies than the general elderly population. There is also a significant need for registry-based studies to evaluate many age-related comorbidities in VWD patients.
Keywords: von Willebrand disease, acquired von Willebrand syndrome, bleeding disorders, aging
Introduction
Von Willebrand disease (VWD) was initially described in 1924 by Erik von Willebrand in a small family cohort with unusual bleeding and deaths resulting from menorrhagia. In the century since the initial clinical pathology was described, multiple genetic and phenotypic variants of VWD have been identified and treatment methods have been developed. VWD is now characterized as an autosomal bleeding disorder with a prevalence in up to 1% of the population,1,2 although the symptomatic prevalence is approximately 1:10,000.3–5 The bleeding patterns of VWD most often involve bleeding on mucosal surfaces such as epistaxis, dental bleeding, menorrhagia, and significant bruising. Bleeding patterns are different based on age. In children, bleeding is most often characterized by bruising and epistaxis, while adults more commonly develop hematomas, menorrhagia, and wound bleeding.6,7 The severity of bleeding depends greatly on the quantity and quality of von Willebrand factor (VWF). Some patients experience only minor bleeding with procedures or menses, while others have significant regular hemorrhages requiring prophylaxis with hemostatic agents.8 As they age, patients with VWD experience age-related health comorbidities and may require interventions for heart disease, cancer, orthopedic fractures, and other diseases. In addition, changes in hemostasis occur as part of normal aging and may influence the risk of bleeding and thrombosis in these patients. Increases in procoagulant factors and fibrinogen, decreases in endogenous anticoagulant factors, and altered platelet activation have all been described in aging.9
The purpose of this review is to describe age-related complications in VWD, how they impact the disease, and how these can be mitigated.
VWD overview
VWD is a disorder of VWF, a large multimeric glycoprotein with molecular weights that range from approximately 500–20,000 kDa. VWF binds to the glycoprotein GPIbα on the surface of platelets that results in platelet tethering in addition to platelet adherence to vessel wall collagen.10 It also functions as a carrier molecule for FVIII, protecting FVIII from proteolysis and prolonging its half-life in circulation.10,11 Mutations in VWF have been described that affect platelet binding, collagen binding, secretion, and synthesis. However, mutations do not always identify patients with VWD. Approximately 30% of patients with type 1 VWD do not have a candidate mutation identified.12,13 Conversely, low levels of VWF have been identified in patients without a clinical bleeding disorder, making the diagnosis of VWD especially challenging in less severe cases.14 The current classification systems of VWD involve a description of the VWF antigen and activity levels, platelet binding tests, VWF-propeptide levels, and VWF multimer analysis (Table 1).15,16 It should also be noted that many types of acquired von Willebrand syndrome (AVWS) exist and can be related to diseases more commonly encountered in the elderly, which is described below in more detail.
 | Table 1 Summary of classification of VWD (based on NHLBI classification) |
The genetics and synthesis of VWF
The genetics of VWD are complicated and not fully predictive of bleeding phenotypes. The VWF molecule consists of 178 kb located on the short arm of chromosome 12 and contains 52 exons. The mature secreted glycoprotein consists of 2,050 amino acids and 4 types of repeat domains to which binding to FVIII, platelet glycoprotein GPIbα, collagen, and integrin αIIbβ3 have been mapped.17 As a functional assay, VWF ristocetin cofactor (VWF:RCo) levels <30 U/dL are strongly associated with a clinical bleeding phenotype and the presence of an identified VWF mutation in type 1 VWD.12 Other genetic factors that influence VWF levels, including the “platelet type” of VWD18 (GP1BA mutation) and the ABO blood groups.19 Newer associations between VWF antigen levels and polymorphisms in non-VWF genes have been identified by genome wide association studies, including polymorphisms in secretory protein genes, such as STAB5, STX2, and STXBP5.20 Some of these variations correlate with patient reported bleeding.21,22 For example, annexin A2 (ANXA2) is involved in trafficking of Weibel–Palade bodies (WPBs) by a cAMP-dependent mechanism.23 ANXA2 is also a key mediator in the trafficking of VWF from the WPB to the endothelial cell surface, and its absence results in impaired endothelial secretion in response to histamine.24 ANXA2’s role as a modifier of clinical disease requires further research.
Both endothelial cells and megakaryocytes synthesize VWF. Endothelial cells store VWF in the WPB, and megakaryocytes store VWF in α-granules. Initial synthesis of VWF occurs in the endoplasmic reticulum, where VWF is synthesized as a propolypeptide. During activation, the signal peptide is cleaved, allowing VWF subunits to dimerize and enter the Golgi network. VWF then assembles into multimers that are directed to the storage organelles.25 The large and ultra-large multimers that are stored in WPBs or platelet α-granules are then secreted by either a constituent or agonist-controlled mechanism.26,27 In circulation, large VWF multimers are cleaved by the metalloprotease ADAMTS13 (A Disintegrin-like and Metalloprotease with Thrombospondin Type one repeat motifs, member 13) into smaller molecular forms.28 The importance of ADAMTS13 in pathophysiology is demonstrated in both congenital and acquired forms of thrombotic thrombocytopenic purpura (TTP), where failure of appropriate VWF multimer cleavage from a deficiency of ADAMTS13 can lead to vessel thrombosis, hemolytic anemia, and platelet consumption.29
Diagnosis and assessing bleeding severity in VWD
The diagnosis of VWD requires both laboratory and clinical evaluations (Table 1).16 An initial evaluation consists of prothrombin time and activated partial thromboplastin time, assessment of VWF activity (ristocetin cofactor; VWF:RCo), VWF antigen (VWF:Ag), and factor VIII activity. Further testing includes multimer analysis, the VWF-collagen binding assay, and VWF propeptide antigen levels, which can assist in refining the diagnosis. The clinician encountering a patient with a history of VWD will want to identify the VWD subtype, the bleeding phenotype, and obtain past and current coagulation studies of the patient. Bleeding may still be difficult to predict based on the laboratory analysis, and VWF:RCo can be poorly predictive of bleeding.14,30 Bleeding assessment scores have been utilized in both the hemophilia and VWD populations to address the limits of laboratory testing and more appropriately quantify the bleeding risk.31–33 Bleeding scores have the potential to predict bleeding phenotype, determine need for treatment, and distinguish between subtypes.34,35 Most bleeding scores are able to distinguish between healthy patients and those with symptomatic VWD.31,33 The International Society of Hemostasis and Thrombosis (ISTH) has also developed a bleeding assessment tool (BAT) that has been used in patients with bleeding disorders.36 Additional advances in laboratory testing, such as whole blood ristocetin-induced platelet aggregometry also have the potential to improve the diagnosis of VWD and inform the bleeding risk.37
At this time, there is no validated bleeding assessment score designed specifically for the elderly population, and although patients over 65 have been included in clinical studies, they have not been represented as a unique population. Development of a bleeding assessment score in elderly patients is an area for further exploration.
Treatment of VWD
Treatment of VWD is generally based on symptoms and bleeding severity. Most VWD patients fall into milder subtypes like VWD type 1, and thus most often require episodic treatment. Treatment goals are to increase circulating VWF activity and reduce bleeding (Table 2). Desmopressin (DDAVP), a vasopressin analog, binds to vasopressin receptors on the surface of endothelial cells, thereby increasing intracellular calcium, promoting trafficking of WPB to the endothelial cell plasma membrane, which then triggers the release of VWF, FVIII, and other coagulation proteins. The net result is an increase in circulating VWF and FVIII, and improved systemic hemostasis. The release of VWF and FVIII normally occurs within 30–60 minutes of its administration, and usually provides a durable hemostatic response that lasts for 4–6 hours. The optimal increase in VWF is associated with bleeding control in 70% of cases of VWD, albeit with some interpatient variability.38,39 DDAVP also releases tissue plasminogen activator (tPA) from the WPB.40 DDAVP therefore increases circulating hemostatic factors but may also increase fibrinolysis. Antifibrinolytic treatments are often used in conjunction with DDAVP to improve hemostasis. This approach has been used successfully in multiple congenital bleeding disorders.41
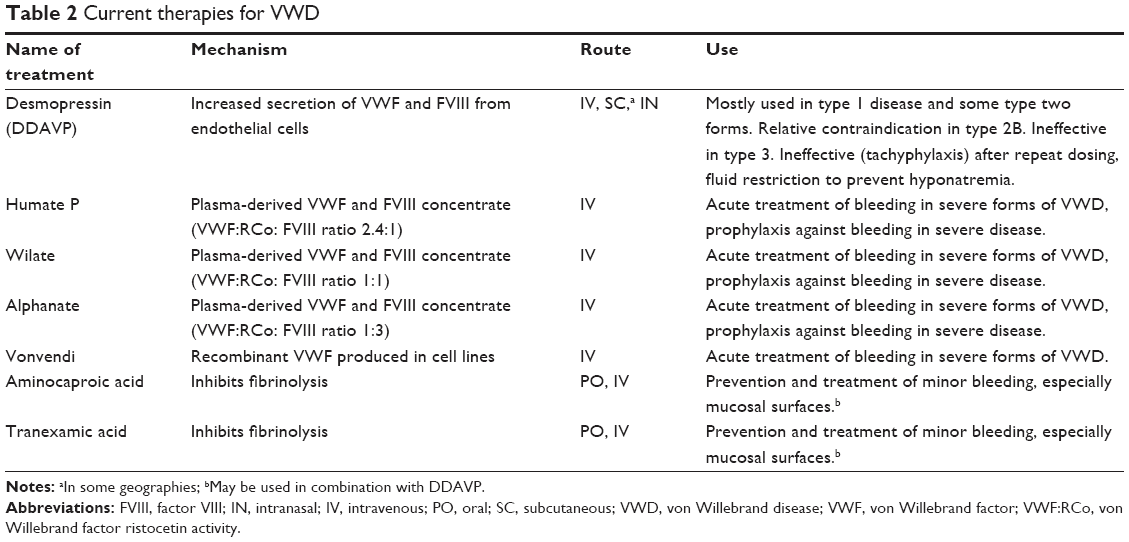 | Table 2 Current therapies for VWD |
In more severe cases of VWD, prophylaxis with a VWF-containing concentrate is desirable to prevent bleeding (Table 2). The prophylactic strategy involves administration of VWF replacement factors on a regular basis to maintain active trough levels of VWF and FVIII in circulation. The strategy is similar to hemophilia treatment, where factor concentrates are regularly administered to reduce joint bleeding and its associated long-term morbidity.42,43 Symptomatology in VWD is more variable than hemophilia; joint bleeding is less common and mucosal surface bleeding can be the predominant bleeding pattern.44 Prophylaxis with VWF concentrates significantly reduces mucosal and joint bleeding rates.44–46 A recombinant VWF concentrate has also been developed that is effective at stabilizing FVIII47 and controlling bleeding events.48
VWF and aging
It has been demonstrated that VWF levels increase with age by approximately 1% per year49–51 in the healthy population and in mild forms of VWD. In addition, patients older than 55 have increases in VWF circulating half-lives and increased secretion.19 The highest levels of circulating VWF can be seen in healthy centenarians.52
VWF levels do not change with age in severe forms of VWD, including types 2 and 3. In addition, it is not clear whether age-related increases in VWF observed in mild VWD affect the bleeding phenotype.53,54 VWD is therefore considered a lifelong bleeding condition that may require ongoing treatment.
In general, patients without bleeding disorders accumulate risk factors based on organ systems (cardiovascular disease, osteoporosis, hypothyroidism), molecular cell signaling and stem cell clonal populations (malignancies), and motor functions (fall risk, tremors) as they age. In addition, age represents an independent risk factor for venous thrombosis. All of these comorbidities combined with the common consequences of aging have significant impact on aging patients with VWD.
VWD and cardiovascular events
Cardiovascular disease represents a leading cause of morbidity and mortality in the adult population in most developed countries.55 Risk factors for arterial disease in the general population have also been associated with VWF including smoking, glycemic control,56 elevated triglycerides, and blood pressure, and aortic stiffness.57
A large number of animal models have shown that VWF deficiency may have a protective effect against atherosclerosis and arterial thrombosis risk in both small and large arteries.58 Studies in humans have been less clear, however, with most trials showing only modest reductions in atherosclerosis.59–63 While arterial thrombosis has been reported in VWD,64 its incidence is low, especially in younger populations.65 Cohort studies have yielded conflicting results on the degree of protection against arterial disease in VWD.65,66 Some studies have also demonstrated that patients with VWD have a reduced risk of both cerebrovascular and peripheral vascular diseases.67
The treating clinician should not assume that a patient is at low risk for cardiovascular events because of a history of VWD, as both male and female patients with VWD have developed acute coronary syndrome (ACS).68 ACS should be managed as it would be in otherwise healthy patients. Percutaneous interventions and coronary artery bypass grafting have been performed69–71 although a hematologist should be available for consultation as perioperative bleeding is a known risk in this setting.68 VWD patients with ACS can also be managed medically without increased mortality in select patients.68 Aspirin, statins, and other therapies for cardiovascular disease can be given in VWD, although caution must be exercised with antiplatelet agents, direct oral acting anticoagulants (DOACs), and warfarin, as these drugs carry an additional bleeding risk.
VWD and venous thrombosis
Elevated levels of VWF have been associated with increased risk for venous thromboembolism (VTE) in healthy populations, leading to speculation that VWD patients may be protected from VTEs.72 However, the degree of risk reduction is not clear and some reports have suggested patients with VWD are not protected against VTE.65 Both deep vein thrombosis and pulmonary emboli have been reported in all types of VWD.64,73 In clinical practice, the risk of VTE should not be considered negligible, even in the setting of an inherited bleeding disorder like VWD. When planning for a major surgery, patients with VWD should be appropriately evaluated for thromboprophylaxis to reduce the risk of post-operative VTE.74
Age is also a risk factor for VTE, and it should be suspected in the appropriate clinical context in elderly patients with VWD. DOACs have been demonstrated to have equal efficacy to vitamin K antagonists (VKA) like warfarin and with improved safety in the elderly thrombosis population.75 Each VTE treatment should be individualized to the patient. There are no consensus guidelines for the selection and use of anticoagulation in patients with VWD, but DOACs have been suggested for use in other bleeding disorders because of their shorter half-life and improved safety profile compared to VKAs.76
VWD and malignancy
To date, the available literature on VWD and malignancy risk is limited. In general, these patients should be considered to be similar to age and risk factor-adjusted healthy individuals in the general population, provided they did not contract viruses associated with contaminated blood products such as hepatitis C virus (HCV) and human immunodeficiency virus (HIV), and that the diagnosis of VWD is a primary diagnosis and not AVWS.
Cohort studies suggest that solid tumors are significantly more common than hematologic malignancies in VWD patients. More severe cases of VWD were also associated with exposure to HCV, likely from VWF concentrate exposure, and with an increased risk of hepatocellular carcinoma and non-Hodgkin lymphoma.77 Interestingly, increased rates of malignancy were seen in type 2 VWD compared to other types, leading the authors to speculate that a qualitative difference in VWF may be an important component in malignancy. In support of this hypothesis, experimental animal data has suggested that a reduction in VWF reduces metastases of tumor cells.78 This has yet to be confirmed in a clinical setting. In small clinical studies, it does not appear that cancer risk is related to VWF antigen levels but may be related to qualitative VWF defects and perturbed angiogenesis that could affect cancer metastasis.77
Hemophilia patients exposed to HCV and HIV have increased risk of malignancies related to these exposures, such as hepatocellular carcinoma and lymphoma.79,80 Treatment plans for malignancies in patients with hemophilia can be successfully customized to reduce bleeding risk related to chemotherapy-induced thrombocytopenia.81 VWD patients are exposed less frequently to VWF-containing concentrates compared with hemophilia patients, and therefore have historically had lower exposure to HCV and HIV, although the natural history of chronic viral infection and subsequent risk of malignancies are similar.77,82 The bleeding risks in VWD associated with treatments of malignancies can be minimized when performed in conjunction with specialty centers.77 The incidence of chronic viral infections with HIV and HCV from plasma concentrate exposure and the subsequent malignancy risk will decrease in current generations of patients who are receiving high purity concentrates or recombinant products. Longitudinal studies are needed to provide more insight into VWD malignancy risks.
Acquired AVWS and aging
It is important to distinguish a patient with congenital VWD who has developed a malignancy, especially a hematologic malignancy, from a patient who has developed AVWS from a malignancy. The distinction is relevant to treatment strategies, since treating an underlying malignancy may also restore functional VWF in cases of AVWS.
AVWS is a syndrome that predominantly occurs in older patients and is almost always encountered in the setting of an underlying disorder (Table 3).83 Most patients with AVWS have a hematologic neoplasm or a form of cardiac disease where perturbed blood flow leads to loss of VWF multimers. In addition, medications including ciprofloxacin84 and valproic acid85 have been implicated in reducing VWF secretion. A longitudinal bleeding history is important to distinguish a new AVWS from a patient with congenital VWD and an accompanying diagnosis of cancer, both of which can be seen more commonly in populations over 60. The diagnosis of AVWS can be challenging, since different etiologies result in different mechanisms whereby VWF is lost and can be broadly divided into immune-mediated clearance of VWF, adsorption, and shear stress.86 In the acute setting, bleeding can be managed by treatments similar to congenital VWD. Treating the underlying condition often restores circulating VWF.
 | Table 3 Pathophysiology associated with acquired von Willebrand syndrome |
As lymphoproliferative and myeloproliferative disorders are more common in elderly populations, bleeding symptoms should be closely evaluated in patients with these diagnoses. AVWS is associated with lymphoproliferative diseases, and most commonly monoclonal gammopathy of uncertain significance (MGUS). The mechanism of action in this AVWS subtype is the development of an anti-VWF antibody that can induce both a loss of function and increased clearance of VWF.87 Both IgG and IgM types of MGUS have been described in relation to AVWS. DDAVP and VWF concentrates agents have been used to control bleeding, although these treatments can be subject to rapid clearance. Immunosuppression has also been used to treat the underlying MGUS syndrome.88 The MGUS AVWS subtype appears to respond to intravenous immunoglobulins (IVIG) that binds the autoantibody. IVIG has been demonstrated to restore plasma VWF levels and control bleeding.89,90
Additional lymphoproliferative disorders have been described, including Waldenstrom’s macroglobulinemia (WM) which responded to chemotherapy and plasmapheresis, and marginal zone lymphoma, which responds to chemotherapy.91 In WM, the IgM paraprotein binds VWF and clears it from circulation. Bleeding symptoms do appear to improve with plasmapheresis.
Myeloproliferative disorders have also been described in association with AVWS. In essential thrombocythemia, characterized by an excess of platelets, high molecular weight multimers (HMWM) adsorb onto the surface of activated platelets removing VWF from circulation. Cytoreduction with hydroxyurea has been shown to reverse this deficit in some cases.91 Chronic myeloid leukemia has also been associated with AVWS and loss of HMWMs. The pathophysiology of this disorder is likely also related to thrombocytosis and has demonstrated responsiveness to tyrosine kinase inhibitor therapy.
The reports of solid tumors and AVWS are few and constitute a small minority of AVWS (5%). The mechanism of AVWS in solid tumors remains incompletely understood. Several solid tumor subtypes have been associated with tumor expression of GP Ib or IIb/IIIa receptors, suggesting the pathology could be related to VWF binding onto the cell surfaces.92 AVWS has also been reported in nephroblastoma and Wilms tumor, where hyaluronic acid has been postulated with loss of VWF.93 A case of AVWS in prostate cancer was also reported without a clear pathophysiologic explanation.94 In general, AVWS in these cases has responded to the chemotherapy treatments used for the underlying malignancy.
Acquired VWS and left ventricular assist devices (LVADs)
The diagnosis of Heyde’s syndrome, characterized by gastrointestinal bleeding from angiodysplasia in the setting of native valve aortic stenosis (AS), is a shear-related loss of VWF that causes both bleeding, and loss of vessel wall integrity. The severity of AS also correlates with more significant loss of VWF and predicts mortality and requirement for surgical valve replacement.95 AVWS related to valve shear forces may resolve with valve replacement although there can be recurrences.96 In addition, new onset gastrointestinal bleeding from angiodysplasia in older adults has been associated with AVWS.97 Gastrointestinal bleeding from angiodysplasia in an elderly patient should prompt an evaluation for AVWS, especially in the setting of cardiac valve disease.
At least two cases of octogenarians with AS and epistaxis related to loss of VWF multimers have been reported in the literature, demonstrating that AS bleeding may occur on different mucosal surfaces.98 AVWS related to shear stress has also been reported in congenital heart diseases.99 Circulatory assistance devices such as LVADs can also cause loss of HMWM and result in bleeding and angiodysplasia. LVAD bleeding is similar to that observed in Heyde’s syndrome.100,101 In many cases, both VWF:RCo and VWF:Ag levels remain normal.100,102,103 Furthermore, the degree of pulsatility is linked to bleeding events.104 It is currently recommended that LVAD settings be altered to improve shear and therefore bleeding,100,105 although it is to be noted that there is no robust data in the published literature to support this recommendation.101
AVWS and thyroid disease
The elderly population is susceptible to hypothyroidism, with an estimated prevalence as high as 4.4%.106 Hypothyroidism accompanied by AVWS can occur at any age, although it has been described as a more common issue in older adults and the prevalence of AVWS in hypothyroidism is approximately 33%.107 Bleeding symptoms correlate with both VWF:RCo and VWF:Ag resembling type 1 VWD. Interestingly, thyroid hormone levels do not appear to correlate with severity of disease. In one prospective cohort, treatment with thyroid hormone replacement over a median of 5 months resulted in normalization of VWF and resolution of bleeding symptoms in almost all cases.107 When it is urgent to raise VWF levels prior to completion of thyroid replacement, DDAVP can be used to increase VWF in circulation.108 Laboratory evaluation of thyroid function should accompany any new case of VWD or AVWS to rule out easily reversible causes.
VWD and osteoporosis, fracture risk, and falls
The discovery of the RANK (Receptor Activator of Nuclear Factor-kappa B), RANK ligand (RANKL), osteoprotegerin (OPG) axis led to a greater understanding of bone growth and remodeling by osteoblasts and osteoclasts. In addition, coagulation proteins have been identified in this physiologic axis. The FVIII-VWF complex inhibits osteoclastogenesis in vitro,109 and a deficit of FVIII in WPBs results in increased RANKL activity which in turn leads to increased bone resorption. In a FVIII deficiency mouse model, animals have reduced bone mineral density and impaired biomechanical strength even in the absence of bleeding.110 Furthermore, population studies suggest hemophilia and VWD patients to have an increased risk of fractures and lower bone mineral density,111,112 and that this relationship is proportional to the degree of the factor deficiency. Although VWF deficiency has not been extensively studied in relation to bone mineral density, both animal models and clinical observations suggest coagulation factor deficiencies contribute to bone loss. Exercise programs, balance exercise, and home safety programs have been shown to reduce rates of falling in the general elderly population,113,114 and these recommendations should be applied to patients with bleeding disorders. An ongoing study has suggested both physical therapy and occupational therapy programs in older patients with hemophilia are feasible, and would be reasonable options for patients with VWD.115 An interesting study in hemophilia patients indicated overall a reduced risk of falls, presumably because of an increased level of caution when ambulating and avoidance of fall risks.116 More studies on the risk and management of osteoporosis and falls in elderly VWD patients are needed.
Dental procedures
Dental procedures are important in elderly patients, including denture replacement and extractions. Proper dental hygiene in geriatric patients improves nutrition, functional status, and quality of life. VWD is a significant risk factor for bleeding with dental work and appropriate use of antifibrinolytics, DDAVP, and VWF concentrates as well as good intraoperative hemostasis is of particular importance.41 In general, dental surgery can be accomplished in elderly patients using a similar approach to younger individuals.117 Dental issues remain an increasing concern in older patients,118 and oral health is a significant component of quality of life in elderly patients, therefore standards of care for general patients should be leveraged wherever possible for elderly patients with VWD.119
Conclusion
Elderly patients are often under-represented in clinical trials, and trials involving patients with bleeding disorders are not different in this respect.120,121 VWD and AVWS are bleeding disorders with diverse relationships to aging populations. A significant amount of data exists comparing VWF parameters between pediatric patients and adults, but data in older adult age cohorts are lacking. While some milder types of VWD may demonstrate increases in VWF levels over time, the more severe phenotypes do not appear to change, which highlights the point that VWD is a lifelong diagnosis and treatment plans need to be carefully established. The use of VWD prophylaxis in severe disease forms has been associated with a decrease in annual bleeding rates. Chronic conditions that increase over an individual’s lifespan also alter bleeding and thrombotic risk, as well as the risk for different types of trauma, surgery, and medications. VWD patients are not entirely protected from thrombotic conditions or atherosclerosis, and wherever possible standards of care for cardiovascular disorders, malignancies, and other diseases should be followed. Elderly patients are also more likely to develop AVWS, given the age-association of most malignancies, AS, and the need for circulatory support devices. The clinician should therefore remain alert to new bleeding symptoms in elderly patients as it may be related to an underlying change in VWF.
This article sought to address some of the open questions in the elderly VWD and AVWS patients. In addition, many questions exist for the elderly population with bleeding disorders that will require longitudinal cohort studies. For example, the burden of cognitive impairment has not been studied. Common frailty measurements have not been validated in inherited bleeding disorders. Finally, in older adults with limited mobility, the incidence, healing rates, and management of pressure ulcers have not been evaluated. It remains important for longitudinal registry studies of bleeding disorders include larger numbers of older adults with VWD to more fully understand the disease complexity related to age.
Acknowledgments
The author would like to acknowledge Bruce Ewenstein for helpful comments and revisions to the manuscript.
Disclosure
The author is an employee and stock holder of Shire. The author reports no other conflicts of interest in this work.
References
Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69(2):454–459. | ||
Werner EJ, Broxson EH, Tucker EL, Giroux DS, Shults J, Abshire TC. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr. 1993;123(6):893–898. | ||
Favaloro EJ, Grispo L, Dinale A, Koutts J, Berndt M. Von willebrand’s disease: laboratory investigation using an improved functional assay for von willebrand factor. Pathology. 1993;25(2):152–158. | ||
Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14(2):171–232. | ||
Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000;84(08):160–174. | ||
Sanders YV, Fijnvandraat K, Boender J, et al. Bleeding spectrum in children with moderate or severe von Willebrand disease: relevance of pediatric-specific bleeding. Am J Hematol. 2015;90(12):1142–1148. | ||
de Wee EM, Sanders YV, Mauser-Bunschoten EP, et al. Determinants of bleeding phenotype in adult patients with moderate or severe von Willebrand disease. Thromb Haemost. 2012;108(10):683–692. | ||
Berntorp E, Petrini P. Long-term prophylaxis in von Willebrand disease. Blood Coagul Fibrinolysis. 2005;16(Suppl 1):S23–S26. | ||
Favaloro EJ, Franchini M, Lippi G. Aging hemostasis: changes to laboratory markers of hemostasis as we age – a narrative review. Semin Thromb Hemost. 2014;40(6):621–633. | ||
Ruggeri ZM, Mendolicchio GL. Interaction of von Willebrand factor with platelets and the vessel wall. Hämostaseologie. 2015;35(3):211–224. | ||
Cosemans JMEM, Schols SEM, Stefanini L, et al. Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood. 2011;117(2):651–660. | ||
Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand Disease (MCMDM-1VWD). Blood. 2007;109(1):112–121. | ||
James PD, Notley C, Hegadorn C, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109(1):145–154. | ||
Sadler JE. Low von Willebrand factor: sometimes a risk factor and sometimes a disease. Hematology. 2009;2009(1):106–112. | ||
Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4(10):2103–2114. | ||
Nichols WL, Rick ME, Ortel TL, et al. Clinical and laboratory diagnosis of von Willebrand disease: a synopsis of the 2008 NHLBI/NIH guidelines. Am J Hematol. 2009;84(6):366–370. | ||
Lillicrap D. von Willebrand disease: advances in pathogenetic understanding, diagnosis, and therapy. Blood. 2013;122(23):3735–3740. | ||
Othman M, Kaur H, Favaloro EJ, et al. Platelet type von Willebrand disease and registry report: communication from the SSC of the ISTH. J Thromb Haemost. 2016;14(2):411–414. | ||
Albánez S, Ogiwara K, Michels A, et al. Aging and ABO blood type influence von Willebrand factor and factor VIII levels through interrelated mechanisms. J Thromb Haemost. 2016;14(5):953–963. | ||
van Loon J, Dehghan A, Weihong T, et al. Genome-wide association studies identify genetic loci for low von Willebrand factor levels. Eur J Hum Genet. 2016;24(7):1096. | ||
Smith NL, Chen M-H, Dehghan A, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand Factor: the CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) consortium. Circulation. 2010;121(12):1382–1392. | ||
van Loon JE, Sanders YV, de Wee EM, Kruip MJHA, de Maat MPM, Leebeek FWG. Effect of genetic variation in STXBP5 and STX2 on von Willebrand Factor and bleeding phenotype in type 1 von Willebrand disease patients. PLoS One. 2012;7(7):e40624. | ||
Brandherm I, Disse J, Zeuschner D, Gerke V. cAMP-induced secretion of endothelial von Willebrand factor is regulated by a phosphorylation/dephosphorylation switch in annexin A2. Blood. 2013;122(6):1042–1051. | ||
Knop M, Aareskjold E, Bode G, Gerke V. Rab3D and annexin A2 play a role in regulated secretion of vWF, but not tPA, from endothelial cells. EMBO J. 2004;23(15):2982–2992. | ||
Leebeek FWG, Eikenboom JCJ. Von Willebrand’s Disease. N Engl J Med. 2017;376(7):701–702. | ||
Wagner DD, Marder VJ. Biosynthesis of von Willebrand protein by human endothelial cells. Identification of a large precursor polypeptide chain. J Biol Chem. 1983;258(4):2065–2067. | ||
Sporn LA, Marder VJ, Wagner DD. von Willebrand factor released from Weibel-Palade bodies binds more avidly to extracellular matrix than that secreted constitutively. Blood. 1987;69(5):1531–1534. | ||
Dong J-F. Cleavage of ultra-large von Willebrand factor by ADAMTS-13 under flow conditions. J Thromb Haemost. 2005;3(8):1710–1716. | ||
Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood. 2017;130(10):1181–1188. | ||
Trossaert M, Lienhart A, Nougier C, et al. Diagnosis and management challenges in patients with mild haemophilia A and discrepant FVIII measurements. Haemophilia. 2014;20(4):550–558. | ||
Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD). J Thromb Haemost. 2006;4(4):766–773. | ||
Cumming A, Grundy P, Keeney S, et al. An investigation of the von Willebrand factor genotype in UK patients diagnosed to have type 1 von Willebrand disease. Thromb Haemost. 2006;96(5):630–641. | ||
Federici AB, Bucciarelli P, Castaman G, et al. The bleeding score predicts clinical outcomes and replacement therapy in adults with von Willebrand disease. Blood. 2014;123(26):4037–4044. | ||
Castaman G, Missiaglia E, Federici AB, Schneppenheim R, Rodeghiero F. An additional unique candidate mutation (G2470A; M740I) in the original families with von Willebrand disease type 2 M Vicenza and the G3864A (R1205H) mutation. Thromb Haemost. 2000;84(2):350–351. | ||
Castaman G, Federici AB, Tosetto A, et al. Different bleeding risk in type 2A and 2M von Willebrand disease: a 2-year prospective study in 107 patients. J Thromb Haemost. 2012;10(4):632–638. | ||
Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063–2065. | ||
Schmidt DE, Bruzelius M, Majeed A, Odeberg J, Holmström M, Ågren A. Whole blood ristocetin-activated platelet impedance aggregometry (Multiplate) for the rapid detection of Von Willebrand disease. Thromb Haemost. 2017;117(08):1528–1533. | ||
Castaman G, Lethagen S, Federici AB, et al. Response to desmopressin is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European Study MCMDM-1VWD. Blood. 2008;111(7):3531–3539. | ||
Federici AB, et al. Biologic response to desmopressin in patients with severe type 1 and type 2 von Willebrand disease: results of a multicenter European study. Blood. 2004;103(6):2032–2038. | ||
Kaufmann JE, Vischer UM. Cellular mechanisms of the hemostatic effects of desmopressin (DDAVP). J Thromb Haemost. 2003;1(4):682–689. | ||
Chapin J, Bamme J, Hsu F, Christos P, Desancho M. Outcomes in patients with hemophilia and von Willebrand disease undergoing invasive or surgical procedures. Clin Appl Thromb Hemost. 2017;23(2):148–154. | ||
Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. | ||
Manco-Johnson MJ, Lundin B, Funk S, et al. Effect of late prophylaxis in hemophilia on joint status: a randomized trial. J Thromb Haemost. 2017;15(11):2115–2124. | ||
Holm E, Abshire TC, Bowen J, et al. Changes in bleeding patterns in von Willebrand disease after institution of long-term replacement therapy: results from the von Willebrand Disease Prophylaxis Network. Blood Coagul Fibrinolysis. 2015;26(4):383–388. | ||
Abshire TC, Federici AB, Alvárez MT, et al. Prophylaxis in severe forms of von Willebrand’s disease: results from the von Willebrand Disease Prophylaxis Network (VWD PN). Haemophilia. 2013;19(1):76–81. | ||
Abshire T, Cox-Gill J, Kempton CL, et al. Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. J Thromb Haemost. 2015;13(9):1585–1589. | ||
Mannucci PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood. 2013;122(5):648–657. | ||
Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015;126(17):2038–2046. | ||
van Loon JE, Kavousi M, Leebeek FW, et al. von Willebrand factor plasma levels, genetic variations and coronary heart disease in an older population. J Thromb Haemost. 2012;10(7):1262–1269. | ||
Kokame K, Sakata T, Kokubo Y, Miyata T. von Willebrand factor-to-ADAMTS13 ratio increases with age in a Japanese population. J Thromb Haemost. 2011;9(7):1426–1428. | ||
Rydz N, Grabell J, Lillicrap D, James PD. Changes in von Willebrand factor level and von Willebrand activity with age in type 1 von Willebrand disease. Haemophilia. 2015;21(5):636–641. | ||
Mari D, Coppola R, Provenzano R. Hemostasis factors and aging. Exp Gerontol. 2008;43(2):66–73. | ||
Laffan M. Can you grow out of von Willebrand disease? Haemophilia. 2017;23(6):807–809. | ||
Sanders YV, Giezenaar MA, Laros-van Gorkom BA, et al. von Willebrand disease and aging: an evolving phenotype. J Thromb Haemost. 2014;12(7):1066–1075. | ||
Gcod C. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1151–1210. | ||
Kumari M, Marmot M, Brunner E. Social determinants of von Willebrand Factor: the Whitehall II Study. Arterioscler Thromb Vasc Biol. 2000;20(7):1842–1847. | ||
Nikolic SB, Adams MJ, Otahal P, Edwards LM, Sharman JE. Association of von Willebrand factor blood levels with exercise hypertension. Eur J Appl Physiol. 2015;115(5):1057–1065. | ||
van Galen KPM, Tuinenburg A, Smeets EM, Schutgens REG. Von Willebrand factor deficiency and atherosclerosis. Blood Rev. 2012;26(5):189–196. | ||
Bilora F, Dei Rossi C, Girolami B, et al. Do hemophilia A and von Willebrand disease protect against carotid atherosclerosis? A comparative study between coagulopathics and normal subjects by means of carotid echo-color Doppler scan. Clin Appl Thromb Hemost. 1999;5(4):232–235. | ||
Bilora F, Boccioletti V, Zanon E, Petrobelli F, Girolami A. Hemophilia A, von Willebrand disease, and atherosclerosis of abdominal aorta and leg arteries: factor VIII and von Willebrand factor defects appear to protect abdominal aorta and leg arteries from atherosclerosis. Clin Appl Thromb Hemost. 2001;7(4):311–313. | ||
Srámek A, Reiber JHC, Gerrits WBJ, Rosendaal FR. Decreased coagulability has no clinically relevant effect on atherogenesis: observations in individuals with a hereditary bleeding tendency. Circulation. 2001;104(7):762–767. | ||
Srámek A, et al. Patients with type 3 severe von Willebrand disease are not protected against atherosclerosis: results from a multicenter study in 47 patients. Circulation. 2004;109(6):740–744. | ||
Bilora F, Zanon E, Casonato A, et al. Type IIb von Willebrand disease: role of qualitative defects in atherosclerosis and endothelial dysfunction. Clin Appl Thromb Hemost. 2007;13(4):384–390. | ||
Girolami A, Tezza F, Scapin M, Vettore S, Casonato A. Arterial and venous thrombosis in patients with von Willebrand’s disease: a critical review of the literature. J Thromb Thrombolysis. 2006;21(2):175–178. | ||
Qureshi W, Hassan S, Dabak V, Kuriakose P. Thrombosis in VonWillebrand disease. Thromb Res. 2012;130(5):e255–e258. | ||
Sanders YV, Eikenboom J, de Wee EM, et al. Reduced prevalence of arterial thrombosis in von Willebrand disease. J Thromb Haemost. 2013;11(5):845–854. | ||
Seaman CD, Yabes J, Comer DM, Ragni MV. Does deficiency of von Willebrand factor protect against cardiovascular disease? Analysis of a national discharge register. J Thromb Haemost. 2015;13(11):1999–2003. | ||
Fogarty PF, Blair A, Vega R, Matthai WH, Gimotty PA. Interventional therapies and in-hospital outcomes in acute coronary syndromes complicated by von Willebrand disease. Haemophilia. 2017;23(3):400–407. | ||
Rathore S, Deleon D, Akram H, Sane D, Ball T. Percutaneous coronary intervention and the management of acute coronary syndromes in patients with von Willebrand disease. J Invasive Cardiol. 2013;25(4):E81–E86. | ||
Hassan SA, Amer S, Qureshi W, Alirhayim Z, Kuriakose P. Treating symptomatic coronary artery disease in patients with Von Willebrand disease. Hematol Oncol Stem Cell Ther. 2013;6(3–4):101–104. | ||
Arjomand H, Aquilina P, Mccormick D. Acute myocardial infarction in a patient with von Willebrand disease: pathogenetic dilemmas and therapeutic challenges. J Invasive Cardiol. 2002;14(10):615–618. | ||
Martinelli I. von Willebrand factor and factor VIII as risk factors for arterial and venous thrombosis. Semin Hematol. 2005;42(1):49–55. | ||
Girolami A, Tasinato V, Sambado L, Peroni E, Casonato A. Venous thrombosis in von Willebrand disease as observed in one centre and as reported in the literature. Blood Coagulation & Fibrinolysis. 2015;26(1):54–58. | ||
Kim JYS, Khavanin N, Rambachan A, et al. Surgical duration and risk of venous thromboembolism. JAMA Surg. 2015;150(2):110–117. | ||
van Es N, Coppens M, Schulman S, Middeldorp S, Büller HR. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: evidence from phase 3 trials. Blood. 2014;124(12):1968–1975. | ||
Martin K, Key NS. How I treat patients with inherited bleeding disorders who need anticoagulant therapy. Blood. 2016;128(2):178–184. | ||
Franchini M, di Perna C, Santoro C, et al. Cancers in patients with von Willebrand Disease: a survey from the Italian Association of Haemophilia Centres. Semin Thromb Hemost. 2016;42(1):36–41. | ||
Terraube V, Marx I, Denis CV. Role of von Willebrand factor in tumor metastasis. Thromb Res. 2007;120(Suppl 2):S64–S70. | ||
Miesbach W, Alesci S, Krekeler S, Seifried E. Comorbidities and bleeding pattern in elderly haemophilia A patients. Haemophilia. 2009;15(4):894–899. | ||
Miesbach W, Seifried E. Does haemophilia influence cancer-related mortality in HIV-negative patients? Haemophilia. 2011;17(1):55–60. | ||
Rutherford SC, Martin P, Chapin J. Management strategies in patients with diffuse large B-cell lymphoma and severe haemophilia. Haemophilia. 2015;21(5):e445–e447. | ||
Federici AB, Santagostino E, Rumi MG, et al. The natural history of hepatitis C virus infection in Italian patients with von Willebrand’s disease: a cohort study. Haematologica. 2006;91(4):503–508. | ||
Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. 2000;84(2):345–349. | ||
Castaman G, Rodeghiero F. Acquired transitory von Willebrand syndrome with ciprofloxacin. Lancet. 1994;343(8895):492. | ||
Eberl W, Budde U, Bentele K, et al. Acquired von Willebrand syndrome as side effect of valproic acid therapy in children is rare. Hamostaseologie. 2009;29(2):137–142. | ||
Kruse-Jarres R. Acquired bleeding disorders in the elderly. Hematology. 2015;2015(1):231–236. | ||
Sucker C, Michiels JJ, Zotz RB, Causes ZRB. Causes, etiology and diagnosis of acquired von Willebrand disease: a prospective diagnostic workup to establish the most effective therapeutic strategies. Acta Haematol. 2009;121(2–3):177–182. | ||
Federici AB, Budde U, Castaman G, Rand JH, Tiede A. Current diagnostic and therapeutic approaches to patients with acquired von Willebrand syndrome: a 2013 update. Semin Thromb Hemost. 2013;39(2):191–201. | ||
Federici AB, Stabile F, Castaman G, Canciani MT, Mannucci PM. Treatment of acquired von Willebrand syndrome in patients with monoclonal gammopathy of uncertain significance: comparison of three different therapeutic approaches. Blood. 1998;92(8):2707–2711. | ||
Lavin M, Ryan K, White B, Byrne M, O’Connell NM, O’Donnell JS. A role for intravenous immunoglobulin in the treatment of acquired von Willebrand syndrome associated with IgM gammopathy. Haemophilia. 2018;24(1):e22–e25. | ||
Fidalgo T, Ferreira G, Oliveira AC, et al. Acquired von Willebrand syndrome in haematologic malignancies – how the clinical-laboratory correlation improves a challenging diagnosis – a case series. Haemophilia. 2017;23(4):e361–e365. | ||
Facon T, Caron C, Courtin P, et al. Acquired type II von Willebrand’s disease associated with adrenal cortical carcinoma. Br J Haematol. 1992;80(4):488–494. | ||
Baxter PA, Nuchtern JG, Guillerman RP, et al. Acquired von Willebrand syndrome and Wilms tumor: not always benign. Pediatr Blood Cancer. 2009;52(3):392–394. | ||
Claus P-E, van Haute I, Verhoye E, Deeren D, Moreau E. Diagnostic challenges in acquired von Willebrand syndrome: a complex case of prostate carcinoma associated-acquired von Willebrand syndrome. Semin Thromb Hemost. 2017;43(01):101–104. | ||
Blackshear JL, Wysokinska EM, Safford RE, et al. Indexes of von Willebrand factor as biomarkers of aortic stenosis severity (from the Biomarkers of Aortic Stenosis Severity [BASS] Study). Am J Cardiol. 2013;111(3):374–381. | ||
Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med Overseas Ed. 2003;349(4):343–349. | ||
Veyradier A, Balian A, Wolf M, et al. Abnormal von Willebrand factor in bleeding angiodysplasias of the digestive tract. Gastroenterology. 2001;120(2):346–353. | ||
Schödel J, Obergfell A, Maass AH. Severe aortic valve stenosis and nosebleed. Int J Cardiol. 2007;120(2):286–287. | ||
Waldow HC, Westhoff-Bleck M, Widera C, Templin C, von Depka M. Acquired von Willebrand syndrome in adult patients with congenital heart disease. Int J Cardiol. 2014;176(3):739–745. | ||
Aggarwal A, Pant R, Kumar S, et al. Incidence and management of gastrointestinal bleeding with continuous flow assist devices. Ann Thorac Surg. 2012;93(5):1534–1540. | ||
Nascimbene A, Neelamegham S, Frazier OH, Moake JL, Dong JF. Acquired von Willebrand syndrome associated with left ventricular assist device. Blood. 2016;127(25):3133–3141. | ||
Crow S, Milano C, Joyce L, et al. Comparative analysis of von Willebrand factor profiles in pulsatile and continuous left ventricular assist device recipients. ASAIO J. 2010;56(5):441–445. | ||
Reich HJ, Morgan J, Arabia F, et al. Comparative analysis of von Willebrand factor profiles after implantation of left ventricular assist device and total artificial heart. J Thromb Haemost. 2017;15(8):1620–1624. | ||
Wever-Pinzon O, Selzman CH, Drakos SG, et al. Pulsatility and the risk of nonsurgical bleeding in patients supported with the continuous-flow left ventricular assist device HeartMate II. Circ Heart Fail. 2013;6(3):517–526. | ||
Susen S, Rauch A, van Belle E, Vincentelli A, Lenting PJ. Circulatory support devices: fundamental aspects and clinical management of bleeding and thrombosis. J Thromb Haemost. 2015;13(10):1757–1767. | ||
Magri F, Muzzoni B, Cravello L, et al. Thyroid function in physiological aging and in centenarians: possible relationships with some nutritional markers. Metabolism. 2002;51(1):105–109. | ||
Stuijver DJ, Piantanida E, van Zaane B, et al. Acquired von Willebrand syndrome in patients with overt hypothyroidism: a prospective cohort study. Haemophilia. 2014;20(3):326–332. | ||
Franchini M, de Gironcoli M, Lippi G, et al. Efficacy of desmopressin as surgical prophylaxis in patients with acquired von Willebrand disease undergoing thyroid surgery. Haemophilia. 2002;8(2):142–144. | ||
Baud’huin M, Duplomb L, Téletchéa S, et al. Factor VIII-von Willebrand factor complex inhibits osteoclastogenesis and controls cell survival. J Biol Chem. 2009;284(46):31704–31713. | ||
Recht M, Liel MS, Turner RT, Klein RF, Taylor JA. The bone disease associated with factor VIII deficiency in mice is secondary to increased bone resorption. Haemophilia. 2013;19(6):908–912. | ||
Mansouritorghabeh H, Rezaieyazdi Z. Bone density status in bleeding disorders: where are we and what needs to be done? J Bone Metab. 2017;24(4):201–206. | ||
Gay ND, Lee SC, Liel MS, Sochacki P, Recht M, Taylor JA. Increased fracture rates in people with haemophilia: a 10-year single institution retrospective analysis. Br J Haematol. 2015;170(4):584–586. | ||
Gillespie L. Preventing falls in older people: the story of a Cochrane review. Cochrane Database Syst Rev. 2013;2(2):ED000053. | ||
Sherrington C, Whitney JC, Lord SR, Herbert RD, Cumming RG, Close JC. Effective exercise for the prevention of falls: a systematic review and meta-analysis. J Am Geriatr Soc. 2008;56(12):2234–2243. | ||
Boccalandro E, Mancuso ME, Riva S, et al. Ageing successfully with haemophilia: a multidisciplinary programme. Haemophilia. 2018;24(1):57–62. | ||
Sammels M, Vandesande J, Vlaeyen E, Peerlinck K, Milisen K. Falling and fall risk factors in adults with haemophilia: an exploratory study. Haemophilia. 2014;20(6):836–845. | ||
Stubbs M, Lloyd J. A protocol for the dental management of von Willebrand’s disease, haemophilia A and haemophilia B. Aust Dent J. 2001;46(1):37–40. | ||
Yellowitz JA, Schneiderman MT. Elder’s oral health crisis. J Evid Based Dent Pract. 2014;14 Suppl(14 Suppl):191–200. | ||
Rouxel P, Tsakos G, Chandola T, Watt RG. Oral health-a neglected aspect of subjective well-being in later life. J Gerontol B Psychol Sci Soc Sci. 2018;73(3):382–386. | ||
Avorn J. Including elderly people in clinical trials. Br Med J. 1997;315(7115):1033–1034. | ||
Britton A, Mckee M, Black N, Mcpherson K, Sanderson C, Bain C. Threats to applicability of randomised trials: exclusions and selective participation. J Health Serv Res Policy. 1999;4(2):112–121. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.