Back to Archived Journals » Oncolytic Virotherapy » Volume 9
Virus–Receptor Interactions and Virus Neutralization: Insights for Oncolytic Virus Development
Authors Jayawardena N, Poirier JT ![]() , Burga LN, Bostina M
, Burga LN, Bostina M ![]()
Received 10 May 2019
Accepted for publication 9 February 2020
Published 6 March 2020 Volume 2020:9 Pages 1—15
DOI https://doi.org/10.2147/OV.S186337
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tommy Alain
Nadishka Jayawardena,1 John T Poirier,2 Laura N Burga,1 Mihnea Bostina1,3
1Department of Microbiology and Immunology, University of Otago, Dunedin, New Zealand; 2Department of Medicine and Molecular Pharmacology Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA; 3Otago Micro and Nano Imaging, University of Otago, Dunedin, New Zealand
Correspondence: Laura N Burga; Mihnea Bostina Tel +64 2 244 5583
Email [email protected]; [email protected]
Abstract: Oncolytic viruses (OVs) are replication competent agents that selectively target cancer cells. After penetrating the tumor cell, viruses replicate and eventually trigger cell lysis, releasing the new viral progeny, which at their turn will attack and kill neighbouring cells. The ability of OVs to self-amplify within the tumor while sparing normal cells can provide several advantages including the capacity to encode and locally produce therapeutic protein payloads, and to prime the host immune system. OVs targeting of cancer cells is mediated by host factors that are differentially expressed between normal tissue and tumors, including viral receptors and internalization factors. In this review article, we will discuss the evolution of oncolytic viruses that have reached the stage of clinical trials, their mechanisms of oncolysis, cellular receptors, strategies for targeting cancers, viral neutralization and developments to bypass virus neutralization.
Keywords: oncolytic viruses, virus-receptor interaction, virus neutralization
Introduction
Cancer remains one of the most prevalent non-communicable diseases worldwide.1 While traditional cancer therapies including chemotherapy, radiotherapy, surgery and radiosurgery can result in a beneficial outcome, they often cause severe off-target cytotoxicity. The necessity to specifically aim at cancer cells, while sparing healthy cells, has encouraged the development of targeted cancer treatment paradigms. In recent years significant progress has been made in developing viruses as a therapeutic strategy against cancer.2 Oncolytic viruses (OVs) are replication competent viral strains that specifically infect and lyse cancer cells. Many of the advantages of using OVs for cancer therapy arise from the fact they can be considered self-amplifying anti-cancer agents. Following tumor cell entry, OVs replicate and eventually trigger cell lysis, releasing new viral progeny, which in turn will invade and kill neighboring cells. The fact that viral amplification occurs within the tumour is likely to play an important role in tumor control through cell-cell spread.3 Additionally, viruses released from lysed cells can be transported by the circulatory system to tumors residing remotely from the original site.
The first documented case of using viruses as a potential cancer treatment dates back to 1910s, when a patient diagnosed with cervical carcinoma experienced remission after vaccination with a live-attenuated rabies vaccine.4 This incident prompted further clinical studies using rabies vaccine as an anti-cancer agent and exploitation of many other oncolytic viral strains such as Egypt 101 virus,5 adenoidal-pharyngeal-conjunctival virus,6 Newcastle disease virus7,8 and mumps virus.9 However, it should be noted that these initial trials were fraught with unethical practices. In recent years, there has been a resurgence of studies focused on possible roles for viruses in killing cancer cells. At the moment, more than 570 clinical trials using OVs are either active or recruiting patients,10 while many other viruses are in pre-clinical trials. This interest was ignited in part by the approval of Talimogene Laherparepvec (T-VEC), a modified oncolytic herpes simplex virus-111 for clinical use in USA, Europe and Australia,12 along with the clinical use of adenovirus derived Oncorine for head and neck cancers treatment in China13 and native Echovirus 7 under the name of Rigvir14 for the treatment of melanoma in several European countries.15
This review is focused on the evolution of oncolytic viruses that have reached the stage of clinical trials, their mechanisms of oncolysis and interactions with cellular receptors. In addition, limitations associated with oncovirotherapy such as antiviral immune response (viral neutralization) will be discussed along with recent developments towards overcoming such obstacles.
Mechanisms of Oncolysis
Most oncolytic viruses exert anti-tumor activity by penetrating the tumor cells, establishing a lytic cycle and ultimately causing the activation of cell death pathways. While some OVs have the natural capacity to infect specific tumors through receptor-mediated internalization,14,16-18 most OVs have been engineered to enhance their tumor selectivity and to reduce virulence in normal cells.12,13,19,20 Even though natural receptors responsible for oncolytic viral entry are expressed on non-malignant cells thereby allowing a successful infection,21–23 OVs often require a defect in innate immunity to successfully infect and propagate, which is only present in tumor cells but not in healthy cells.24 Alterations in transcriptional and cell signaling pathways, such as increased expression of B-cell lymphoma-extra-large (Bcl-xL) and activation of mitogen-activated protein kinases (MAPK) signaling can lead cancer cells to be more susceptible to OVs.25,26 In addition to direct cell lysis, OV infection and subsequent cell lysis trigger the release of danger-associated molecular patterns (DAMPs) that contribute to a long-lasting adaptive antitumor immune response.27–29 In fact, substantial effort has been made to develop OVs that encode transgenes designed to induce an immunogenic cell death (ICD) with the goal of priming the immune system against tumors.30–33 ICD releases DAMPs, which are recognized by antigen-presenting cells (APCs) such as macrophages and dendritic cells in the tumor microenvironment to elicit an innate immune response.34 As viral replication and tumor lysis progress, APCs produce cytokines, eventually recruiting other immune cells. The ultimate goal of this immune priming process is to activate T lymphocytes against specific tumor antigens in order to establish an adaptive immunity.35 Evidence for OV-induced innate and adaptive immune responses comes from several clinical trials. For instance, increased abundance of CD8+ and CD4+ T cells has been reported in patients with advanced melanoma, who received T-VEC or coxsackievirus in separate clinical trials.31,36-38 Patients with metastatic pancreatic adenocarcinoma showed increased B cells and natural killer cells when treated with a combination of reovirus and paclitaxel/carboplatin and these responses were linked to an increased disease control rate (DCR) in responding patients.33 Furthermore, increased expression of anti–inflammatory cytokines such as interleukin (IL) 6, IL 10 and tumor necrosis factor-α (TNF-α) was reported in patients with refractory primary or metastatic liver cancer treated with poxvirus strain JX-594,32 with some patients showing a durable objective response according to response evaluation criteria in solid tumors. In addition, OVs have also been reported to directly interfere with tumor perfusion. Engineered forms of adenoviruses39 and vaccinia virus40 have been shown to elicit antiangiogenic effects in mouse models. While not yet clinically documented, the possibility of a single OV to employ all three mechanisms of oncolysis in ongoing trials or in future developments holds promise.
Architecture of Oncolytic Viruses
The nature of the genome and the morphology of OVs are two essential factors that influence their amenability for cancer treatment development. First, oncolytic viruses pose different advantages for oncovirotherapy depending on their DNA or RNA genome. The structural stability of DNA combined with the precision of the DNA polymerase allows double-stranded DNA (dsDNA) viruses to encode a large number of proteins. The dsDNA viruses have the advantage of a stable genome that can be engineered to attenuate a pathogenic viral strain, to increase tumor specificity, to avoid anti-viral immune response or to code for proteins that can act synergistically with existing inflammatory host responses to establish a stronger anti-tumor immune response.2 A classic example is T-VEC, which has been modified to attain reduced neurovirulence41 and to stimulate the immune response.42
On the other hand, oncolytic RNA viruses possess the advantages of tumor specificity and a low off-target cytotoxicity dependent on the affinity to specific receptor usage. Paramyxoviruses and picornaviruses are two virus families that have been extensively studied for their oncolytic potential. Newcastle disease virus and measles virus from Paramyxoviridae family feature a negative-sense, non-segmented single-stranded RNA, which requires conversion into a positive-sense RNA before translation.43 In picornaviruses, the positive-sense single-stranded RNA (ssRNA) genome acts as a messenger RNA (mRNA) and it is translated into the viral polyprotein shortly after penetrating the host cells. This provides a mechanism for oncolytic picornaviruses to replicate and propagate faster.44,45 RNA viruses inherit a high error rate in genome replication and therefore are genetically unstable.46 From an evolutionary point of view, such mechanisms allow them to outmaneuver the host’s antiviral response due to viral diversity but can raise challenges for oncovirotherapy, especially when the virus is pathogenic in nature.
In addition to the classification of oncolytic viruses based on their genetic materials, they can be further sub-divided into two groups depending on virion architecture. Herpes simplex virus, vaccinia virus and rhabdoviruses fall into the enveloped DNA virus group, whereas adenovirus and reovirus belong to the non-enveloped DNA virus group. In the oncolytic RNA virus group, paramyxoviruses such as Newcastle disease virus and measles virus are enveloped, whereas picornaviruses are in the non-enveloped group. In terms of viral morphology, enveloped viruses are easily amenable to modification for use as OVs. Their morphology implies a relatively direct mode of infection in which the viral and cellular membranes fuse in order to deliver the nucleocapsid to the cytoplasmic space.47 The mechanism is mediated by the presence of fusion proteins evolved to recognize specific cellular receptors (Figure 1). Triggered by special cues, either changes in pH or the binding of co-receptors, fusion proteins undergo major conformational changes to bring the two membranes in close proximity and eventually causing them to merge.47 Non-enveloped virus architecture consists of a protein cage with icosahedral or helical symmetry harbouring the genome. Their mechanisms of cell entry are less understood, but it is known to involve the binding of a specific cellular receptor (Figure 1) that could trigger a signaling process leading to capsid endocytosis.48 Alternatively, receptor binding could function just as an attachment strategy to be followed by entry.49 The requirement of a specific receptor for tumor recognition and infection has been intensively investigated in picornaviruses and adenoviruses (Figure 1).
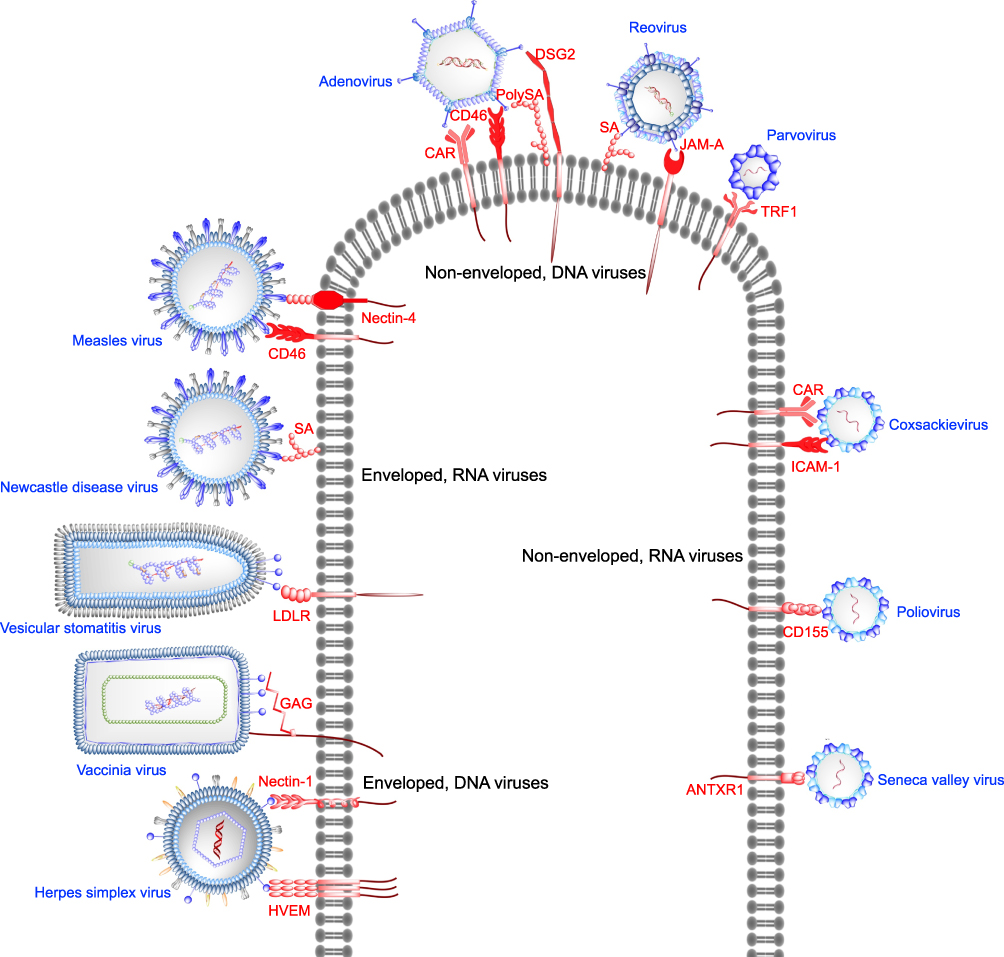 |
Figure 1 Targeting of receptors overexpressed in cancers with oncolytic viruses. Enveloped DNA viruses utilize their surface glycoproteins to bind receptors overexpressed in cancers. Herpes simplex virus glycoprotein D binds herpes virus entry mediator (HVEM) or nectin-1 prior to initiation of host membrane fusion by HSV glycoprotein B. Vaccinia virus and vesicular stomatitis virus bind cell surface glycosaminoglycans (GAGs) and low-density lipoprotein (LDLR) receptor, respectively. Enveloped, RNA viruses Newcastle disease virus and measles virus interact with cell surface sialic acid (SA) and CD46 or nectin-4, respectively, to facilitate entry into host cells. Sialic acid or poly-sialic acid (PolySA) serves as an attachment receptor for non-enveloped DNA viruses such as reovirus and human adenovirus. Junction adhesion molecule-A (JAM-A) acts as the entry receptor for reovirus, whereas coxsackievirus-adenovirus receptor (CAR), CD46, desmoglein-2 (DSG) have been shown to be the entry receptors for adenoviruses. Parvovirus (ssDNA) exploits cell surface transferrin receptor 1 as the entry receptor. Among the non-enveloped RNA viruses Seneca Valley virus, poliovirus, coxsackievirus bind anthrax toxin receptor-1 (ANTXR1), CD155, intercellular adhesion molecule-1 (ICAM-1) or CAR, respectively. |
The virion size and morphology are two important factors controlling OVs applicability. In order to spread and elicit their antitumor effect, oncolytic viruses must be able to overcome numerous physical barriers in the tumor microenvironment such as tight cell-cell junctions, extracellular matrix deposits, stromal cells and interstitial fluid pressure.50 Some oncolytic viruses, such as picornaviruses, have a small size (~30 nm) and can overcome such physical barriers.51,52
A serious obstacle for both enveloped and non-enveloped oncolytic viruses is to pass the genome across the cellular membrane. This can be done either by disrupting the membrane continuity or by using a channel formed by viral and/or cellular proteins.48
Strategies for Targeting Cancer Cells and Reducing off-Target Cytotoxicity
Selective targeting of tumors is of utmost importance and perhaps the most frequently discussed topic in the field of oncovirotherapy due to the use of human pathogenic viruses to treat cancers.53 These strategies could either involve exploiting inherent properties of a wild-type oncolytic virus such as specific receptor/dis-regulated cellular mechanisms usage and/or manipulating specific viral genes and surface properties to render tumor specificity.
Utilizing the Natural Tropism of OVs
Natural tropism is the capacity of a population of viruses to exploit extracellular markers expressed in cancer cells or to utilize intracellular pathways or immune-avoidance mechanisms to target tumors. Receptors responsible for oncoviral permissivity in tumors often play an essential role in tumor growth and progression or in providing protection from anti-tumor immune mechanisms.24 For instance, cluster of differentiation (CD) 155, CD46, and integrin α2β1 overexpressed in tumors, providing innate immunity for cancer cells, serve as entry ports for oncolytic poliovirus, measles virus and echovirus, respectively.54–56 On the other hand, herpes virus entry mediator (HVEM) and nectin-1, anthrax toxin receptor 1 (ANTXR1), laminin receptor, intracellular adhesion molecule-1 (ICAM1) and decay-accelerating factor (DAF), which all have a functional role in tumor growth and progression, have been identified as the cellular receptors for herpes simplex virus, Seneca Valley virus-001, Sindbis virus and coxsackievirus.17,57-61 In addition, numerous oncogenic pathways involved in carcinogenesis overlap with requirements for a successful infection and replication in some native oncolytic viruses. For example, tumor selectivity of Reolysin is dependent on a number of endogenous tumor factors such as RAS activation, downregulation of interferon (IFN) antiviral response and p53 pathway.26,62,63 Furthermore, Newcastle disease virus has been shown to target cancer cells overexpressing an anti-apoptotic protein Bcl-xL,25 while the high specificity of vesicular stomatitis virus for cancer cells is governed by its high sensitivity to type I IFNs, a system defective in most cancer types.64
Engineered Tropism
There are several strategies dictated by their viral architecture for modifying OVs to specifically target cancer cells. For enveloped viruses, a direct method is the insertion of glycoproteins from other viruses that recognize the targeted receptor (Figure 2). For instance, it was shown that a modified lentiviral vector, that possesses E2 glycoprotein in the envelope, can target a P-glycoprotein in melanoma.65 The neurotoxicity of vesicular stomatitis virus (VSV) has been abrogated by the substitution of its glycoprotein G with a glycoprotein variant of the lymphocytic choriomeningitis virus (LMCV-GP).66 Similarly, coat proteins can be modified with peptide ligands or antibody fragments recognized by the desired receptors.24,67 Adenovirus capsid fibers have been modified with an insertion of arginine-glycine-aspartic acid (RGD) moiety acting as the binding site of integrin receptors overexpressed in tumors (Figure 2).68 An alternative retargeting approach that does not involve the modification of the virus is the use of bispecific soluble adapters designed to bind both the OV and any given targeted antigen on cell membrane, mimicking a bona fide virus-receptor engagement (Figure 2). This strategy was employed to redirect herpes simplex virus-1 binding from nectin-1 to epidermal growth factor receptor (EGFR) by using a soluble adaptor protein P-V528LH that harbors a gD-binding domain of nectin-1 fused to virus and a single-chain antibody with affinity to EGFR.69
 |
Figure 2 Strategies for retargeting cancers with oncolytic viruses. Oncolytic viral architecture can be modified primarily in three different ways to target cancer-specific receptors. Pseudotyping of lentiviral (LV) envelope glycoproteins with a variant of Sindbis virus envelope protein has enabled successful targeting of P-glycoprotein expressed on melanoma cells. Substitution of vesicular stomatitis virus envelope glycoprotein with a variant glycoprotein from lymphocytic choriomeningitis virus glycoprotein (LCMV-GP) has enhanced the tumor specificity of the recombinant vesicular stomatitis virus (rVSV). Recombinant adenovirus strains have been developed (Ad5lucRGD) by incorporating an RGD moiety required for interaction with integrin receptors overexpressed in cancers. Finally, bispecific soluble adaptors (P-V528LH) have been used in the case of herpes simplex virus (HSV), that includes gD-binding domain of nectin-1 fused to virus and a single-chain antibody with affinity to epidermal growth factor receptor (EGFR). |
Several genetic modifications have been introduced in some oncolytic viruses to warrant tumor selectivity and to monitor the biodistribution of such viruses after administration (Table 1). Mutations introduced into herpes simplex virus-1 (HSV-1), vaccinia virus (VV), adenovirus (HAdV) and poliovirus (PV) strains have been shown to restrict the replication of these viruses to cancer cells and to reduce the toxicity associated with wild-type strains. The selectivity of T-VEC, a modified HSV-1 strain, for tumors is regulated by inherently low expression of protein kinase R (PKR) in cancers, that otherwise serves as an upstream target in normal cells to phosphorylate eukaryotic translation initiation factor 2 (eIF2) to terminate host cell protein synthesis.70 However, HSV infected cell protein (ICP) 34.5 has the capacity to reverse this mechanism by dephosphorylating eIF2.71 In addition, HSV ICP47 can inhibit the transporter associated with antigen presentation (TAP), ultimately reducing the expression levels of the antigen-major histocompatibility complex (MHC) type I.72 Therefore, both ICP34.5 and 47 genomic sites have been deleted in T-VEC and replaced with two copies of hematopoietic granulocyte-macrophage colony-stimulating factor (GM-CSF), that promotes the recruitment of dendritic cells and antigen-presenting cells (APC) to the tumor site.73 The deletion of ICP47 also serves in translocating the herpes virus protein US11 to decrease the activity of PKR in cancer cells.41
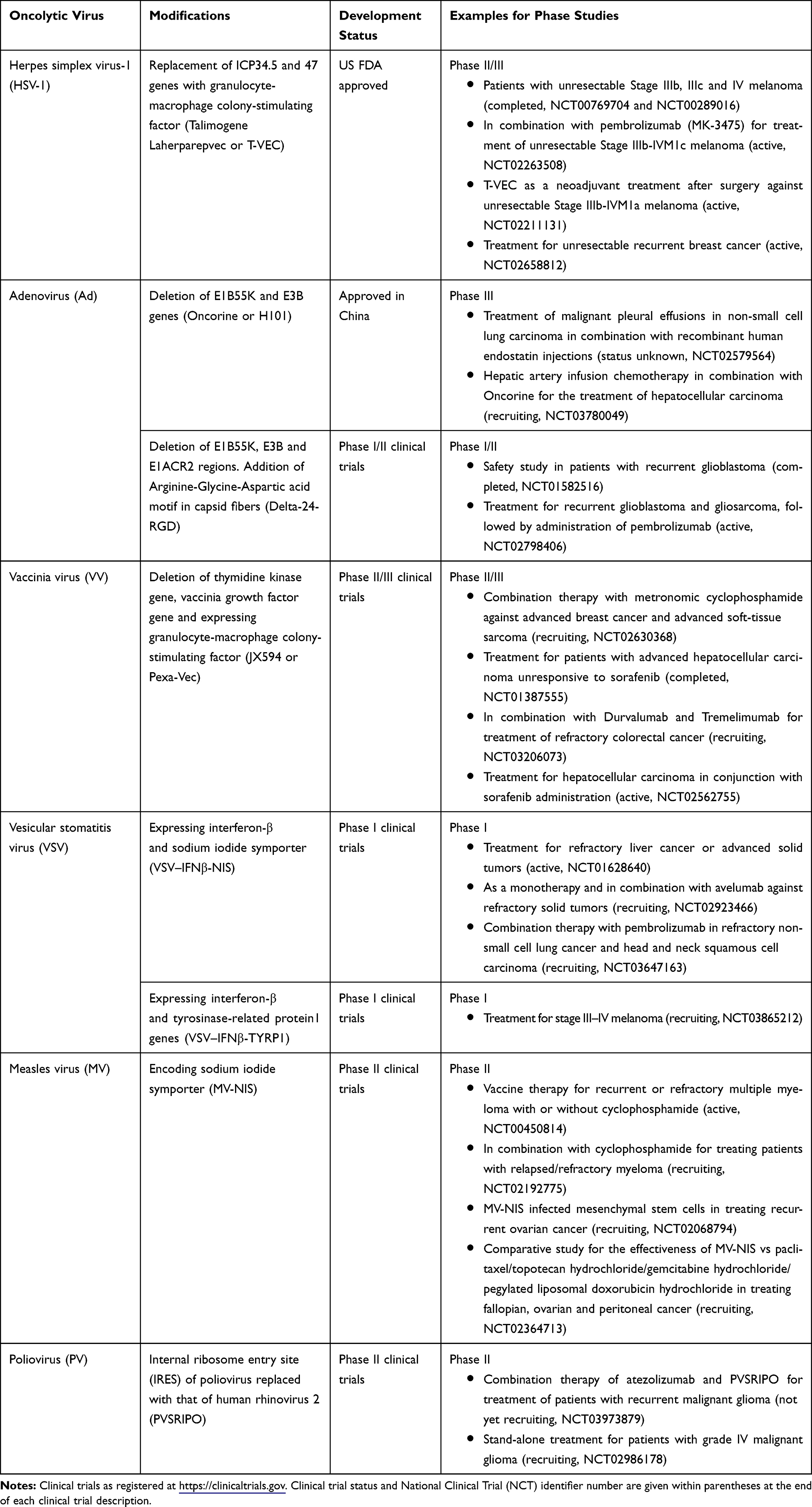 |
Table 1 Modified Oncolytic Viruses in Clinical Trials |
Numerous HAdV strains have been genetically engineered to overcome healthy tissue damage and to selectively target tumors. ONYX-015, one of the first strains of genetically engineered HAdV, was designed to target p53 gene-deficient tumors. A deletion in E1B region prevents the expression of E1B55K protein, that inactivates p53-dependent apoptosis in normal cells.74 In the absence of E1B55K protein, normal cells undergo p53-dependent apoptosis, thereby halting the viral life cycle. By contrast, ONYX-015 has the capacity to replicate in tumors with p53 deficiency, given the function of E1B55K can be compensated by other mechanisms in tumor cells. An in vivo study suggested that ONYX-015 can exert greater antitumor activity when combined with radiotherapy.75 Furthermore, in a Phase II clinical trial in patients with recurrent squamous cell cancer of the head and neck, intratumoral administration of ONYX-015 in conjunction with 5-fluorouracil and cisplatin showed a more significant effect when compared to monotherapy.76 H101, a successor of ONYX-015, has been further modified with a deletion in E3B gene. H101 was the first HAdV to be approved for cancer treatment in China in 2006 under the name of Oncorine.13 Next generation modified adenoviral strains harbor small deletions in E1A gene (E1ACR2 mutants) to suppress the release of E2F transcription factor by ablating the interactions between retinoblastoma protein (pRb) and E1A.77 This modification further restricts adenoviral replication only in tumor cells with activated E2F expression.
In order to make vaccinia virus safer for cancer therapy, two deletions have been made: thymidine kinase gene (TK) and vaccinia growth factor gene (VGF).78 Further attenuated viral strains have been developed by introducing mutations in F14.5L and A56R genes.20 These genes are responsible for encoding a secretory signal peptide and hemagglutinin, respectively. JX-594, a TK-mutant, expressing GM-CSF has been tested in Phase I and Phase II clinical studies in hepatocellular carcinoma and liver cancer as stand-alone treatment or in combination with sorafenib. Collectively, these studies showed a safe yet profound anti-tumor response in JX-594 monotherapy and combination treatment groups in comparison to sorafenib alone.79–81
Several strategies have been tested in reducing PV neurotoxicity: (1) use of live-attenuated poliovirus vaccines,82 (2) delivery of engineered PV genome deficient of P1 coding region (replicons), thereby preventing the formation of new viral progenies and spread,83 (3) A133G mutation in cis-acting replication element (CRE) and relocation of CRE to a spacer region,84 (4) replacement of internal ribosome entry site (IRES) of PV (PVSRIPO) with that of human rhinovirus 2 (HRV2).85 Ribonucleoprotein complex formed in PVSRIPO is incompatible with HRV2 IRES-mediated translation in normal human central nervous system, therefore, rendering the neuronal incompetence of PVSRIPO.86 PVSRIPO has completed a Phase I dose-finding clinical study in patients with grade IV malignant glioma with no neurotoxicity reported.87
Virus Neutralization
A major limitation of the extensive use of OVs in cancer treatment is the rapid neutralization by the immune system, which can restrain the viral spread and reduce the efficacy of repeat administrations.88 Antiviral immune response could hinder viral infection or replication at several stages: 1) Neutralization, opsonization and sequestration prior to cell entry, 2) Inhibition of virus replication by induction of antiviral responses such as type I interferons in infected cells, and 3) Lysis of infected cells by innate immune cells prior to viral-induced lysis of cells. Many of the viruses used in cancer therapy are human pathogens and pre-existing antiviral antibodies obstruct the systemic delivery to the tumor, limiting the potential routes for viral delivery to intratumoral injection. Various pre-clinical and Phase I clinical studies have shown decreased oncolytic viral replication, viral clearance and reduced anti-tumor activity in immunocompetent hosts.89–96
Evidence for pre-existing immunity against oncolytic viruses has been well documented for vaccinia virus due to its use in eradicating the smallpox and also for reovirus, that is universally abundant in the environment.97 In vaccinia virus, neutralizing antibodies have been shown to target H3L envelope protein, that plays an essential role in viral-host cell membrane fusion.98 Structural insights into antibody neutralization of reovirus suggested that neutralizing antibodies sterically hinder the JAM-A receptor binding to reovirus.99 Pre-clinical studies on prostate-specific attenuated replication competent adenovirus (ARCA) showed a decreased antitumor activity in the presence of pre-existing antibodies.92 In the case of measles virus (MV), pre-existing antibodies act as a major limitation in treating cancers in previously vaccinated patients.100 Therefore, MV oncovirotherapy may only be limited for patients with certain cancers such as advanced multiple myeloma, where the immunosuppressed patients have a low level of anti-measles antibodies.101 Furthermore, the administration of T-VEC is limited to intralesional injections for melanoma treatment due to high prevalence of anti-HSV1 antibodies in humans.102 Even in the absence of pre-existing antibodies, the immune system will eventually mount a response and severely reduce the period of virus efficacy to between a few days and couple of weeks, requiring multiple/increased doses of the virus.97,103,104 Even more troublesome is the induction of primary antibody response or augmentation of a low pre-existing antiviral response upon initial administration of low seroprevalence viruses.105,106 Such evidence arises from neutralization of low human seroprevalence viruses such as vesicular stomatitis virus (VSV) in non-immune human serum as early as one hour after exposure.107,108
On the other hand, a major advantage of the stimulating effect raised by OVs on immune system against viruses is that it could enhance epitope spread to tumor antigens. One pre-clinical study on recombinant measles virus VLPs expressing the tumor-specific antigen claudin-6 triggered claudin-6-specific immune responses in melanoma mouse models, ultimately inhibiting tumor metastasis.109 While generally pre-existing immunity for oncolytic viruses reduces their efficacy, a contrary finding was reported in the case of Newcastle disease viruses (NDV) where an augmented therapeutic effect was observed in melanoma mouse models in the presence of NDV-specific antibodies through potentiation of systemic anti-tumor immunity.110
Solutions to Virus Neutralization
The presence of pre-existing anti–viral immunity or the development of neutralizing antibodies upon systemic administration of oncolytic viruses highlights the importance of developing novel strategies to prolong their availability to access tumors. At proof-of-concept level, novel OV delivery methods have been proposed and can be categorized into four distinct groups (Figure 3 and Table 2).
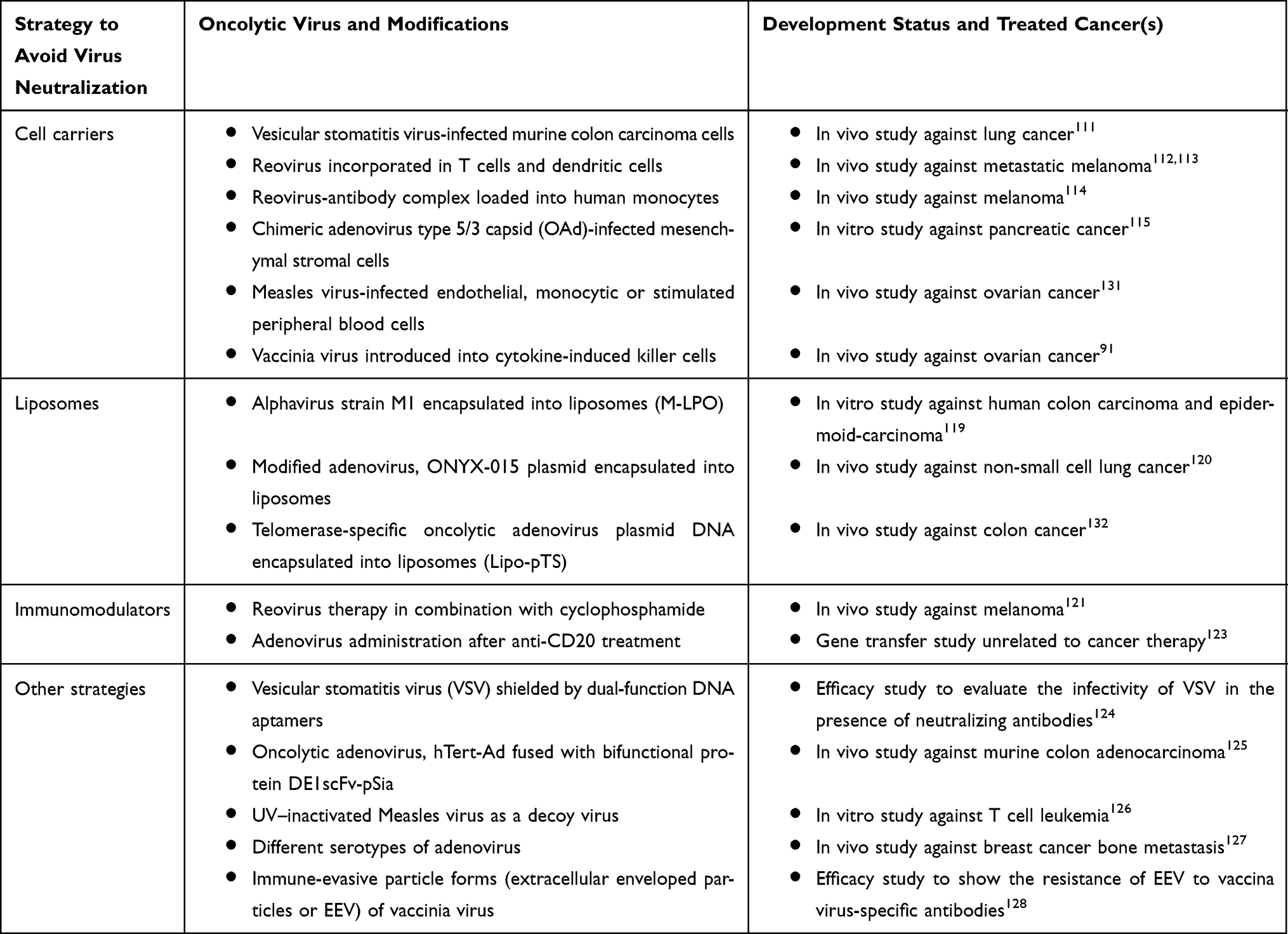 |
Table 2 Strategies to Avoid Oncolytic Virus Neutralization |
 |
Figure 3 Strategies to avoid virus neutralization. (A) Cell carriers such as monocytes (Reovirus-neutralizing antibody complex), dendritic cells (reovirus), endothelial cells (measles virus), stromal cells (adenovirus) and killer cells (vaccinia virus) remain as most extensively researched solutions to bypass the recognition by neutralizing antibodies. (B) Liposomes have been used to incorporate plasmids of oncolytic viruses such as Telomerase-specific oncolytic adenovirus (pTS). (C) Anti-CD20 and cyclophosphamide (immunomodulators) aid in suppressing the antiviral immune response associated with adenovirus and reovirus treatment. (D) Other solutions to virus neutralization include the use of different serotypes of adenovirus strains, sequestration of pre-existing antibodies using UV–inactivated measles virus (decoy virus), and shielding of vesicular stomatitis virus and adenovirus with coadministration of DNA aptamers and bifunctional protein DE1scFv-pSia, respectively. |
Cell Carriers
Encapsulation of virus particles in a carrier is a logical approach as a strategy to conceal the antigenicity of native virions. Several molecular and cellular carriers have been investigated. Murine colon carcinoma cells infected with vesicular stomatitis virus (VSV) homed to cancer cells but not to normal cells, when delivered intravenously in a mouse lung tumor model.111 Reovirus incorporated into dendritic cells and T cells can efficiently deliver the virus into cancer cells in the presence of neutralizing antibodies in vitro112 and in vivo.113 Furthermore, antibody-neutralized reovirus complex can be introduced into human monocytes, where internalized complexes were processed to release infectious particles, ultimately targeting cancer cells.114 Mesenchymal stem cells (MSCs) have been used as a delivery system for chimeric human adenovirus5/3 (HAdV5/3) with the primary purpose of masking the virus from immune attack.115 The cellular receptor for HAdV5 entry, coxsackievirus and adenovirus receptor (CAR) is poorly expressed in MSCs.116 This has been circumvented by swapping the receptor binding fiber knob domain of HAdV5 with that of HAdV3, allowing a CAR-independent cell binding.115
Liposomes
Liposomes are large hydrophilic spherical vesicles, which have been widely used for encapsulation and delivery of diverse range of drugs since they act as a shield from cellular and humoral responses.117,118 In a pre-clinical study, liposomes were used to encapsulate oncolytic alphavirus strain M1 (M-LPO) with anti-tumor efficacy in vitro and a reduced immunogenicity in mice when administered intravenously.119 A similar approach was used to encapsulate a replication-competent, ONYX-015-based plasmid in liposomes.120 Liposomes harboring the plasmids were resistant to antibodies neutralizing the parent strain, while the plasmids could only transfect tumor cells which are p53 deficient.
Immunomodulators
With the aid of conventional immunosuppressants used in the treatment of autoimmune disorders, the host immune response can be partially reduced to favor the targeted delivery of oncolytic viruses to tumors. Combination therapy of cyclophosphamide and reovirus has been shown to rescue reovirus when administered intravenously in mice.121 Currently, there are seven clinical trials that are either completed, active or in recruiting stage for combination or neoadjuvant therapy of metronomic cyclophosphamide with oncolytic viruses such as ONCOS-102 (adenovirus), rQNestin34.5v.2 and T-VEC (HSV-1), MV-NIS (measles virus), and JX-594 (vaccinia virus).122 In a non-cancer related study, T cells and B cells activated by repeated administration of adenovirus have been inhibited by anti-CD 20 en route to assist a successful immunosuppressive regime of liver gene transfer.123
Other Strategies
The use of DNA aptamers to shield viruses from neutralizing antibodies has been shown as a proof-of-concept in vitro study with vesicular stomatitis virus (VSV).124 In this study, aptamers were developed to bind virus surface as well as the antigen-binding fragment (Fab) of anti-VSV antibodies, providing a dual protection mechanism when used concurrently with VSV. Another example for use of bifunctional adapters arises from a recent study on oncolytic adenovirus hTert-Ad treatment in combination with DE1scFv-pSia protein containing a DE1 domain of adenovirus hexon and a polysialic acid-specific single-chain variable fragment (scFv) to capture neutralizing antibodies and for tumor cell recognition, respectively.125
In a different approach, prior treatment of cancer cells with UV–inactivated measles virus prevented the neutralization of the active virus, suggesting the possibility of using a “decoy virus” to sequester pre-existing antibodies.126
Another strategy to counteract host anti-viral immunity is to use different serotypes of the virus (native or modified) or immune-evasive particle forms of the same virus. The feasibility of using different serotypes of adenovirus has been shown in a pre-clinical study, where intravenous administration inhibited the formation of bone metastases.127 Vaccinia virus produces extracellular enveloped particles (EEV) that possess a cell-derived envelope capable of evading neutralizing antibodies.128 Therefore, high EVV-producing strains of vaccinia virus can be engineered to improve the spread of the virus upon systemic delivery.129 In the case of measles virus, N-linked glycosylation of hemagglutinin resulted in strain resistance to a mixture of monoclonal antibodies.130
Conclusion
Oncolytic virotherapy is a promising field of cancer treatment with selective targeting of tumors. However, the antiviral immune response is still a limiting factor hindering the outcome of the treatment. While many OVs have a rapid replication in tumors and direct oncolysis, it is often the antitumor immunity induced by oncolytic activity that contributes to preventing the disease progression and recurrence. When OVs are originally pathogenic to humans, specific targeting of tumors has been achieved either through the manipulation of viral genome to exploit de-regulated signaling pathways in tumors or by modifying viral coat proteins to bind receptors overexpressed in cancer cells. However, the expression levels of attachment/entry receptors specific to OVs differ depending on the type of cancer and/or patient, highlighting the importance of understanding of OV-receptor interactions to modify capsid architecture and re-target cancers. The systemic administration still remains a less effective mean of OV delivery due to the existence of pre-existing neutralizing antibodies or rapid anti-viral immune response after initial treatments. To address this issue, novel treatment strategies have been developed and showed promise in various proof-of-concept and pre-clinical studies: encapsulation of OV in carriers, modification of capsid or envelope proteins, the use of decoy viruses to sequester pre-existing antibodies, multiple administration of different viral serotypes and adjuvant therapy with immunomodulators.
Disclosure
Dr John T. Poirier reports personal fees from Perceiver Pharmaceuticals LLC, outside the submitted work; in addition, Dr John T. Poirier has a patent WO2017096201A1 licensed to Perceiver Pharmaceuticals, LLC. The authors report no other conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Seymour LW, Fisher KD. Oncolytic viruses: finally delivering. Br J Cancer. 2016;114(4):357–361. doi:10.1038/bjc.2015.481
3. Komarova NL, Wodarz D. ODE models for oncolytic virus dynamics. J Theor Biol. 2010;263(4):530–543. doi:10.1016/j.jtbi.2010.01.009
4. Sinkovics JG, Horvath JC. Natural and genetically engineered viral agents for oncolysis and gene therapy of human cancers. Arch Immunol Ther Exp (Warsz). 2008;56(S1):1–59. doi:10.1007/s00005-008-0047-9
5. Southam CM, Moore AE. Clinical studies of viruses as antineoplastic agents with particular reference to Egypt 101 virus. Cancer. 1952;5(5):1025–1034. doi:10.1002/(ISSN)1097-0142
6. Georgiades J, Zielinski T, Cicholska A, Jordan E. Research on the oncolytic effect of APC viruses in cancer of the cervix uteri; preliminary report. Biul Inst Med Morsk Gdansk. 1959;10:49–57.
7. Lorence RM, Roberts MS, Groene WS, Rabin H. Replication-competent, oncolytic newcastle disease virus for cancer therapy. Monogr Virol. 2001;22:160–182.
8. Csatary LK, Eckhardt S, Bukosza I, et al. Attenuated veterinary virus vaccine for the treatment of cancer. Cancer Detect Prev. 1993;17(6):619–627.
9. Asada T. Treatment of human cancer with mumps virus. Cancer. 1974;34(6):1907–1928. doi:10.1002/(ISSN)1097-0142
10. Peters C, Grandi P, Nigim F. Updates on oncolytic virus immunotherapy for cancers. Mol Ther Oncolytics. 2019;12:259–262. doi:10.1016/j.omto.2019.01.008
11. Hu JCC, Coffin RS, Davis CJ, et al. A Phase I Study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737. doi:10.1158/1078-0432.CCR-06-0759
12. Greig SL. Talimogene laherparepvec: first global approval. Drugs. 2016;76(1):147–154. doi:10.1007/s40265-015-0522-7
13. Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98(5):298–300. doi:10.1093/jnci/djj111
14. Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: the Rigvir(R) story. Eur J Pharmacol. 2018;837:117–126. doi:10.1016/j.ejphar.2018.08.042
15. Babiker HM, Riaz IB, Husnain M, Borad MJ. Oncolytic virotherapy including Rigvir and standard therapies in malignant melanoma. Oncolytic Virother. 2017;6:11–18. doi:10.2147/OV.S100072
16. Bradley S, Jakes AD, Harrington K, Pandha H, Melcher A, Errington-Mais F. Applications of coxsackievirus A21 in oncology. Oncolytic Virother. 2014;3:47–55. doi:10.2147/OV.S56322
17. Miles LA, Burga LN, Gardner EE, Bostina M, Poirier JT, Rudin CM. Anthrax toxin receptor 1 is the cellular receptor for seneca valley virus. J Clin Invest. 2017;127(8):2957–2967. doi:10.1172/JCI93472
18. Angelova AL, Aprahamian M, Balboni G, et al. Oncolytic rat parvovirus H-1PV, a candidate for the treatment of human lymphoma: in vitro and in vivo studies. Mol Ther. 2009;17(7):1164–1172. doi:10.1038/mt.2009.78
19. Desjardins A, Sampson J, Peters K, et al. Final results of a Phase 1 trial of an oncolytic Polio/Rhinovirus Recombinant (Pvsripo) against Recurrent Glioblastoma (Gbm). Neuro-Oncology. 2014;16(suppl 5):v13–v13.
20. Zhang Q, Liang C, Yu YA, Chen N, Dandekar T, Szalay AA. The highly attenuated oncolytic recombinant vaccinia virus GLV-1h68: comparative genomic features and the contribution of F14.5L inactivation. Mol Genet Genomics. 2009;282(4):417–435. doi:10.1007/s00438-009-0475-1
21. Di Giovine P, Settembre EC, Bhargava AK, et al. Structure of herpes simplex virus glycoprotein d bound to the human receptor nectin-1. PLoS Pathog. 2011;7(9):e1002277. doi:10.1371/journal.ppat.1002277
22. Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci USA. 2013;110(18):7306. doi:10.1073/pnas.1214441110
23. Carter GC, Law M, Hollinshead M, Smith GL. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol. 2005;86(5):1279–1290. doi:10.1099/vir.0.80831-0
24. Jhawar SR, Thandoni A, Bommareddy PK, et al. Oncolytic viruses-natural and genetically engineered cancer immunotherapies. Front Oncol. 2017;7:202. doi:10.3389/fonc.2017.00202
25. Mansour M, Palese P, Zamarin D. Oncolytic specificity of newcastle disease virus is mediated by selectivity for apoptosis-resistant cells. J Virol. 2011;85(12):6015. doi:10.1128/JVI.01537-10
26. Strong JE, Coffey MC, Tang D, Sabinin P, Lee PWK. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17(12):3351. doi:10.1093/emboj/17.12.3351
27. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642. doi:10.1038/nrd4663
28. Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14:559. doi:10.1038/nrc3770
29. Kepp O, Galluzzi L, Martins I, et al. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. 2011;30(1):61–69. doi:10.1007/s10555-011-9273-4
30. Andtbacka RHI, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. doi:10.1200/JCO.2014.58.3377
31. Puzanov I, Milhem MM, Minor D, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(22):2619–2626. doi:10.1200/JCO.2016.67.1529
32. Park B-H, Hwang T, Liu T-C, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9(6):533–542. doi:10.1016/S1470-2045(08)70107-4
33. Noonan AM, Farren MR, Geyer SM, et al. Randomized phase 2 trial of the oncolytic virus pelareorep (Reolysin) in upfront treatment of metastatic pancreatic adenocarcinoma. Mol Ther. 2016;24(6):1150–1158. doi:10.1038/mt.2016.66
34. Guo ZS, Liu Z, Kowalsky S, et al. Oncolytic immunotherapy: conceptual evolution, current strategies, and future perspectives. Front Immunol. 2017;8:555. doi:10.3389/fimmu.2017.00555
35. Marelli G, Howells A, Lemoine NR, Wang Y. Oncolytic viral therapy and the immune system: a double-edged sword against cancer. Front Immunol. 2018;9:866. doi:10.3389/fimmu.2018.00866
36. Andtbacka RHI, Curti B, Hallmeyer S, et al. Phase II CALM extension study: enhanced immune-cell infiltration within the tumour micro-environment of patients with advanced melanoma following intralesional delivery of Coxsackievirus A21. Eur J Cancer. 2015;51:S677–S677. doi:10.1016/S0959-8049(16)31854-8
37. Silk AW, Kaufman H, Gabrail N, et al. Abstract CT026: phase 1b study of intratumoral Coxsackievirus A21 (CVA21) and systemic pembrolizumab in advanced melanoma patients: interim results of the CAPRA clinical trial. Cancer Res. 2017;77(13 Supplement):CT026.
38. Andtbacka RHI, Curti BD, Hallmeyer S, et al. Phase II calm extension study: coxsackievirus A21 delivered intratumorally to patients with advanced melanoma induces immune-cell infiltration in the tumor microenvironment. J ImmunoTher Cancer. 2015;3(Suppl 2):P343. doi:10.1186/2051-1426-3-S2-P343
39. Zhang Z, Zou W, Wang J, et al. Suppression of tumor growth by oncolytic adenovirus-mediated delivery of an antiangiogenic gene, soluble Flt-1. Mol Ther. 2005;11(4):553–562. doi:10.1016/j.ymthe.2004.12.015
40. Gholami S, Marano A, Chen NG, et al. A novel vaccinia virus with dual oncolytic and anti-angiogenic therapeutic effects against triple-negative breast cancer. Breast Cancer Res Treat. 2014;148(3):489–499. doi:10.1007/s10549-014-3180-7
41. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22(5):1048. doi:10.1158/1078-0432.CCR-15-2667
42. Conry RM, Westbrook B, McKee S, Norwood TG. Talimogene laherparepvec: first in class oncolytic virotherapy. Hum Vaccin Immunother. 2018;14(4):839–846. doi:10.1080/21645515.2017.1412896
43. Cox RM, Plemper RK. Structure and organization of paramyxovirus particles. Curr Opin Virol. 2017;24:105–114. doi:10.1016/j.coviro.2017.05.004
44. Wodarz D, Hofacre A, Lau JW, Sun Z, Fan H, Komarova NL. Complex spatial dynamics of oncolytic viruses in vitro: mathematical and experimental approaches. PLoS Comput Biol. 2012;8(6):e1002547. doi:10.1371/journal.pcbi.1002547
45. Wodarz D. Use of oncolytic viruses for the eradication of drug-resistant cancer cells. J R Soc Interface. 2009;6(31):179–186. doi:10.1098/rsif.2008.0191
46. Duffy S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018;16(8):e3000003. doi:10.1371/journal.pbio.3000003
47. Helenius A. Virus entry: looking back and moving forward. J Mol Biol. 2018;430(13):1853–1862. doi:10.1016/j.jmb.2018.03.034
48. Kumar CS, Dey D, Ghosh S, Banerjee M. Breach: host membrane penetration and entry by nonenveloped viruses. Trends Microbiol. 2018;26(6):525–537. doi:10.1016/j.tim.2017.09.010
49. Jolly CL, Sattentau QJ. Attachment factors. In: Pöhlmann S, Simmons G, editors. Viral Entry into Host Cells. New York: Springer New York; 2013:1–23.
50. Vähä-Koskela M, Hinkkanen A. Tumor restrictions to oncolytic virus. Biomedicines. 2014;2(2):163–194. doi:10.3390/biomedicines2020163
51. McCarthy C, Jayawardena N, Burga LN, Bostina M. Developing picornaviruses for cancer therapy. Cancers. 2019;11(5):685. doi:10.3390/cancers11050685
52. Bostina M. Monoclonal antibodies point to achilles’ heel in picornavirus capsid. PLoS Biol. 2019;17(4):e3000232. doi:10.1371/journal.pbio.3000232
53. Howells A, Marelli G, Lemoine NR, Wang Y. Oncolytic viruses-interaction of virus and tumor cells in the battle to eliminate cancer. Front Oncol. 2017;7:195. doi:10.3389/fonc.2017.00195
54. He Y, Mueller S, Chipman PR, et al. Complexes of Poliovirus serotypes with their common cellular receptor, CD155. J Virol. 2003;77(8):4827. doi:10.1128/JVI.77.8.4827-4835.2003
55. Anderson BD, Nakamura T, Russell SJ, Peng K-W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64(14):4919. doi:10.1158/0008-5472.CAN-04-0884
56. Bergelson JM, Shepley MP, Chan BM, Hemler ME, Finberg RW. Identification of the integrin VLA-2 as a receptor for echovirus 1. Science. 1992;255(5052):1718–1720. doi:10.1126/science.1553561
57. Yu Z, Adusumilli PS, Eisenberg DP, et al. Nectin-1 expression by squamous cell carcinoma is a predictor of herpes oncolytic sensitivity. Mol Ther. 2007;15(1):103–113. doi:10.1038/sj.mt.6300009
58. Carfı́ A, Willis SH, Whitbeck JC, et al. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell. 2001;8(1):169–179. doi:10.1016/S1097-2765(01)00298-2
59. Xiao C, Bator-Kelly CM, Rieder E, et al. The crystal structure of Coxsackievirus A21 and its interaction with ICAM-1. Structure. 2005;13(7):1019–1033. doi:10.1016/j.str.2005.04.011
60. Yoder JD, Cifuente JO, Pan J, Bergelson JM, Hafenstein S. The crystal structure of a Coxsackievirus B3-RD variant and a refined 9-angstrom cryo-electron microscopy reconstruction of the virus complexed with Decay-Accelerating Factor (DAF) Provide a new footprint of DAF on the virus surface. J Virol. 2012;86(23):12571. doi:10.1128/JVI.01592-12
61. Rea VEA, Rossi FW, Paulis AD, Ragno P, Selleri C, Montuori N. 67 kDa laminin receptor: structure, function and role in cancer and infection. Infez Med. 2012;20(Suppl 2):8–12.
62. Gong J, Mita MM. Activated Ras signaling pathways and reovirus oncolysis: an update on the mechanism of preferential reovirus replication in cancer cells. Front Oncol. 2014;4:167. doi:10.3389/fonc.2014.00167
63. Norman KL, Hirasawa K, Yang A-D, Shields MA, Lee PWK. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci U S A. 2004;101(30):11099. doi:10.1073/pnas.0404310101
64. Stojdl DF, Lichty BD, tenOever BR, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4(4):263–275. doi:10.1016/S1535-6108(03)00241-1
65. Morizono K, Xie Y, Ringpis G-E, et al. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat Med. 2005;11:346. doi:10.1038/nm1192
66. Muik A, Stubbert LJ, Jahedi RZ, et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014;74(13):3567–3578. doi:10.1158/0008-5472.CAN-13-3306
67. Nakamura T, Peng K-W, Harvey M, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23(2):209–214. doi:10.1038/nbt1060
68. Dmitriev I, Krasnykh V, Miller CR, et al. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J Virol. 1998;72(12):9706–9713. doi:10.1128/JVI.72.12.9706-9713.1998
69. Nakano K, Asano R, Tsumoto K, et al. Herpes simplex virus targeting to the EGF receptor by a gD-specific soluble bridging molecule. Mol Ther. 2005;11(4):617–626. doi:10.1016/j.ymthe.2004.12.012
70. He B, Gross M, Roizman B. The γ1 34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA. 1997;94(3):843.
71. Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet. 2001;357(9267):1513–1518. doi:10.1016/S0140-6736(00)04638-9
72. Tomazin R, van Schoot NEG, Goldsmith K, et al. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J Virol. 1998;72(3):2560. doi:10.1128/JVI.72.3.2560-2563.1998
73. Kaufman HL, Ruby CE, Hughes T, Slingluff CL. Current status of granulocyte–macrophage colony-stimulating factor in the immunotherapy of melanoma. J ImmunoTher Cancer. 2014;2(1):11. doi:10.1186/2051-1426-2-11
74. Dix BR, O’Carroll SJ, Myers CJ, Edwards SJ, Braithwaite AW. Efficient induction of cell death by adenoviruses requires binding of E1B55k and p53. Cancer Res. 2000;60(10):2666.
75. Rogulski KR, Freytag SO, Zhang K, et al. In vivo antitumor activity of ONYX-015 is influenced by p53 status and is augmented by radiotherapy. Cancer Res. 2000;60(5):1193.
76. Khuri FR, Nemunaitis J, Ganly I, et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6:879. doi:10.1038/78638
77. Martínez-Vélez N, Garcia-Moure M, Marigil M, et al. The oncolytic virus delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun. 2019;10(1):2235. doi:10.1038/s41467-019-10043-0
78. McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61(24):8751.
79. Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99. doi:10.1038/nature10358
80. Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329. doi:10.1038/nm.3089
81. Heo J, Breitbach CJ, Moon A, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther. 2011;19(6):1170–1179. doi:10.1038/mt.2011.39
82. Atsumi S, Matsumine A, Toyoda H, et al. Oncolytic virotherapy for human bone and soft tissue sarcomas using live attenuated poliovirus. Int J Oncol. 2012;41(3):893–902. doi:10.3892/ijo.2012.1514
83. Ansardi DC, Porter DC, Jackson CA, Gillespie GY, Morrow CD. RNA replicons derived from poliovirus are directly oncolytic for human tumor cells of diverse origins. Cancer Res. 2001;61(23):8470–8479.
84. Toyoda H, Yin J, Mueller S, Wimmer E, Cello J. Oncolytic treatment and cure of neuroblastoma by a novel attenuated poliovirus in a novel poliovirus-susceptible animal model. Cancer Res. 2007;67(6):2857–2864. doi:10.1158/0008-5472.CAN-06-3713
85. Jahan N, Wimmer E, Mueller S. A host-specific, temperature-sensitive translation defect determines the attenuation phenotype of a human rhinovirus/poliovirus chimera, PV1(RIPO). J Virol. 2011;85(14):7225–7235. doi:10.1128/JVI.01804-09
86. Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A. 1996;93(6):2370–2375. doi:10.1073/pnas.93.6.2370
87. Desjardins A, Gromeier M, Herndon JE
88. Filley AC, Dey M. Immune system, friend or foe of oncolytic virotherapy? Front Oncol. 2017;7:106. doi:10.3389/fonc.2017.00106
89. Nemunaitis J, Cunningham C, Tong AW, et al. Pilot trial of intravenous infusion of a replication-selective adenovirus (ONYX-015) in combination with chemotherapy or IL-2 treatment in refractory cancer patients. Cancer Gene Ther. 2003;10:341. doi:10.1038/sj.cgt.7700585
90. Parato KA, Senger D, Forsyth PAJ, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965. doi:10.1038/nrc1750
91. Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311(5768):1780. doi:10.1126/science.1121411
92. Chen Y, Yu D-C, Charlton D, Henderson DR. Pre-existent adenovirus antibody inhibits systemic toxicity and antitumor activity of CN706 in the nude mouse LNCaP xenograft model: implications and proposals for human therapy. Hum Gene Ther. 2000;11(11):1553–1567. doi:10.1089/10430340050083289
93. White CL, Twigger KR, Vidal L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (dearing type 3) during a phase I clinical trial. Gene Ther. 2008;15:911. doi:10.1038/gt.2008.21
94. Rudin CM, Poirier JT, Senzer NN, et al. Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res. 2011;17(4):888–895. doi:10.1158/1078-0432.CCR-10-1706
95. Tsai V, Johnson DE, Rahman A, et al. Impact of Human neutralizing antibodies on antitumor efficacy of an oncolytic adenovirus in a murine model. Clin Cancer Res. 2004;10(21):7199. doi:10.1158/1078-0432.CCR-04-0765
96. Sumida SM, Truitt DM, Lemckert AAC, et al. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J Immunol. 2005;174(11):7179. doi:10.4049/jimmunol.174.11.7179
97. Ferguson MS, Lemoine NR, Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv Virol. 2012;2012:1–14. doi:10.1155/2012/805629
98. Davies DH, McCausland MM, Valdez C, et al. Vaccinia virus H3L envelope protein is a major target of neutralizing antibodies in humans and elicits protection against lethal challenge in mice. J Virol. 2005;79(18):11724–11733. doi:10.1128/JVI.79.18.11724-11733.2005
99. Dietrich MH, Ogden KM, Katen SP, et al. Structural insights into reovirus σ1 interactions with two neutralizing antibodies. J Virol. 2017;91(4):e01621–e01616. doi:10.1128/JVI.01621-16
100. Ong HT, Hasegawa K, Dietz AB, Russell SJ, Peng KW. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2006;14:324. doi:10.1038/sj.gt.3302880
101. Dingli D, Peng K-W, Harvey ME, et al. Interaction of measles virus vectors with auger electron emitting radioisotopes. Biochem Biophys Res Commun. 2005;337(1):22–29. doi:10.1016/j.bbrc.2005.08.261
102. Chakradhar S. Viral vanguard: designing cancer-killing viruses to chase metastatic tumors. Nat Med. 2017;23(6):652–655. doi:10.1038/nm0617-652
103. Russell SJ, Peng K-W, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658. doi:10.1038/nbt.2287
104. Wong HH, Lemoine NR, Wang Y. Oncolytic viruses for cancer therapy: overcoming the obstacles. Viruses. 2010;2(1):78–106. doi:10.3390/v2010078
105. Sun JY, Anand-Jawa V, Chatterjee S, Wong KK. Immune responses to adeno-associated virus and its recombinant vectors. Gene Ther. 2003;10(11):964–976. doi:10.1038/sj.gt.3302039
106. Lang SI, Giese NA, Rommelaere J, Dinsart C, Cornelis JJ. Humoral immune responses against minute virus of mice vectors. J Gene Med. 2006;8(9):1141–1150. doi:10.1002/(ISSN)1521-2254
107. Tesfay MZ, Kirk AC, Hadac EM, et al. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J Virol. 2013;87(7):3752. doi:10.1128/JVI.02832-12
108. Tesfay MZ, Ammayappan A, Federspiel MJ, et al. Vesiculovirus neutralization by natural IgM and complement. J Virol. 2014;88(11):6148. doi:10.1128/JVI.00074-14
109. Hutzler S, Erbar S, Jabulowsky RA, et al. Antigen-specific oncolytic MV-based tumor vaccines through presentation of selected tumor-associated antigens on infected cells or virus-like particles. Sci Rep. 2017;7(1):16892. doi:10.1038/s41598-017-16928-8
110. Ricca JM, Oseledchyk A, Walther T, et al. Pre-existing immunity to oncolytic virus potentiates its immunotherapeutic efficacy. Mol Ther. 2018;26(4):1008–1019. doi:10.1016/j.ymthe.2018.01.019
111. Power AT, Wang J, Falls TJ, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15(1):123–130. doi:10.1038/sj.mt.6300039
112. Ilett EJ, Bárcena M, Errington-Mais F, et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clin Cancer Res. 2011;17(9):2767. doi:10.1158/1078-0432.CCR-10-3266
113. Ilett EJ, Prestwich RJ, Kottke T, et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009;16:689. doi:10.1038/gt.2009.29
114. Berkeley RA, Steele LP, Mulder AA, et al. Antibody-neutralized reovirus is effective in oncolytic virotherapy. Cancer Immunol Res. 2018;6(10):1161. doi:10.1158/2326-6066.CIR-18-0309
115. Hammer K, Kazcorowski A, Liu L, et al. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int J Cancer. 2015;137(4):978–990. doi:10.1002/ijc.v137.4
116. Pereboeva L, Komarova S, Mikheeva G, Krasnykh V, Curiel DT. Approaches to utilize mesenchymal progenitor cells as cellular vehicles. Stem Cells. 2003;21(4):389–404. doi:10.1634/stemcells.21-4-389
117. Shikano T, Kasuya H, Sahin TT, et al. High therapeutic potential for systemic delivery of a liposome-conjugated herpes simplex virus. Curr Cancer Drug Targets. 2011;11(1):111–122. doi:10.2174/156800911793743673
118. Wan Y, Han J, Fan G, Zhang Z, Gong T, Sun X. Enzyme-responsive liposomes modified adenoviral vectors for enhanced tumor cell transduction and reduced immunogenicity. Biomaterials. 2013;34(12):3020–3030. doi:10.1016/j.biomaterials.2012.12.051
119. Wang Y, Huang H, Zou H, et al. Liposome encapsulation of oncolytic virus M1 to reduce immunogenicity and immune clearance in vivo. Mol Pharm. 2019;16(2):779–785. doi:10.1021/acs.molpharmaceut.8b01046
120. Yotnda P, Davis AR, Hicks MJ, Templeton NS, Benner MK. Liposomal enhancement of the antitumor activity of conditionally replication-competent adenoviral plasmids. Mol Ther. 2004;9(4):489–495. doi:10.1016/j.ymthe.2004.01.018
121. Qiao J, Wang H, Kottke T, et al. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin Cancer Res. 2008;14(1):259. doi:10.1158/1078-0432.CCR-07-1510
122. ClinicalTrials.gov. Cyclophosphamide, oncolytic virus. Cancer. 2019.
123. Fontanellas A, Hervás-Stubbs S, Mauleón I, et al. Intensive pharmacological immunosuppression allows for repetitive liver gene transfer with recombinant adenovirus in nonhuman primates. Mol Ther. 2010;18(4):754–765. doi:10.1038/mt.2009.312
124. Muharemagic D, Zamay A, Ghobadloo SM, et al. Aptamer-facilitated protection of oncolytic virus from neutralizing antibodies. Mole Ther Nucleic Acids. 2014;3(6):e167–e167. doi:10.1038/mtna.2014.19
125. Niemann J, Woller N, Brooks J, et al. Molecular retargeting of antibodies converts immune defense against oncolytic viruses into cancer immunotherapy. Nat Commun. 2019;10(1):3236. doi:10.1038/s41467-019-11137-5
126. Xu C, Goß AV, Dorneburg C, Debatin K-M, Wei J, Beltinger C. Proof-of-principle that a decoy virus protects oncolytic measles virus against neutralizing antibodies. Oncolytic Virother. 2018. doi:10.2147/OV.S150637
127. Zhang Z, Krimmel J, Zhang Z, Hu Z, Seth P. Systemic delivery of a novel liver-detargeted oncolytic adenovirus causes reduced liver toxicity but maintains the antitumor response in a breast cancer bone metastasis model. Hum Gene Ther. 2011;22(9):1137–1142. doi:10.1089/hum.2011.003
128. Ichihashi Y. Extracellular enveloped vaccinia virus escapes neutralization. Virology. 1996;217(2):478–485. doi:10.1006/viro.1996.0142
129. Kirn DH, Wang Y, Liang W, Contag CH, Thorne SH. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 2008;68(7):2071. doi:10.1158/0008-5472.CAN-07-6515
130. Lech PJ, Tobin GJ, Bushnell R, et al. Epitope dampening monotypic measles virus hemagglutinin glycoprotein results in resistance to cocktail of monoclonal antibodies. PLoS One. 2013;8(1):e52306. doi:10.1371/journal.pone.0052306
131. Iankov ID, Blechacz B, Liu C, et al. Infected Cell carriers: a new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol Ther. 2007;15(1):114–122. doi:10.1038/sj.mt.6300020
132. Aoyama K, Kuroda S, Morihiro T, et al. Liposome-encapsulated plasmid DNA of telomerase-specific oncolytic adenovirus with stealth effect on the immune system. Sci Rep. 2017;7(1):14177. doi:10.1038/s41598-017-14717-x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.