Back to Journals » International Journal of Nanomedicine » Volume 15
Virus-Like Particles Presenting the FGF-2 Protein or Identified Antigenic Peptides Promoted Antitumor Immune Responses in Mice
Authors Shu C, Sun P, Xie H, Huang W, Qi J, Ma Y
Received 3 November 2019
Accepted for publication 25 February 2020
Published 24 March 2020 Volume 2020:15 Pages 1983—1996
DOI https://doi.org/10.2147/IJN.S237182
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Congyan Shu 1,2, Pengyan Sun 3, Hanghang Xie 1, Weiwei Huang 1, Jialong Qi 1, Yanbing Ma 1
1Institute of Medical Biology, Chinese Academy of Medical Science & Peking Union Medical College, Kunming 650118, People’s Republic of China; 2Sichuan Institute for Food and Drug Control, Chengdu, 611731, People’s Republic of China; 3Yunnan Center for Disease Control and Prevention, Kunming 650022, People’s Republic of China
Correspondence: Yanbing Ma
Laboratory of Molecular Immunology, Institute of Medical Biology, Chinese Academy of Medical Sciences & Peking Union Medical College, Kunming 650118, People’s Republic of China
Tel +86 871 68339287
Fax +86 871 68334483
Email [email protected]
Background: Fibroblast growth factor (FGF)-2 is overexpressed in various tumor tissues. It affects tumor cell proliferation, invasion and survival, promotes tumor angiogenesis and is tightly involved in the development of systemic and local immunosuppressive tumor mechanisms.
Purpose: This study aimed to develop an effective vaccine against FGF-2 and to investigate the effects of anti-FGF-2 immunization on tumor growth and antitumor immune responses.
Methods: A set of thirteen synthesized overlapping peptides covering all possible linear B-cell epitopes of murine FGF-2 and a recombinant FGF-2 protein were conjugated to virus-like particles (VLPs) of recombinant hepatitis B core antigen (HBcAg). The VLPs were immunized through a preventive or therapeutic strategy in a TC-1 or 4T1 grafted tumor model.
Results: Immunization with FGF-2 peptides or full-length protein-coupled VLPs produced FGF-2-specific antibodies with a high titer. Peptide 12, which is located in the heparin-binding site of FGF-2, or protein-conjugated VLPs presented the most significant effects on the suppression of TC-1 tumor growth. The levels of IFN-γ-expressing splenocytes and serum IFN-γ were significantly elevated; further, the immune effector cells CD8+ IFN-γ+ cytotoxic T lymphocytes (CTLs) and CD4+ IFN-γ+ Th1 cells were significantly increased, whereas the immunosuppressive cells CD4+ CD25+ FOXP3+ Treg cells and Gr-1+ CD11b+ myeloid-derived suppressor cells (MDSCs) were decreased in the immunized mice. In addition, VLP immunization significantly suppressed tumor vascularization and promoted tumor cell apoptosis. In mice bearing 4T1 breast tumor, preventive immunization with FGF-2-conjugated VLPs suppressed tumor growth and lung metastasis, and increased effector cell responses.
Conclusion: Active immunization against FGF-2 is a new possible strategy for tumor immunotherapy.
Keywords: fibroblast growth factor-2, virus-like particle, VLP, anticytokine active immunization, tumor, immune response
Introduction
Neoangiogenesis is necessary for delivering nutrients and oxygen into tumor tissues to support the fast growth of tumor cells, to transport metabolic waste out of the tumor, and to support the occurrence of tumor invasion and metastasis.1,2 In addition, abnormal proliferation of vessels in tumors leads to a hypoxic microenvironment,3,4 which facilitates the production of immunosuppressive cells, including myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), tumor-associated macrophages (TAMs) and immature DCs;5 additionally, abnormal vessel proliferation promotes cell migration, ultimately leading to the cells accumulating in tumor tissues. The immunosuppressive cells secrete some important cytokines to further promote tumor neovascularization, which directly stimulates the proliferation and migration of endothelial cells6 or cooperates with the cytokines produced by tumor cells7 and other tumor microenvironment cells. On the other hand, immunosuppressive cells can downregulate the functions of antitumor effector cells through direct action8 or secretion of some suppressive mediators,9 facilitating the development of tumor immune suppression and escape.10 Therefore, targeting tumor angiogenesis represents an important strategy for tumor immunotherapy that aims at cutting off the tumor nutrition supply and modifying the immunosuppressive tumor microenvironment.
Bevacizumab is the first humanized monoclonal antibody approved by the FDA to clinically treat cancer by suppressing tumor angiogenesis, which binds to VEGF and blocks its pathological functions in tumors.11 The combined use of bevacizumab and other chemotherapeutic drugs has attained significant efficacy in some cancers, such as metastatic colon carcinoma.12–14 Bevacizumab has been approved for the treatment of many cancers, including colon carcinoma, breast cancer,15 nonsquamous non-small-cell lung cancer,16 ovarian cancer,17 renal cancers,18 head and neck cancer,19 and glioblastoma multiforme of the brain.20 However, drug resistance to bevacizumab or even tumor recurrence was found after clinical use in some cancers, which may be attributed to the compensatory upregulation of some other angiogenesis factors.21–23
Fibroblast growth factor-2 (FGF-2) is one of the most important protumor angiogenesis factors24 and can promote tumor neovascularization independent of VEGF.25 It was reported that FGF-2 is a key contributor to bevacizumab resistance26 and the drug resistance of chemotherapy in breast cancer and melanoma.27–29 FGF-2 directly stimulates the survival and proliferation of tumor cells and indirectly facilitates tumor growth and metastasis by promoting the proliferation and migration of endothelial cells.30 Many kinds of FGF-2 signaling inhibitors have been developed in preclinical studies or clinical trials, including nonspecific small-molecule tyrosine kinase inhibitors, such as dovitinib31 and nintedanib,32 as well as specific antagonistic large protein drugs, such as monoclonal antibodies33 and soluble receptor fusion proteins.34,35
This current study sought to reveal the potential of active immunization targeting FGF-2 to modify immunosuppressive mechanisms in tumors and to promote antitumor immune responses, which may also provide a new optional approach to block tumor neovascularization and overcome drug resistance to anti-VEGF treatment.
Materials and Methods
Mouse and Cell Lines
Female C57BL/6 and BALB/C mice (6–8 weeks; 16–18 g; SCXK [Jing] 2012–0001) were purchased from Vital River Laboratory Animal Technology, Ltd. (Beijing, China). All mice were kept in the Central Animal Care Services of Institute of Medical Biology, CAMS and PUMC under specific pathogen-free (SPF) conditions. All mice were randomly divided into 5 per group. All protocols followed in the animal experiments were approved by the Animal Ethics Committee of Institute of Medical Biology ([2017]16), and in accordance with the principles of “Guide for the Care and Use of Laboratory Animals” and “The Guidance to Experimental Animal Welfare and Ethical Treatment”. All efforts were made to minimize animal suffering.
TC-1 tumor cells and 4T1 breast tumor cells were provided by the Tumor Center of the Chinese Academy of Medical Sciences. The cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium with 10% fetal bovine serum. TC-1 cells were derived from primary lung epithelial cells from C57BL/6 mice co-transformed with the HPV-16 oncoproteins E6 and E7 and the c-Ha-ras oncogene.
Peptides and Vaccine Preparation
The sequence of the mature mouse FGF-2 molecule (154 amino acids, GenBank accession number: 14173) was divided into 7 connecting peptides with lengths varying from 18 to 26 aa, and another set of 6 peptides was designed to overlap half of the two adjacent peptides from the above 7 peptides (Figure 1A). A total of 13 peptides were synthesized by Sangon Biotech (Shanghai, People’s Republic of China). The mature FGF-2 protein was prepared by recombinant technology with thioredoxin (Trx) fused at the N terminus. The expressed fusion protein Trx-FGF-2 was purified with a successive chromatography procedure consisting of cationic exchange, heparin affinity, and size exclusion chromatography. Preparation of the carrier HBcAg VLPs was performed as previously described.36 A lysine was inserted between 78 and 79 amino acids of truncated HBcAg (1–149 amino acid, GenBank accession number: GQ 377581) using gene engineering methods. The vaccine was prepared by conjugating peptides and a recombinant protein to HBcAg VLPs with the coupling agent 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) (Sigma-Aldrich, USA), following the manufacturer’s instructions.
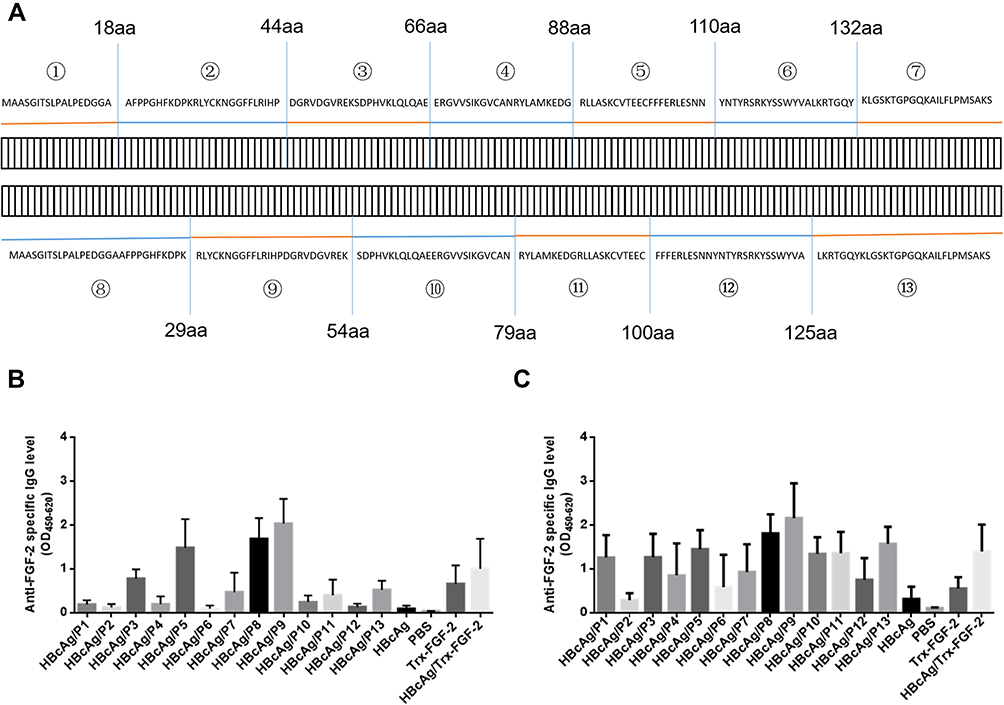 |
Figure 1 Immunization with FGF-2 peptide- or protein-conjugated VLPs elicited specific antibody responses. A set of overlapping peptides was designed to cover all possible linear B cell epitopes of the mouse FGF-2 molecule. The mice were immunized subcutaneously with FGF-2 protein- or peptide-conjugated HBcAg VLPs three times at two-week intervals. (A) Amino acid sequences of the FGF-2 protein and synthetic peptides; (B) IgG responses induced at one week after the second immunization; (C) IgG responses induced at one week after the third immunization (n = 5/group). |
Tumor Challenge and Mouse Immunization
A grafted TC-1 tumor model was established as previously described. Briefly, 1×105 TC-1 tumor cells mixed with Basement Membrane Matrix (BD Biosciences, San Jose, CA, USA) were injected subcutaneously (s.c.) into the right flank of the C57BL/6 mice. For grafted 4T1 breast tumor model, 1×105 4T1 cells were orthotopically injected into the fourth mammary gland fat pad of BALB/c mice under isoflurane inhalation anesthesia.
The protocol for the preventive experiment is shown in Figure 2A. Mice were first immunized subcutaneously (s.c.) with 50 μg of FGF-2 protein or peptide-conjugated VLPs, and carrier VLPs or PBS was used as controls. The immunization was performed three times at an interval of two weeks and then challenged with TC-1 cells two weeks after the last immunization. For the treatment experiment, mice were immunized once in advance and then received TC-1 inoculation one week later. When the tumor diameter reached 5–6 mm, two booster immunizations were given at an interval of two weeks. Tumor growth was monitored periodically (twice a week) using a slide caliper, and tumor volume was calculated as1/2 length × width2. Mice were euthanized when the tumor diameter reached approximately 15 mm, which was set as the humane endpoint of the experiments based on the animal care regulations. The protocol is shown in Figure 3A.
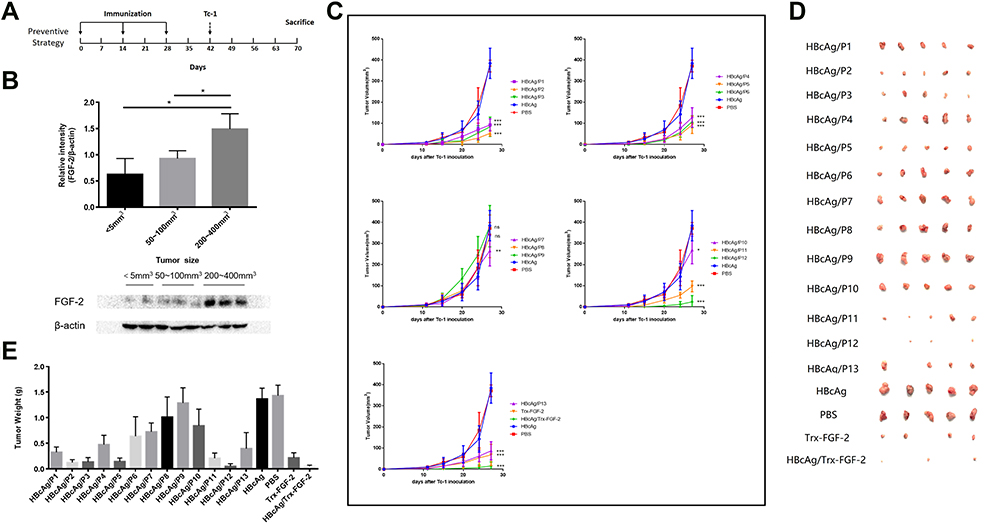 |
Figure 2 Preventive immunization with FGF-2 protein- or peptide-conjugated VLPs significantly inhibits the growth of TC-1 graft tumors. A graft tumor model of TC-1 cells was employed, and FGF-2-specific IgG responses were induced before the inoculation of tumor cells. (A) The protocol applied in the mouse model; (B) the expression level of FGF-2 in tumor tissues; (C) tumor growth dynamically monitored twice a week. *Represents a comparison with the HBcAg control (ns no significant, *p < 0.05, **p < 0.01, and ***p < 0.001; n = 5/group); (D) representative pictures of the size of tumor masses; (E) statistical analyses of tumor weight. |
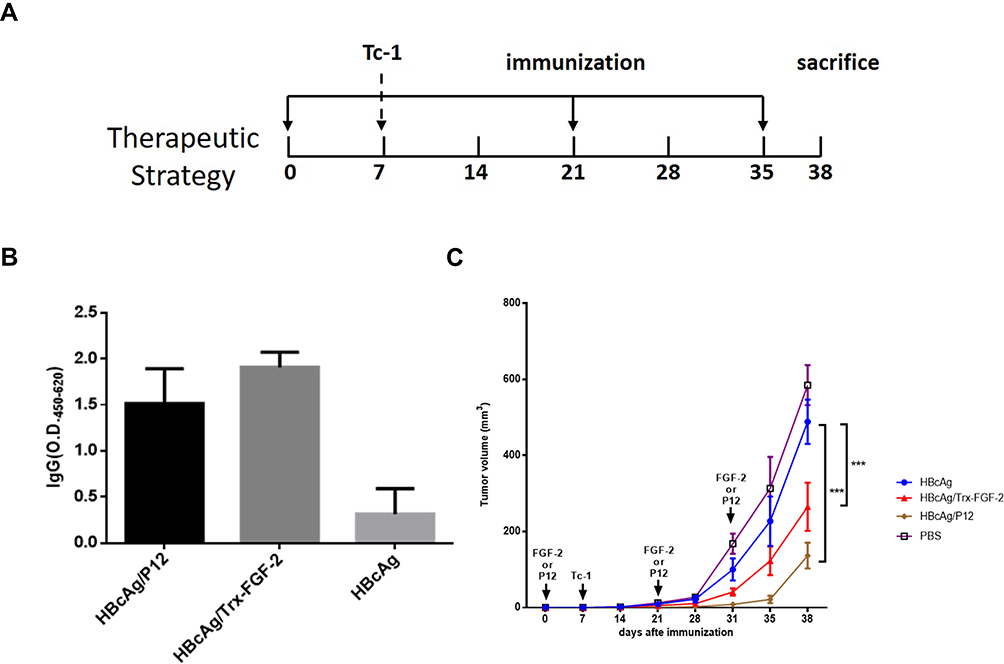 |
Figure 3 Therapeutic immunization with FGF-2 protein- or peptide 12-conjugated VLPs suppresses the growth of established tumors. Effective IgG antibody responses were induced after the tumor size reached a diameter of 5–6 mm, and the carrier or PBS was used instead of FGF-2 protein- or peptide 12-conjugated VLPs as the controls. (A) The protocol applied in the mouse model; (B) the FGF-2-specific IgG responses from the third immunization; (C) tumor growth was dynamically monitored once a week (***p < 0.001; n = 5/group). |
Enzyme-Linked Immunosorbent Assay (ELISA)
Measurements of FGF-2-specific IgG antibodies (BioLegend, Inc., San Diego, CA, USA) were performed by an ELISA. Briefly, 1 μg/mL of FGF-2 protein was coated onto the microplates overnight at 4°C, and then, serum samples at a 1:1000 dilution were loaded and incubated for 1 hr at room temperature. Further, HRP-conjugated goat anti-mouse IgG was added and incubated for another 1 hr; finally, the reaction was developed using TMB Single-Component Substrate Solution (Solarbio, Beijing, China). Between each step, 5 microplate washes were performed. OD450 values were detected with an ELISA reader.
The concentration of IFN-γ in the serum was also determined by ELISA using the double antibody sandwich method, according to the manufacturer’s instructions. The paired capture and biotinylated detection antibodies were purchased from (Affymetrix eBioscience, Inc., San Diego, CA, USA). Briefly, the capture antibodies were coated onto microplates at 4°C overnight, and 100 μL of serum samples at a 1:50 dilution were loaded and incubated at 37°C for 1 hr, followed by 1 hr incubation with biotinylated detection antibodies. Then, HRP-labeled streptavidin (Beyotime, Shanghai, China) was added and incubated for 1 hr at 37°C. Finally, TMB Single-Component Substrate Solution (Solarbio, Beijing, China) was used to develop the assay at 37°C, and 1 M HCl was added to stop the reaction. OD450 values were obtained with an ELISA reader.
Enzyme-Linked Immunospot Assay (ELISPOT)
The mice were sacrificed at the end of the experiment, and the spleens were isolated, weighed and dispersed using a 70 μm cell strainer (Becton, Dickinson and Company FalconTM, USA). Then, the lymphocytes were isolated using mouse lymphocyte separation medium (DAKEWE Biotechnology Co., Ltd., Shenzhen, China) according to the manufacturer’s instructions. Antigen-specific IFN-γ-producing lymphocytes were detected using an ELISPOT kit (DAKEWE Biotechnology Co., Ltd., Shenzhen, China) following the manufacturer’s protocol. Briefly, splenocytes were plated into 96-microwell plates at 3×105 cells per well and incubated with 5 μg/mL of an HPV16 E7 peptide (E749-57) or irradiated 4T1 cell lysates (1×105/mL) for 16–20 hr at 37°C and 5% CO2. The cells stimulated with PMA and ionomycin were used as positive controls, and the cells incubated with medium only were used as negative controls. Spots were counted using an ELISPOT reader system (AID Diagnostika GmbH, Straßberg, Germany), and the results were calculated as the mean number of IFN-γ-positive cells per 3×105 splenocytes ± standard error of the mean (SEM).
Flow Cytometry
Surface staining and intracellular staining for spleen lymphocytes were performed according to the manufacturer’s protocols. Briefly, 1×106 lymphocytes were stimulated for 7 hrs with 5 μg/mL E749–57 peptide or 2 μL cell activation cocktail (BioLegend, Inc., San Diego, CA, USA), which is composed of phorbol 12-myristate 13-acetate (PMA) and ionomycin, and cytokine release was blocked by adding brefeldin A (BioLegend, Inc., San Diego, CA, USA) for the last 4 hrs of culture. Cells were harvested, washed and stained with phycoerythrin (PE)-anti-mouse CD8 for cytotoxic T lymphocytes (CTLs), FITC-anti-mouse CD4 for Th1 cells, FITC-anti-mouse CD4 and allophycocyanin (APC)-anti-mouse CD25 for Tregs, or FITC-anti-mouse CD11b and APC-anti-mouse Gr-1 monoclonal antibodies (mAbs) for MDSCs. After surface staining, cells stained for CTL, Th1, and Treg markers were fixed with fixation/permeabilization buffer for IFN-γ staining or transcription factor fix/perm buffer for Foxp3 staining (BioLegend, Inc., San Diego, CA, USA) according to the manufacturer’s protocol. The cells were then followed for intracellular staining with an APC-conjugated anti-mouse IFN-γ mAb or a PE-conjugated anti-mouse Foxp3 mAb. All mAbs were purchased from BioLegend. Cells were analyzed by BD Accuri C6 (BD Biosciences, San Jose, CA, USA), and the data were analyzed using FlowJo Software vX (Tree Star, Inc., San Diego, CA, USA).
Immunofluorescence
The isolated tumor tissues were fixed in formalin, embedded in paraffin and sectioned. After dewaxing and hydration, the slides were immersed in EDTA antigen retrieval buffer. Then, the slides were incubated with a fluorescence-labeled anti-CD31 or anti-caspase-3 antibody (Abcam, plc, Cambridge, UK) overnight at 4°C in a wet box. The objective tissue was covered with secondary antibody (Abcam, plc, Cambridge, UK) and incubated at room temperature for 50 min in dark conditions. After that, the slides were incubated with DAPI solution at room temperature for 10 min in the dark. Finally, images were collected by fluorescence microscopy, and pixels of the positive area were analyzed.
Statistical Analysis
The differences between experimental groups were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test (GraphPad Software, San Diego, CA, USA). Values are reported as the mean ± SEM. Results with p < 0.05 were considered statistically significant. *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Immunization with FGF-2 Peptides or Protein-Conjugated VLPs Elicited Specific Antibody Responses
A set of synthetic peptides covering all possible linear B cell epitopes of the FGF-2 molecule is shown in Figure 1A. The sera collected from the mice immunized with FGF-2 peptides or recombinant protein-conjugated VLPs presented a specific reaction with coated commercially derived FGF-2 protein in ELISA. One week after the second immunization, peptides 9, 8, 5 and 3 elicited specific antibody responses with a level comparable to or even higher than that induced by FGF-2 protein-conjugated VLPs; peptides 13, 11, and 7 also produced apparent antibody responses; compared to the control, the other peptides except for peptide 6 showed slight antibody responses (Figure 1B). After the third immunization, almost all peptides elicited obvious specific antibody responses but with varied reaction strengths (Figure 1C). Most of the peptides, except for peptides 2, 4, 6, and 7, produced a comparable antibody response to that induced by FGF-2 protein-conjugated VLPs. The antibody responses elicited by peptides 13, 11, 12, 10, and 1 were elevated dramatically from a quite low level after the second immunization to a level similar to that produced by protein-conjugated VLPs after the third immunization.
Preventive Immunization with FGF-2 Protein- or Peptide-Conjugated VLPs Significantly Inhibits the Growth of TC-1 Graft Tumors
A grafted tumor model of TC-1 cells that persistently express and present HPV16 E6 and E7 antigens was employed to assess whether anti-FGF-2 active immunization could significantly affect tumor growth and antitumor immune responses (Figure 2A). We first showed that, in this model, the expression level of FGF-2 in tumor tissue increased with tumor growth (Figure 2B), implying possible roles of FGF-2 in the development of TC-1 tumors. All immunized mice were challenged with TC-1 cells two weeks after the last immunization with VLPs. The results showed that the mice immunized with VLPs conjugated with the peptides 12, 5, 2, 3, 11, 1 and 13 generated the most significant suppression on tumor growth, and compared to the carrier or PBS control, the other peptide-conjugated VLPs except for peptide 8 and 9 also produced tumor suppression efficacy to some extent (Figure 2C). The tumor masses were carefully isolated and weighed. Representative pictures of tumor masses are shown in Figure 2D, and the statistical analyses of tumor weight are shown in Figure 2E. The results of the tumor weight measurements were consistent with those of the tumor volume measurements. Among all the peptides, peptide 12 had the most significant tumor-suppressive effect, which was close to that obtained by immunization with FGF-2 protein-conjugated VLPs.
Preventive Immunization with FGF-2 Protein- or Peptide 12-Conjugated VLPs Promotes the Antitumor Cellular Immune Response and Decreases the Level of Immunosuppressive Cells in TC-1 Bearing Mice
The responses of Th1/CTL cells and their relative effector molecules, such as IFN-γ, are key components of adaptive antitumor immunity. The ELISPOT assay showed that immunization with FGF-2 protein- or peptide 12-conjugated VLPs produced significantly increased levels of HPV16 E7 peptide-stimulated IFN-γ-expressing splenocytes than did immunization with the carrier VLPs (Figure 4A and B). Consistent with the ELISPOT results, an increased IFN-γ level was found in the sera of immunized mice by ELISA (Figure 4C). In addition, compared to the carrier, FGF-2 protein- or peptide 12-conjugated VLPs elicited an increased frequency of IFN-γ+ CD8+ CTLs (Figure 4D) and IFN-γ+ CD4+ Th1 cells (Figure 4E). The results strongly indicate that active immunization targeting FGF-2 has the potency to promote antitumor cellular immune responses.
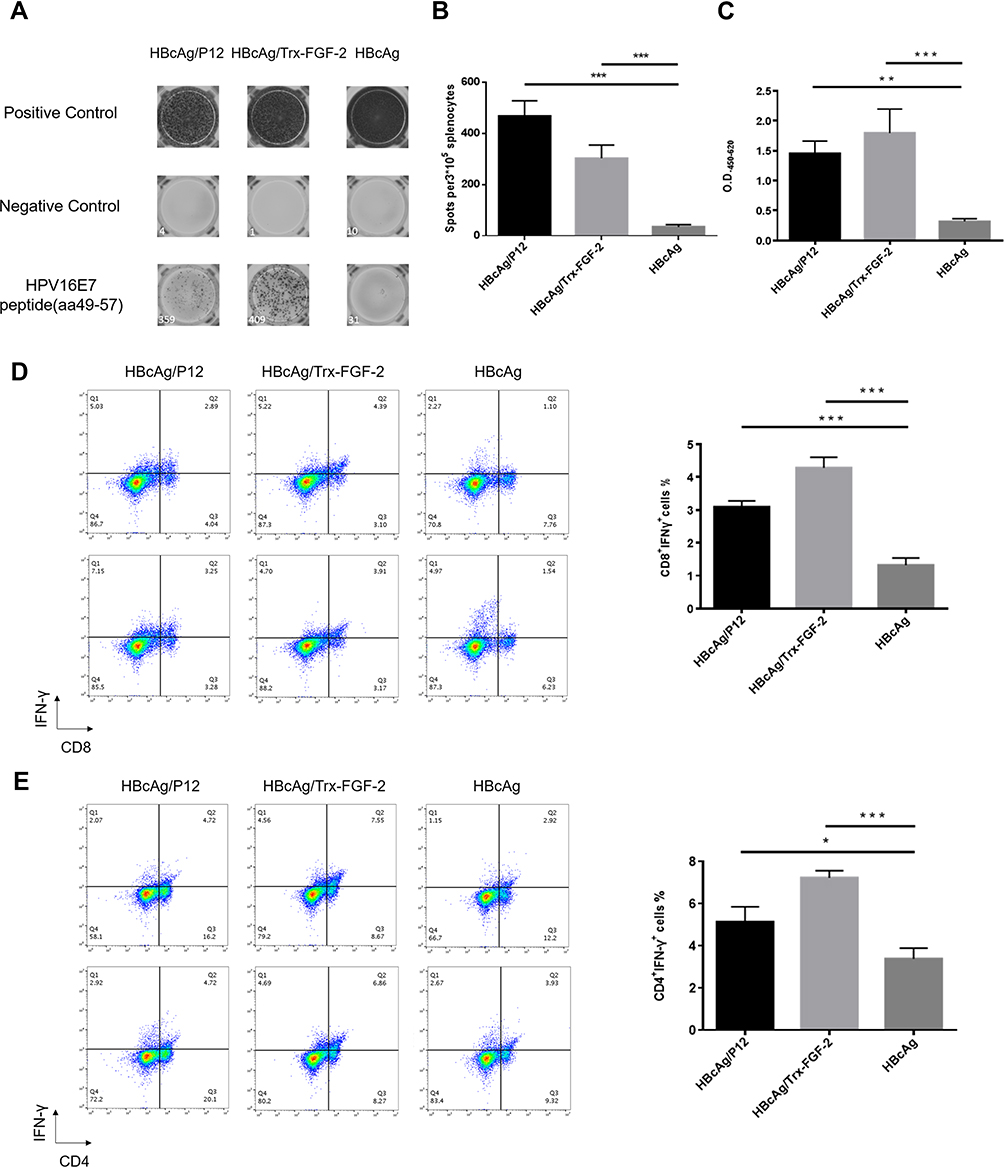 |
Figure 4 Preventive immunization with FGF-2 protein or peptide 12-conjugated VLPs promotes the responses of antitumor cellular immune responses. The isolated splenocytes were cultured and stimulated with the antigenic peptide E749–57, and then ELISPOT and flow cytometry analyses were performed. (A) Representative picture of ELISPOT showing the responses of IFN-γ+-expressing lymphocytes; (B) statistical analyses on the results of ELISPOT; (C) the IFN-γ level in serum detected by ELISA; (D) percentage of CD8+ IFN-γ+ lymphocytes in splenocytes (right) and representative flow cytometry dot plots for each group (left); (E) percentage of CD4+ IFN-γ+ lymphocytes in splenocytes (right) and representative dot plots (left) (*p < 0.05, **p < 0.01, and ***p < 0.001; n = 5/group). |
In addition, we also analyzed the frequency of Tregs and MDSCs, which play key roles in the initiation and development of tumor immune suppression. Compared to immunization with the carrier, immunization with FGF-2 protein- or peptide 12-conjugated VLPs significantly decreased the CD4+ CD25+ Foxp3+ and CD11b+ Gr-1+ cell frequencies (Figure 5), indicating that the targeting of FGF-2 through an active immunization approach may also affect the production of immunosuppressive Tregs and MDSCs.
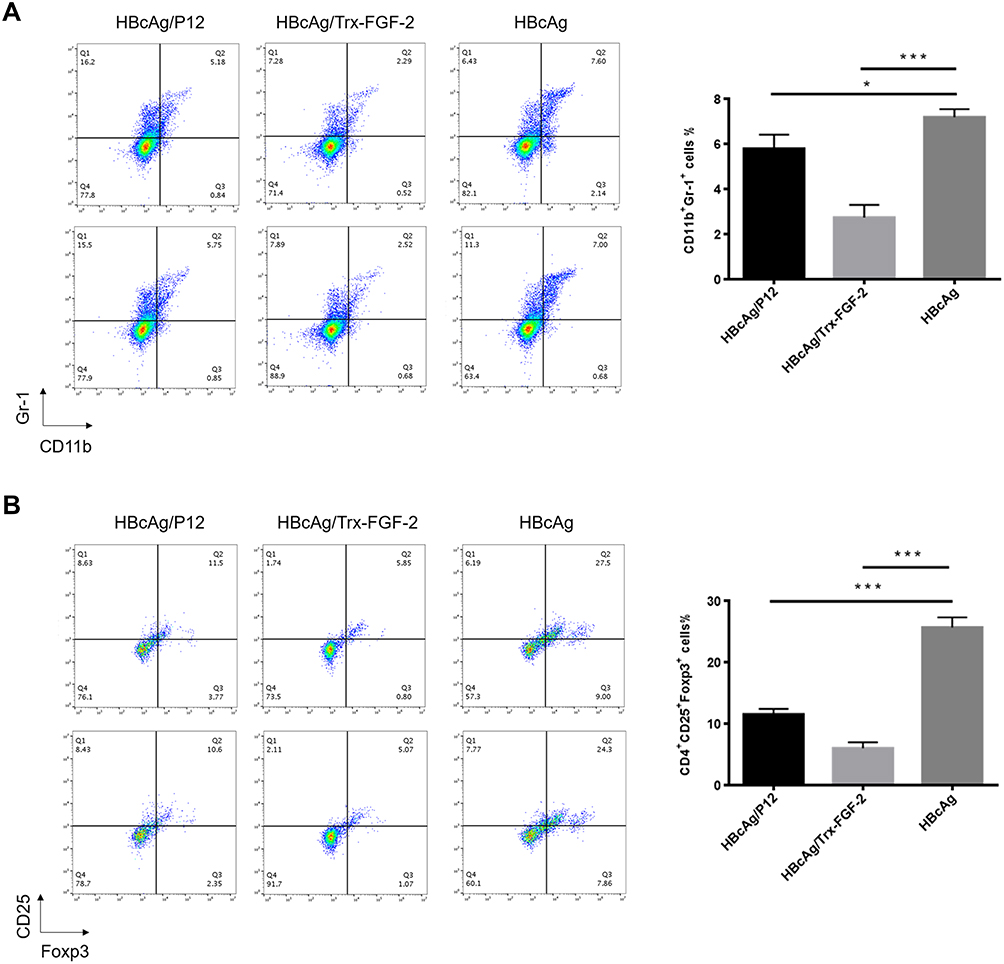 |
Figure 5 Preventive immunization with FGF-2 protein- or peptide 12-conjugated VLPs decreases the level of immunosuppressive cells. (A) Percentage of CD25+ Foxp3+ cells in CD4+ splenocytes (right) and representative dot plots (left); (B) percentage of Gr-1+ CD11b+ MDSCs in isolated splenocytes (right) and representative dot plots (left) (*p < 0.05, and ***p < 0.001; n = 5/group). |
Preventive Immunization with FGF-2 Protein- or Peptide 12-Conjugated VLPs Decreases Vascular Formation in Tumor Tissue and Promotes Tumor Cell Apoptosis in TC-1 Bearing Mice
Tumor growth requires a large amount of neoangiogenesis to provide adequate nutrition, and the excessive abnormal proliferation of blood vessels facilitates the formation of the hypoxic microenvironment of tumor tissue, which further promotes the development of tumor immune suppression and immune escape. FGF-2 is a key tumor angiogenesis factor, and active immunization against FGF-2 has the potential to affect tumor vascularization. Immunofluorescence analyses showed that immunization with FGF-2 protein- or peptide 12-conjugated VLPs significantly decreased the content of CD31-positive cells, which were labeled with a red fluorescent marker, indicating that the amount of vasculature in tumor tissue was reduced in the immunized mice in comparison with that in the mice receiving the carrier (Figure 6A). Suppressing vascularization or increasing antitumor immune responses may induce tumor cell apoptosis. Thus, we also analyzed the apoptosis level of tumor tissue cells by labeling caspase 3 with a green fluorescent marker. The results showed that the area and strength of green fluorescence were significantly increased in the immunized mice compared with those in the control mice, indicating that the apoptosis of tumor cells was significantly increased after targeting FGF-2 through an active immunization approach (Figure 6B).
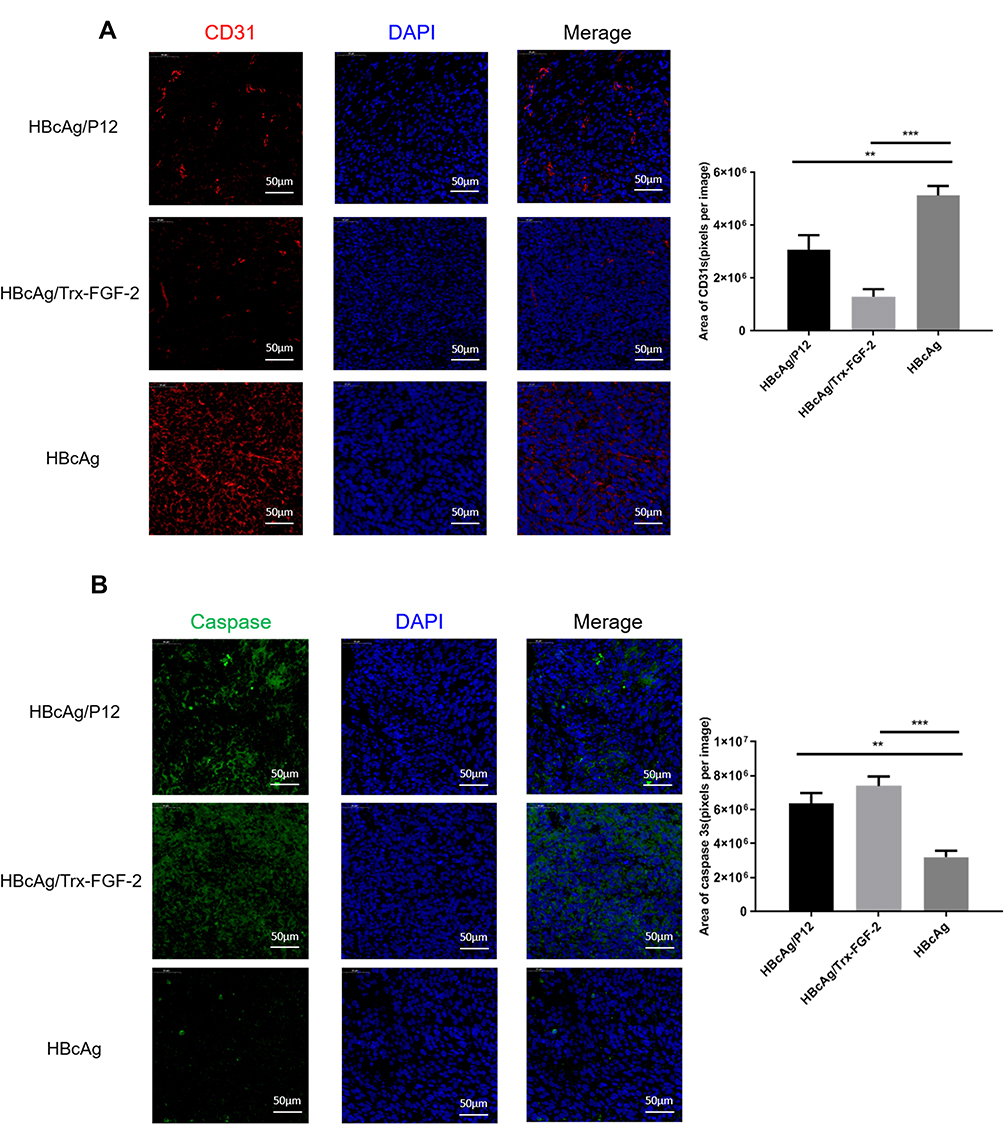 |
Figure 6 Preventive immunization with FGF-2 protein- or peptide 12-conjugated VLPs decreases vascular formation in tumor tissue and promotes tumor cell apoptosis. (A) Representative pictures for anti-CD31-stained tumor tissue sections analyzed by an immunofluorescence assay (40×, bar=50um) (left) and statistical analyses on the area of blood vessels expressing CD31 with ImageJ software (right); (B) Representative pictures for anti-caspase 3-stained tumor tissue sections analyzed by an immunofluorescence assay (40×, bar=50um) (left) and statistical analyses on the caspase 3-positive area with ImageJ software (right) (**p<0.01, and ***p<0.001; n=5/group). |
Therapeutic Immunization with FGF-2 Protein- or Peptide 12-Conjugated VLPs Suppresses the Growth of Established TC-1 Tumors
To further investigate whether active immunization against FGF-2 still has the potential to affect antitumor immune responses when a tumor has been fully established, a brief experiment with a therapeutic immunization approach was conducted. In this experiment, effective antibody responses were induced after the tumor size reached a diameter of 5–6 mm, and the carrier or PBS was used as the controls (Figure 3A). Mice immunized with FGF-2 protein- or peptide 12-conjugated VLPs still produced high tiered FGF-2-specific antibody responses (Figure 3B), and the titer reached 1:64,000. Compared with the control mice, the immunized mice showed a significant suppression of tumor growth (Figure 3C). The results indicated that active immunization against FGF-2 with VLPs was still capable of effectively breaking B cell immune tolerance and eliciting a high level of FGF-2-specific antibody responses; additionally, the antibody responses had effectively intervened in the further growth of established tumors, even when tumor immune suppression had already developed.
Preventive Immunization with FGF-2 Protein-Conjugated VLPs Suppresses the Primary Tumor Growth and Lung Metastasis in the 4T1 Model
We further tested the effects of anti-FGF-2 active immunization in a model of orthotopically grafted 4T1 breast tumor. A preventive immunization was conducted as the protocol shown in Figure 2A. The dynamic measurement of tumor size showed that the mice immunized with FGF-2 protein-conjugated mice significantly suppressed the growth of primary tumors inoculated in the mammary gland fat pad compared to the mice receiving carrier or PBS (Figure 7A). At the experimental endpoint, tumors were carefully isolated and weighed. The results showed that the vaccinated mice had significantly slighter tumors than the control mice (Figure 7B). In addition, the India ink staining of the lungs showed that there were significantly reduced metastatic nodules in the lungs of the vaccinated mice (Figure 7C), and representative pictures were shown (Figure 7D). Further analyses by ELISPOT showed that the level of IFN-γ expression in splenocytes was significantly increased in the immunized mice (Figure 7E), which was supported by the result that the percentage of CD8+IFN-γ+ T lymphocyte in splenocytes was higher in the immunized mice than in the control mice (Figure 7F).
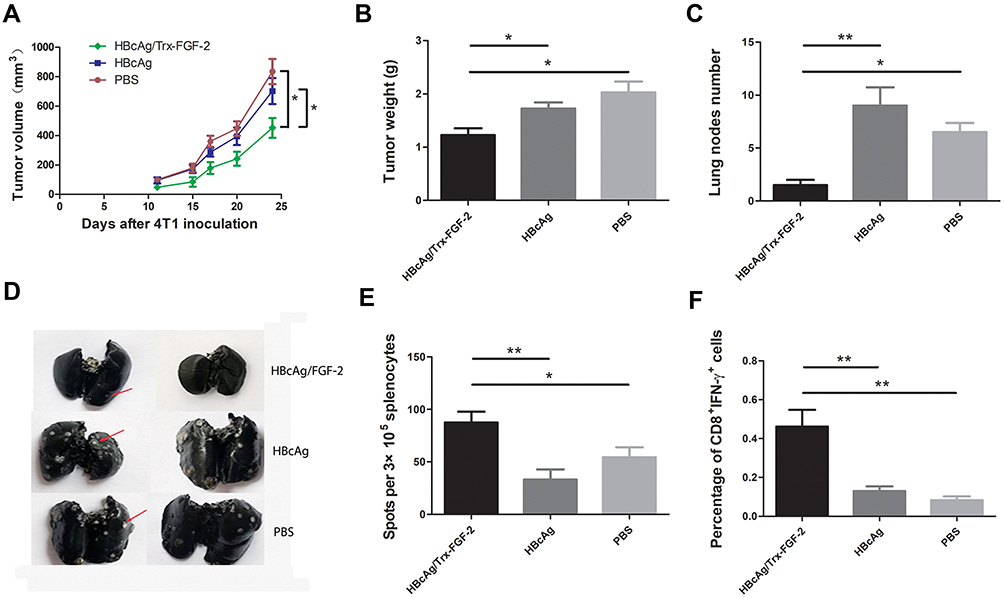 |
Figure 7 Preventive immunization with FGF-2 protein-conjugated VLPs suppresses the primary tumor growth and lung metastasis in the 4T1 model. (A) Dynamic monitoring of the changes in tumor size; (B) tumor weight; (C) lung nodule number; (D) representative images of lung metastatic nodules stained with Indian ink; (E) IFN-γ-expressing splenocytes detected by ELISPOT; (F) the percentage of CD8+IFN-γ+ lymphocytes in splenocytes (*p < 0.05, and **p < 0.01; n = 5/group). |
Discussion
Targeting a pathological self-molecule with monoclonal antibodies or soluble receptors has been proven to be effective in preclinical animal model studies,34,35,37,38 and this strategy has been used to clinically treat severe human diseases.39 Active immunization presents some advantages over passive administration with monoclonal antibodies, especially because it provides persistent antibody responses with only several immunizations.40,41 Active immunization targeting FGF-2 is emerging as a promising strategy of anti-tumor angiogenesis. FGF-2 protein adjuvanted with a liposome-saponins formulation or loaded with biodegradable nanoparticles in thermosensitive hydrogel, or a N- and C- terminally truncated FGF-2 fragment adjuvanted with cationic liposome-DNA complex was reported, respectively, to successfully induce FGF-2 -specific antibody responses in mice and produced suppression on the growth and lung metastasis of B16 melanoma,42 Lewis lung carcinoma,43 or CT26 colonic carcinoma.44 The studies used human-derived FGF-2 as antigen, which has high homogenicity with mouse FGF-2. To utilize carrier effects of immunogenic protein and provide sufficient help of heterogeneous Th epitopes for overcoming self-tolerance, Zhang HL constructed fusion protein consists of a nontoxic mutant of diphtheria toxin CRM197 and human FGF-2 and induced effective anti-tumor effects.45 In this current study, we used mouse-derived FGF-2 antigen instead of exogenous human FGF-2 to elicit a robust antibody response in mice by employing VLPs as a powerful vaccine carrier. For a cytokine target such as FGF-2, an antibody against even a linear epitope likely has the capability of effectively inhibiting the signal transduction of the cytokine by promoting the clearance of abnormally accumulated target molecules or by blocking its binding to the receptor.46 In this study, potential linear B cell epitopes of mouse FGF-2 were screened by synthesizing a set of overlapping peptides covering the whole sequence of FGF-2 molecules. Theoretically, a peptide with 5–6 amino acids can constitute the smallest B cell epitope;47 however, according to our previous experiences,48,49 a peptide with a length of 11–15 amino acids holds a great chance to elicit a high titer of antibodies with a high affinity. In this study, we designed and synthesized peptides with a length between 18 and 29 aa and obtained a set of 13 peptides with a peptide overlapping two adjacent peptides to avoid missing any possible linear epitopes at the joint. With this design, we can quickly obtain useful antigenic peptides and avoid the time and labor-consuming task of directly identifying peptides with 11–15 aa, and we still hold the feasibility of deducing the smaller antigenic peptides if needed. To rapidly identify the candidate peptides, we simply coupled the peptides to a recombinant VLP carrier consisting of modified HBcAg with an introduced lysine present in the immunodominant domain to provide free NH2- groups and to facilitate the coupling of FGF-2 peptides. VLPs have unique structural and immunological characteristics, including nanoscale size, a highly repetitive and ordered presentation of epitopes and an enclosure of nucleic acid immunostimulators, which convey high immunogenicity to the presented peptides and help eliminate B cell tolerance to self-proteins and induce specific antibody responses.50,51 To serve as a control, a full-length recombinant FGF-2 protein was also conjugated to VLPs. To display the importance of employing VLPs as a carrier for self-antigen, in our experiment we compared the antibody response and tumor growth suppression induced by Trx-FGF-2 and HBcAg/Trx-FGF-2 (Figures 1 and 2). The reason why Trx-FGF-2 was used here instead of FGF-2 is that without the help of heterogenous Th epitope which was provided by Trx, mouse FGF-2 was not able to break self-tolerance and induce antibody responses in mice. Compared to the use of a linear epitope, the use of full-length recombinant FGF-2 as an antigen provides an increased opportunity to obtain effective intervening effects since the protein antigen includes spatial conformation epitopes and more potential epitopes. However, full-length FGF-2-based recombinant VLP is difficult to obtain since the capability of VLP assembly was easily damaged by the insertion of exogenous fragments especially long peptides, leading to a dramatical loss of VLP immunogenicity. In addition, the antibodies induced by the peptides located in the important activity domains of FGF-2 are capable of directly blocking the binding of FGF-2 to its receptors, given that the avidity is strong enough. In a summary, VLP presented peptides hold the potentials to mount an immune response even higher than full-length molecules.
In this study, we first used a preventive immunization strategy to assess whether targeting FGF-2 by employing peptides or protein-conjugated VLPs can prevent the growth of subsequently inoculated tumors. The results showed that almost all the peptides induced FGF-2-specific antibody responses, among which peptides 1, 3, 5, 8, 9, 10, 11, and 13 induced comparable antibody levels to those induced by full-length FGF-2 protein-conjugated VLPs. Nevertheless, it was obvious that the efficacy of tumor suppression was not only attributed to the high level of antibody response. For example, peptides 12, 2, 3, 5, and 11 produced the most significant suppression of tumor growth, but these peptides, especially peptide 2, did not induce an antibody level as high as some other peptides did. Among these peptides, peptides 8 and 9 produced the highest antibody responses but failed to induce significant tumor suppression. Published data showed that peptides 12, 2, 3, and 5 are located either in the receptor-binding site or the heparin-like polysaccharide-binding site of FGF-2, both of which are important structural domains for FGF-2 function exertion. In addition, our preliminary analyses indicated that the antibodies elicited by these peptides had a high affinity for binding to FGF-2 molecules. This strongly indicated that the location of antigenic peptides in the original molecule and the affinity of the induced antibody might be pivotal for the induced antibody to effectively block the signaling of the target cytokine.
Our interest in this study is not limited to understanding the anti-angiogenesis effects of targeting FGF-2; we put more emphasis on discussing the effects of blocking FGF-2 signaling on antitumor immune responses. Therefore, we analyzed the level of IFN-γ, the key antitumor effector molecule, and the responses of IFN-γ-expressing effector cells. We showed that active immunization against FGF-2 increased the levels of IFN-γ in the serum and IFN-γ-expressing lymphocytes in isolated splenocytes; in addition, we displayed that the frequency of key antitumor effector cells IFN-γ+ CD4+ Th1 cells and IFN-γ+ CD8+ CTLs in splenocytes was elevated significantly. The above results indicated that immunization against FGF-2 significantly induced antitumor cellular immune responses. Further, we found that an FGF-2 blockade dramatically reduced the production of immunosuppressive CD11b+ Gr-1+ MDSCs and CD4+ CD25+ Foxp3+ Treg cells. The enhanced antitumor cellular immune responses and decreased level of immunosuppressive cells may account for the significant suppression of tumor growth in the vaccinated mice. As mentioned before, FGF-2 is an important pro-angiogenesis factor in tumors; we detected the level of CD31, a key marker of endothelial cells, and the results showed that targeting FGF-2 significantly repressed tumor neovascularization. Correspondingly, tumor cell apoptosis was increased in mice receiving anti-FGF-2 immunization.
Next, we performed a brief therapeutic experiment in which the effective antibody response was induced only after the tumor was fully established. As mentioned above, once tumor immunosuppression is developed, the production, migration and maintenance of the antitumor activity of effector cells (such as Th1/CTLs) might be significantly suppressed, finally leading to tumor growth without effective immunological surveillance. Thus, the therapeutic study mimicked the clinical situation better and was more challenging than the preventive experiment. Furthermore, the therapeutic study may provide more convincing information for evaluating the roles of FGF-2 in established tumors and demonstrating the possible antitumor efficacy of targeting FGF-2 through a strategy of active immunization. The results indicated that even in the presence of fully established tumors, peptides or protein-conjugated VLPs still held the ability to facilitate antitumor immunity and suppress tumor growth.
Finally, by employing another model of mouse tumor, orthotopically grafted 4T1 breast tumor, we further indicated the application potential of anti-FGF-2 active immunization in the immune intervention of various tumors. The results demonstrated that targeting FGF-2 still significantly suppressed both primary tumor growth and lung metastasis of 4T1 tumor in the vaccinated mice, which was accompanied with significantly enhanced effector cell responses.
Conclusion
In summary, this study demonstrated that targeting FGF-2 through active immunization was achieved using recombinant VLPs as an antigen delivery platform. Antigenic peptides providing an effective blockade of FGF-2 signaling were identified; peptides or protein-conjugated VLPs suppressed tumor neovascularization and tumor growth and promoted tumor cell apoptosis; the conjugated VLPs boosted antitumor Th1/CTL and decreased immunosuppressive MDSC and Treg responses. The results confirmed that FGF-2 played important roles in tumor development and that targeting FGF-2 with active immunization might be an effective intervention strategy for the treatment of cancers where FGF-2 plays a pro-tumorigenic role.
It was predictable that targeting FGF-2 through active immunization might be more promising when combined with other treatment strategies, such as anti-VEGF immunotherapy or chemotherapy.
Acknowledgments
We thank Mr. Jinxian Zhou for his excellent assistance on VLP observation using an electron microscope.
Funding
This work was financially supported by the CAMS Initiative for Innovative Medicine (2016-I2M-1-019), the National Natural Science Foundation of China (81773270), the Science and Technology Project of Yunnan Province (2016FA049), and Fundamental Research Funds for Institute of Pathogen Biology of PUMC (2014IPB107), the Science and Technology Project of Yunnan Province (2019FB045), and Yunnan High-level Health and Technical Personnel Project (H-2018069).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146(6):873–887. doi:10.1016/j.cell.2011.08.039
2. Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–2049. doi:10.1056/NEJMra0706596
3. Lagory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol. 2016;18(4):356. doi:10.1038/ncb3330
4. Bergers G, Benjamin LE. Angiogenesis: tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401. doi:10.1038/nrc1093
5. Conejo-garcia JR, Benencia F, Coukos G. Tumor-infiltrating dendritic cell precursors recruited by a β-defensin contribute to vasculogenesis under the influence of VEGF-A in ovarian cancer. J Immunother. 2004;27(6):S58–S59. doi:10.1097/00002371-200411000-00208
6. Facciabene A, Peng X, Hagemann IS, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature. 2011;475(7355):226–230. doi:10.1038/nature10169
7. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi:10.1038/nri3175
8. Parker KH, Beury DW, Ostrandrosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128(8 Supplement):95–139.
9. Li B, Lalani AS, Harding TC, et al. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin Cancer Res. 2006;12(22):6808. doi:10.1158/1078-0432.CCR-06-1558
10. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med. 2015;94(5):509–522. doi:10.1007/s00109-015-1376-x
11. Russo AE, Priolo D, Antonelli G, Libra M, Mccubrey JA, Ferraù F. Bevacizumab in the treatment of NSCLC: patient selection and perspectives. Lung Cancer. 2017;8:259–269. doi:10.2147/LCTT.S110306
12. Herbert H, Louis F, William N, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. J Evid Based Med. 2004;350(23):2335–2342
13. Saltz LB, Clarke S, Díaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized Phase III study. J Clin Oncol. 2008;26(12):2013–2019. doi:10.1200/JCO.2007.14.9930
14. Tebbutt NC, Wilson K, Gebski VJ, et al. Capecitabine, bevacizumab, and mitomycin in first-line treatment of metastatic colorectal cancer: results of the Australasian Gastrointestinal Trials Group Randomized Phase III MAX Study. J Clin Oncol. 2010;28(19):3191–3198. doi:10.1200/JCO.2009.27.7723
15. Mayer EL. Paclitaxel plus Bevacizumab versus Paclitaxel Alone for Metastatic Breast Cancer: miller K, Wang M, Gralow J, et al (Indiana Univ Cancer Ctr, Indianapolis; Dana-Farber Cancer Inst, Boston; Puget Sound Oncology Consortium, Seattle; et al) New Engl J Med 357. Breast Dis a Year Book Q. 2008;19(3):272–273. doi:10.1016/S1043-321X(08)79083-0
16. Reck M, Von pawel J,P, Zatloukal P, et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: aVAil. J Clin Oncol. 2009;27(8):1227. doi:10.1200/JCO.2007.14.5466
17. Muggia F. Bevacizumab in ovarian cancer: unanswered questions. Drugs. 2012;72(7):931–936. doi:10.2165/11633510-000000000-00000
18. Wallin JJ, Bendell JC, Funke R, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624. doi:10.1038/ncomms12624
19. Ahn PH, Machtay M, Anne PR, et al. Phase I trial using induction cisplatin, docetaxel, 5-FU and erlotinib followed by cisplatin, bevacizumab and erlotinib with concurrent radiotherapy for advanced head and neck cancer. Am J Clin Oncol. 2016;41(5):1.
20. Diaz RJ, Ali S, Qadir MG, Fuente MIDL, Ivan ME, Komotar RJ. The role of bevacizumab in the treatment of glioblastoma. J Neurooncol. 2017;133(3):1–13. doi:10.1007/s11060-017-2477-x
21. Ebos JML, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210–221. doi:10.1038/nrclinonc.2011.21
22. Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov. 2016;15(6):385–403. doi:10.1038/nrd.2015.17
23. Zhao M, Yu Z, Li Z, Tang J, Lai X, Liu L. Expression of angiogenic growth factors VEGF, bFGF and ANG1 in colon cancer after bevacizumab treatment in vitro: a potential self-regulating mechanism. Oncol Rep. 2017;37(1):601. doi:10.3892/or.2016.5231
24. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. doi:10.1038/nrc2780
25. Cao Y, Cao R, Hedlund EM. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J Mol Med. 2008;86(7):785–789. doi:10.1007/s00109-008-0337-z
26. Gyanchandani G, Alves MV, Ortega MJN, Kim K. A proangiogenic signature is revealed in FGF-mediated bevacizumab-resistant head and neck squamous cell carcinoma. Mol Cancer Res. 2013;11(12):1585–1596. doi:10.1158/1541-7786.MCR-13-0358
27. Shee K, Yang W, Hinds JW, et al. Therapeutically targeting tumor microenvironment-mediated drug resistance in estrogen receptor-positive breast cancer. J Exp Med. 2018;215(3):
28. Somasundaram R, Zhang G, Fukunagakalabis M, et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat Commun. 2017;8(1):607. doi:10.1038/s41467-017-00452-4
29. Chua V, Orloff M, Teh JL, et al. Stromal fibroblast growth factor 2 reduces the efficacy of bromodomain inhibitors in uveal melanoma. EMBO Mol Med. 2019;11:2. doi:10.15252/emmm.201809081
30. Xiuqin Z, Ibrahimi OA, Olsen SK, Hisashi U, Moosa M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The Complete Mammalian FGF Family. J Biol Chem. 2006;281(23):15694–15700.
31. Andre F, Bachelot T, Campone M, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. 2013;19(13):3693–3702. doi:10.1158/1078-0432.CCR-13-0190
32. Lin B, Song X, Yang D, Bai D, Yao Y, Lu N. Anlotinib inhibits angiogenesis via suppressing the activation of VEGFR2, PDGFRβ and FGFR1. Gene. 2018;654:S0378111918301550. doi:10.1016/j.gene.2018.02.026
33. Wang L, Park H, Chhim S, et al. A novel monoclonal antibody to fibroblast growth factor 2 effectively inhibits growth of hepatocellular carcinoma xenografts. Mol Cancer Ther. 2012;11(4):864–872. doi:10.1158/1535-7163.MCT-11-0813
34. Harding TC, Long L, Palencia S, et al. Blockade of nonhormonal fibroblast growth factors by FP-1039 inhibits growth of multiple types of cancer. Sci Transl Med. 2013;5(178):178ra139. doi:10.1126/scitranslmed.3005414
35. Li D, Wei X, Xie K, Chen K, Li J, Fang J. A novel decoy receptor fusion protein for FGF-2 potently inhibits tumour growth. Br J Cancer. 2014;111(1):68–77. doi:10.1038/bjc.2014.282
36. Chu X, Li Y, Q L, et al. Chimeric HBcAg virus-like particles presenting a HPV 16 E7 epitope significantly suppressed tumor progression through preventive or therapeutic immunization in a TC-1-grafted mouse model. Int J Nanomedicine. 2016;11(default):2417–2429.
37. Tanaka M, Yamaguchi M, Shiota M, et al. Establishment of neutralizing rat monoclonal antibodies for fibroblast growth factor-2. Monoclon Antib Immunodiagn Immunother. 2014;33(4):261–269. doi:10.1089/mab.2013.0085
38. Yang Y, Luo Z, Qin Y, et al. Production of bFGF monoclonal antibody and its inhibition of metastasis in Lewis lung carcinoma. Mol Med Rep. 2017;16(4):4015–4021. doi:10.3892/mmr.2017.7099
39. Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18(7):1855. doi:10.1158/1078-0432.CCR-11-0699
40. Kourilsky P, Truffa-bachi P. Cytokine fields and the polarization of the immune response. Trends Immunol. 2001;22(9):502–509. doi:10.1016/S1471-4906(01)02012-9
41. Zagury D, Buanec HL, Bizzini B, Burny A, Lewis G, Gallo RC. Active versus passive anti-cytokine antibody therapy against cytokine-associated chronic diseases. Cytokine Growth Factor Rev. 2003;14(2):123–137. doi:10.1016/S1359-6101(03)00004-2
42. Zhang X, Li NL, Guo C, et al. A vaccine targeting basic fibroblast growth factor elicits a protective immune response against murine melanoma. Cancer Biol Ther. 2018;19(6):518–524. doi:10.1080/15384047.2018.1435223
43. Wu QJ, Zhu XC, Xiao X, et al. A novel vaccine delivery system: biodegradable nanoparticles in thermosensitive hydrogel. Growth Factors. 2011;29(6):290–297. doi:10.3109/08977194.2011.624517
44. Zhang XP, Yang L, Shi HS, et al. An N-, C-terminally truncated basic fibroblast growth factor and LPD (liposome-polycation-DNA) complexes elicits a protective immune response against murine colon carcinoma. Cancer Biol Ther. 2010;10(3):276–281. doi:10.4161/cbt.10.3.12421
45. Zhang HL, Yuan C, Zhang DM, et al. A novel combined conjugate vaccine: enhanced immunogenicity of bFGF with CRM197 as a carrier protein. Mol Med Rep. 2011;4(5):857–863. doi:10.3892/mmr.2011.521
46. Schneble E, Clifton GT, Hale DF, Peoples GE. Peptide-based cancer vaccine strategies and clinical results. Methods Mol Biol. 2016;1403:797.
47. Trier NH, Hansen PR, Houen G. Production and characterization of peptide antibodies. Methods. 2012;56(2):136–144. doi:10.1016/j.ymeth.2011.12.001
48. Chu X, Li Y, Huang W, et al. Combined immunization against TGF-beta1 enhances HPV16 E7-specific vaccine-elicited antitumour immunity in mice with grafted TC-1 tumours. Artif Cells Nanomed Biotechnol. 2018;46(sup2):1199–1209. doi:10.1080/21691401.2018.1482306
49. Ma Y, HayGlass KT, Becker AB, et al. Novel recombinant interleukin-13 peptide-based vaccine reduces airway allergic inflammatory responses in mice. Am J Respir Crit Care Med. 2007;176(5):439–445. doi:10.1164/rccm.200610-1405OC
50. Birnbaum F, Nassal M. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J Virol. 1990;64(7):3319–3330. doi:10.1128/JVI.64.7.3319-3330.1990
51. Lee BO, Tucker A, Frelin L, et al. Interaction of the hepatitis B core antigen and the innate immune system. J Immunol. 2009;182(11):6670–6681. doi:10.4049/jimmunol.0803683
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.