Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 12
Vesiculobullous diseases in relation to lupus erythematosus
Authors Rutnin S ![]() , Chanprapaph K
, Chanprapaph K ![]()
Received 26 June 2019
Accepted for publication 13 August 2019
Published 4 September 2019 Volume 2019:12 Pages 653—667
DOI https://doi.org/10.2147/CCID.S220906
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Suthinee Rutnin, Kumutnart Chanprapaph
Division of Dermatology, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Kumutnart Chanprapaph
Division of Dermatology, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, 270 Rama VI Road, Ratchatewi, Bangkok 10400, Thailand
Tel +66 2 201 1141
Fax +66 2 201 1211
Email [email protected]
Abstract: Vesiculobullous lesions in lupus erythematosus (LE) are a rare cutaneous manifestation of cutaneous and/or systemic LE with variable presentation. While the minor forms of LE-associated vesiculobullous disease may cause disfigurement and discomfort, the severe forms can present with hyperacute reaction and life-threatening consequences. Specific LE and aspecific cutaneous LE are defined by the presence or absence of interface change on histopathology that can be applied to vesiculobullous diseases in relation to LE. However, the diagnosis of LE-associated vesiculobullous diseases remains difficult, due to the poorly defined nosology and the similarities in clinical and immunohistopathological features among them. Herein, we thoroughly review the topic of vesiculobullous skin disorders that can be encountered in LE patients and organize them into four groups: LE-specific and aspecific vesiculobullous diseases, LE-related autoimmune bullous diseases, and LE in association to non-autoimmune conditions. We sought to provide an updated overview highlighting the pathogenesis, clinical, histological, and immunopathological features, laboratory findings, and treatments and prognosis among vesiculobullous conditions in LE.
Keywords: autoimmune blistering diseases, erythema multiforme, cutaneous lupus erythematosus, systemic lupus erythematosus, Stevens–Johnson syndrome, toxic epidermal necrolysis
Introduction
Lupus erythematosus (LE) is an autoimmune disease with a wide range of clinical manifestations, from limited skin disease to multiorgan systemic involvement.1 The annual incidence of systemic LE (SLE) is approximately one to ten per 100,000. The prevalence of SLE is around 5.5-130 per 100,000.1,2 Between 59% and 85% of SLE patients have cutaneous manifestations; however, <5% develop vesiculobullous lesions, which manifest in the form of vesicles, bullae, erosions, and/or crusts.3 Distinct immunopathogenic factors cause various types of vesiculobullous eruption encountered in patients with LE. There are two mechanisms classified by the presence or absence of LE-specific interface dermatitis in histopathology for LE-associated blistering conditions.4,5 Firstly, LE-specific vesiculobullous diseases are blistering diseases occurring in active and severe chronic cutaneous LE (CLE) lesions from exuberant interface dermatitis, leading to severe hydropic degeneration of the basal-cell layer. Secondly, LE-nonspecific vesiculobullous diseases, an antibody mediated neutrophilic dermatosis, a specific subgroup with distinct clinical finding and immunopathology. Bullous SLE (BSLE) is a prime example of LE-aspecific vesiculobullous disease.3 Other autoimmune bullous disorders have been reported anecdotally to occur in association with LE, such as pemphigus vulgaris (PV), pemphigus foliaceus (PF), pemphigus erythematosus (PE), pemphigus herpetiformis (PH), paraneoplastic pemphigus (PNP), bullous pemphigoid (BP), linear IgA bullous dermatosis (LABD), mucous-membrane pemphigoid (MMP), epidermolysis bullosa acquisita (EBA) and dermatitis herpetiformis (DH).6–13 In addition, nonautoimmune diseases, namely erythema multiforme (EM), Stevens–Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and porphyria cutanea tarda (PCT), have been documented in SLE patients.14,15 Significant association of nonautoimmune diseases with LE is uncertain. Considering the relatively high prevalence of SLE, this might only be a chance association. Patients with CLE/SLE can also develop vesiculobullous skin lesions that have no direct relationship with their underlying CLE/SLE, such as microvesicles secondary to acute eczema. Finally, SLE patients with systemic involvement commencing numerous immunosuppressive agents are prone to developing infection-related blistering conditions, eg, herpes infection. The diagnosis of LE-associated vesiculobullous disease can be difficult and often confusing, due to poorly defined nosology and the clinical, histological, and/or immunological similarities of these conditions. Table 1 summarizes the differential diagnoses of vesiculobullous diseases in the context of LE.
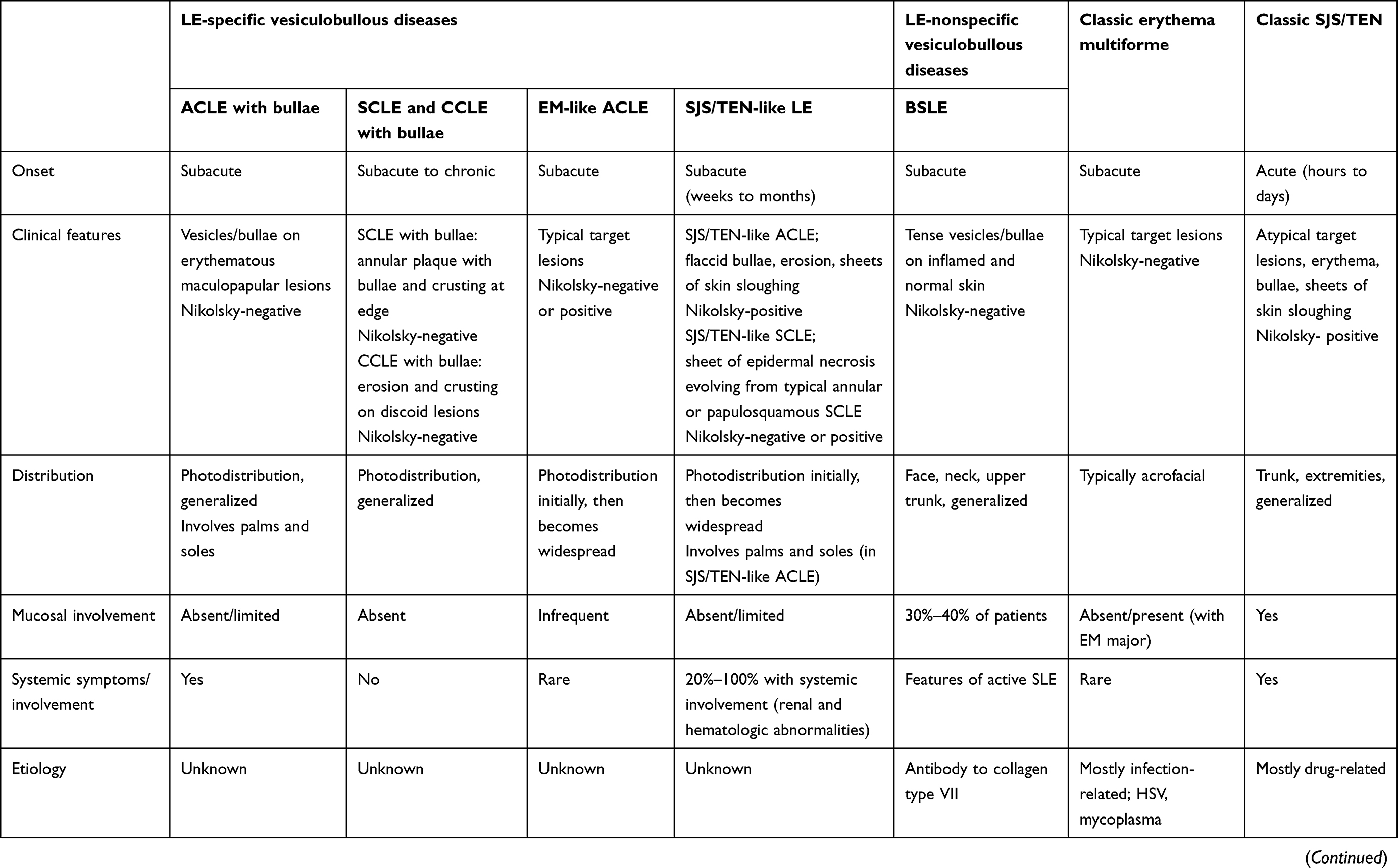 | 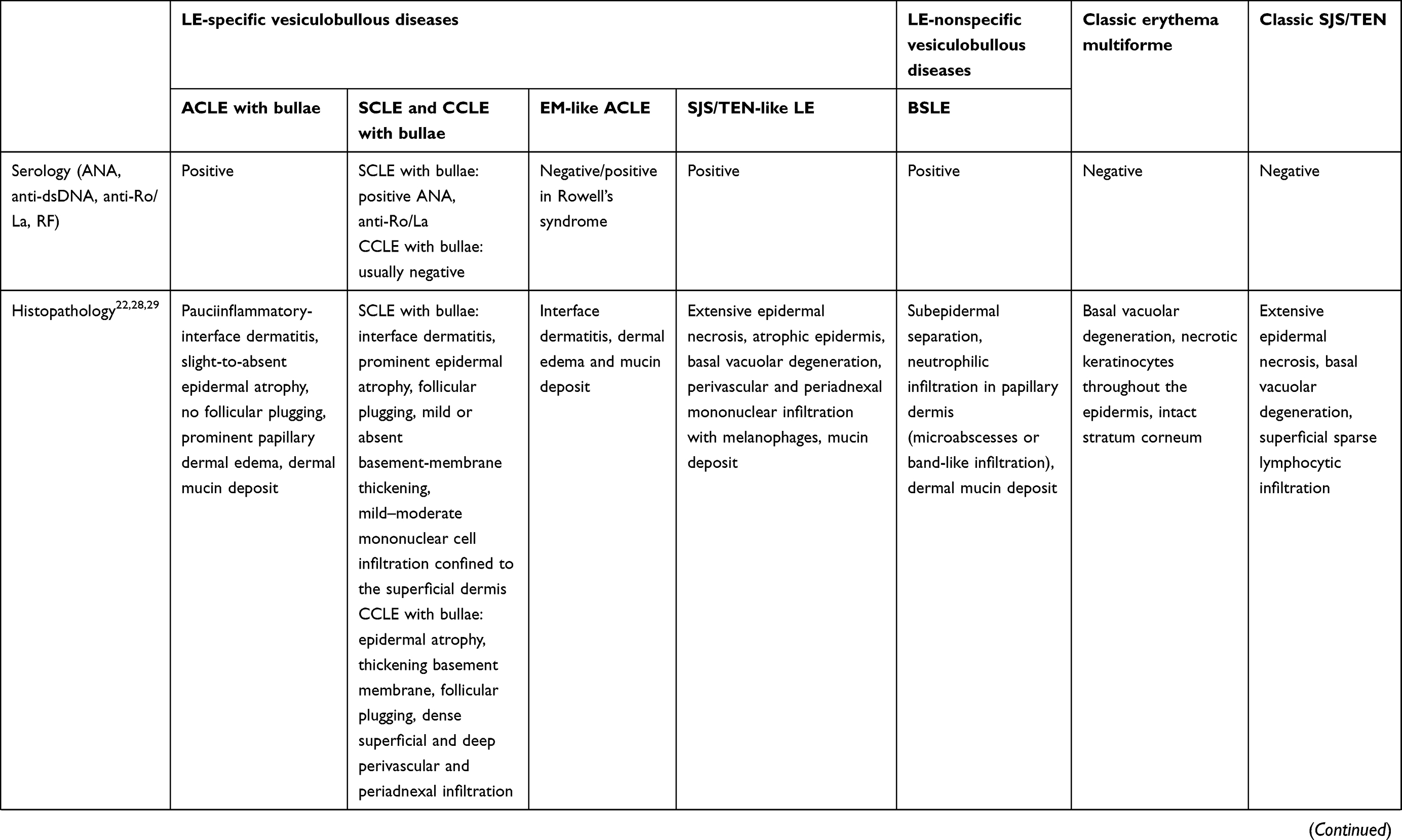 | 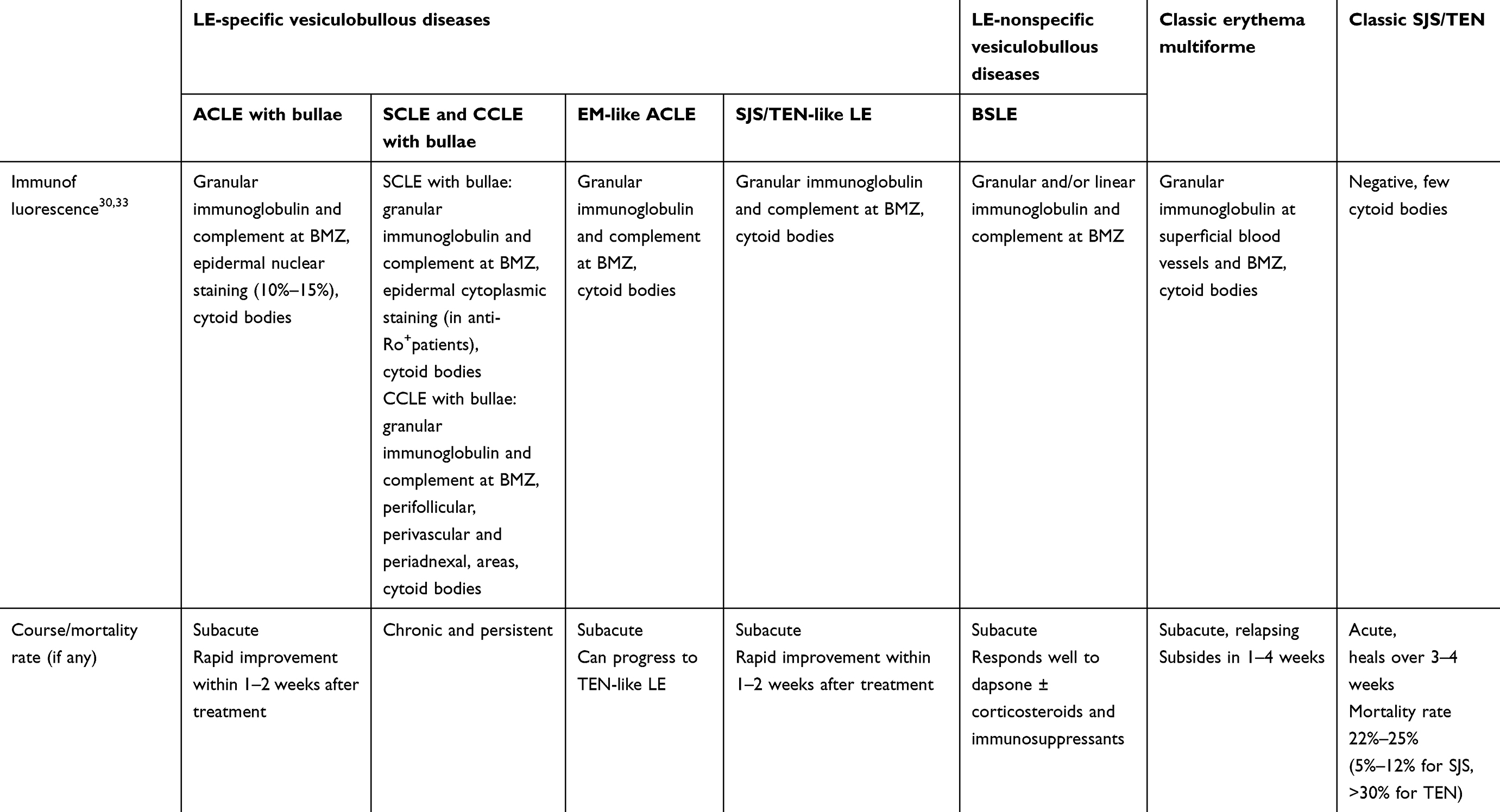 |
Table 1 Diagnostic features of differential diagnosis for LE-specific and -aspecific vesiculobullous diseases, EM, classic SJS, and TEN |
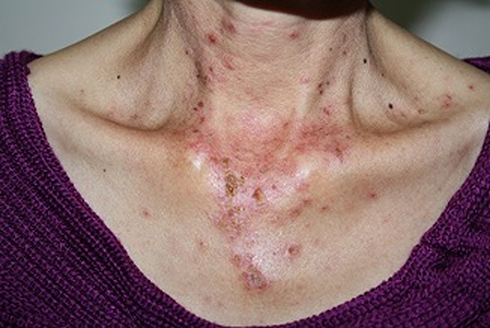 |
Figure 1 Acute cutaneous lupus erythematosus–associated vesiculobullous disease. A few vesicles and erosions arising on erythematous edematous maculopapular patches in a V shape on the neck. |
While minor forms of LE-associated vesiculobullous lesions may cause disfigurement and discomfort, severe forms of hyperacute CLE, eg, SJS- or TEN-like acute CLE (ACLE)/subacute CLE (SCLE) are life-threatening conditions that warrant prompt diagnosis and proper management. Given the broad differential diagnosis and varying degrees of severity, it is important to have a thorough analysis of the patients’ history and complete clinical and histopathological and/or immunopathological information to establish a definitive diagnosis of LE-associated vesiculobullous diseases. As such, this literature review focuses on vesiculobullous diseases in relation to LE, the differentiation between LE-specific and -aspecific blistering conditions, and similarities with other immunobullous diseases and epidermal necrolysis (EM, SJS, and TEN).
A literature search of published articles from the Medline database was performed using the specific search terms “systemic lupus erythematosus and blistering disease”, “cutaneous lupus erythematosus and blistering disease”, “bullous systemic lupus erythematosus, Stevens–Johnson syndrome, and systemic lupus erythematosus”, “toxic epidermal necrolysis and systemic lupus erythematosus”, and “autoimmune bullous disease and systemic lupus erythematosus” (no exclusion criteria were utilized). Then, a manual search of each article’s references was undertaken to ensure a more complete sample of existing case reports/series, review articles, and original articles.
Classification
Symptom-based diagnostic classification divides LE into cutaneous-limited LE, intermediate LE, and SLE. In 1981, Gilliam and Sontheimer described a morphological classification scheme to organize cutaneous LE as lupus-specific or lupus-aspecific based on characteristic findings of interface dermatitis on histopathology in the former group.4 Then, in 1997, Sontheimer adapted the aforementioned classification to distinguish types of vesiculobullous LE lesions. LE-specific vesiculobullous diseases comprised three forms — ACLE (TEN-like ACLE), SCLE (TEN-like SCLE and vesiculobullous annular SCLE), and chronic CLE (CCLE; bullous discoid LE [DLE] — while LE-aspecific vesiculobullous diseases included BSLE and vesiculobullous skin disorders reported to occur concomitantly in LE patients, eg, BP, DH, PV, PF, and PCT.5 Tochia et al suggested including EM-like lesions in the setting of SCLE/SLE, and hypothesized that EM-like lesions represented an independent morphological variant of LE-specific skin lesions.14 As for the severe spectrum of LE-specific vesiculobullous disease, Ting et al classified three forms of TEN-like LE — TEN-like ACLE, TEN-like SCLE, and TEN occurring in SLE patients not having conventional LE-specific skin lesions. They also proposed a spectrum of acute syndrome of apoptotic panepidermolysis to address life-threatening situations of massive epidermal cleavage resulting from hyperacute apoptotic injury, which includes SJS/TEN-like ACLE, drug-induced epidermal necrolysis (TEN), and fulminant cases of graft-versus-host disease or pseudoporphyria.16
LE-specific vesiculobullous diseases
Subtypes
ACLE-associated vesiculobullous diseases
ACLE lesions are described as localized (involving the malar areas) or generalized, referring to widespread morbilliform eruption. When the ACLE-typical interface dermatitis is intense, vesicles or bullae may arise on erythematous edematous maculopapular patches (Figure 1). In the most severe form of ACLE, SJS/TEN-like eruptions, painful flaccid bullae that progress to sheaths of epidermal necrosis, and a positive Nikolsky sign occurs. Bullous lesions in SJS/TEN-like ACLE usually start on photodistributed areas, then coalesce and later become generalized, spreading symmetrically, often involving the palms and soles (Figure 2A and B). The duration between initial rash and the onset of epidermal detachment is normally subacute (weeks to months). This is significantly longer than the acute progression of conventional drug-induced SJS/TEN (hours to days).17 However, TEN-like ACLE has also been reported to occur abruptly in SLE patients that do not have preceding conventional LE-specific skin changes.16 Mucosal involvement is usually absent or minimal, affecting the mouth (mostly hard palate and gingiva), nose, pharynx, and vagina. This is another useful feature to differentiate SJS/TEN-like ACLE from drug-induced SJS-TEN, which has extensive mucosal involvement (Table 1).
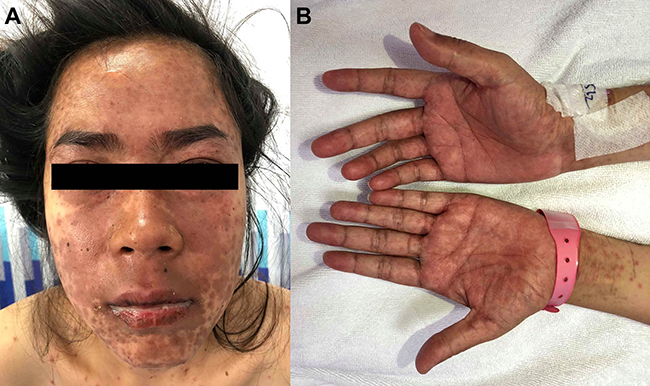 |
Figure 2 A 25-year-old female patient diagnosed with Stevens–Johnson syndrome–like acute cutaneous lupus erythematosus. (A) Multiple erythematous macules and patches with dusky-red center on the face with crusted erosions, predominantly on the lower lip. (B) Involvement of the palms. |
ACLE may present as EM-like target lesions. Despite being virtually indistinguishable from typical target lesions in classic EM, EM-like ACLE is defined by the presence of skin lesions resembling EM and DIF of lesional skin showing an LE-specific pattern similar to findings in ACLE. EM-like ACLE differs from classic EM, and most often does not show the preferential localization of acrofacial sites and mucous-membrane involvement (Figure 3A).14 Moreover, unlike typical EM, which does not carry a risk of developing TEN, subacute progression of EM-like ACLE to TEN has been noted to occur (Figure 3B).18 No obvious infectious or drug culprit has been identified in EM-like ACLE. EM-like cutaneous eruptions in LE in the setting of positive antinuclear antibodies (ANA; speckle pattern), anti-La/SSB antibodies, and rheumatoid factor without an identifiable precipitating cause were first described as Rowell’s syndrome (RS) in 1963.19 Recently, RS-diagnostic criteria were revised to clarify RS from other variants of LE and EM.14 While some authors believe that RS and TEN-like ACLE are components of the same spectrum of illnesses, sharing the same pathogenesis, and that RS may be a limited form of TEN-like ACLE or SCLE, others consider RS an independent CCLE subtype.14ACLE is often induced by excessive ultraviolet exposure with underlying SLE predisposition.16,20 Patients with ACLE-associated vesiculobullous diseases usually meet American College of Rheumatology (ACR) and/or Systemic Lupus International Collaborating Clinics (SLICC) diagnostic criteria for SLE with the presence of extracutaneous involvement.16,21,22
 |
Figure 3 A 33-year-old Thai female diagnosed with erythema multiforme–like acute cutaneous lupus erythematosus (ACLE) and subsequent progress to toxic epidermal necrolysis–like ACLE. (A) The patient presented with fever and typical target-like lesions on the left forearm and left legs for 5 days. (B) Widespread erosions and sloughing of the skin involving 40% of BSA developed 3 weeks later. |
SCLE-associated vesiculobullous diseases
SCLE is characterized by scaly erythematous papules arranged in psoriasiform (papulosquamous) or annular morphology. Vesiculobullous annular SCLE is mainly characterized by erosion and crusting, and more rarely by vesicles and bullae occurring at the active advancing edge of annular SCLE lesions or at the center of the plaque.5,16 Like ACLE, TEN-like sheets of epidermal cleavage evolving from typical photodistributed nonscarring annular or papulosquamous SCLE have rarely been reported.16,23,24 The onset of TEN-like SCLE is gradual. Nikolsky's sign can be either positive or negative, and mucosal involvement is usually absent or minimal.24 Most cases of SCLE-associated vesiculobullous disease lack visceral-organ involvement.22
CCLE-associated vesiculobullous diseases
CCLE comprises DLE, LE profundus, chilblain LE, and LE tumidus. DLE, the most common clinical subtype of CCLE, is characterized by well-demarcated discoid (coin-shaped) scaly purplish plaques with central atrophy and hyperpigmented periphery. Classic DLE is categorized as localized (above neck) or generalized (above and below neck, typically over the extensor forearms and hands).25 Vesiculobullous CCLE is an extremely rare condition that presents as erosions or crusts in typical DLE (Figure 4). As with classic DLE, patients presenting bullous DLE have a low risk of developing clinically significant SLE during the course of disease.
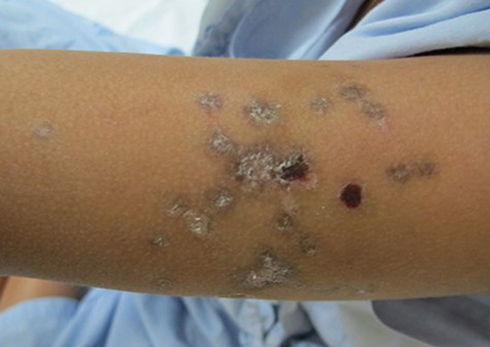 |
Figure 4 Erosions and crusts arising on typical discoid lupus erythematosus. |
The pathophysiology of CLE is likely multifactorial, which includes genetic predisposition, environmental triggers, and ultraviolet aggravation leading to innate and adaptive immunoresponse.1 Activation of the adaptive immune system against “self” antigens leads to the activation of autoantibodies (produced by plasma cells) or self-antigen–specific T cells. Autoreactive cytotoxic T lymphocytes (mixed with histocytes) from dermis and basement-membrane and/or dermal autoantibody deposition cause hydropic degeneration of the basal-cell layer of the epidermis and apoptotic keratinocytes.26 Type 1 interferon induction of proinflammatory cytokines and chemokines supports cellular immunoresponse as the pathogenesis. There is close association between IFN-inducible proteins and the distribution of CXCR3+ lymphocytes. IFN-inducible CXCL10 has been shown to express in the exact areas that cytotoxic lymphocytes invade the basal epidermis, and causes necrotic keratinocytes and further leads to dermoepidermal separation.27 Due to its rarity, there is very limited work to elucidate the exact pathophysiology of LE-specific vesiculobullous disease.
The characteristic histopathology of LE-specific vesiculobullous skin lesions is the presence of dramatic interface dermatitis, leading to vacuolar degeneration of the epidermal basal-cell layer (Figure 5A).14 These are qualitatively similar in each form of LE-specific vesiculobullous diseases.14,25 However, a wide range of disease severity causes variable histologic findings: the degree of epidermal necrosis varies from isolated necrotic keratinocytes in the lower epidermis and basal vacuolar alteration (limited end of the spectrum) to partial- or full-thickness epidermal necrosisleading to epidermal detachment (hyperacute end of the spectrum, Figure 5B). Epidermal atrophy and interface change are classic histopathological findings of CLE. Additional histopathological features are thickening basement membrane, superficial and deep perivascular and periadnexal lymphocytic infiltrates, follicular hyperkeratosis, and mucin deposition, depending on the histopathological criteria for each CLE subtype.22,28,29
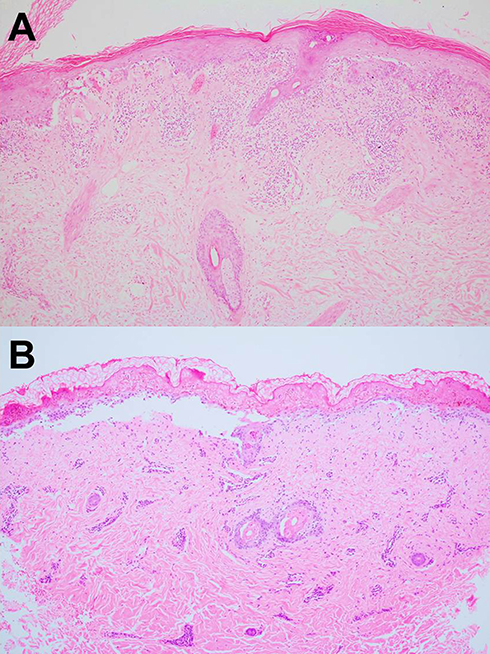 |
Figure 5 (A) Acute interface dermatitis demonstrating vacuolar degeneration of the epidermal basal-cell layer in lupus erythematosus–specific vesiculobullous disease (H&E, 100×). (B) Full-thickness epidermal necrosis leading into epidermal detachment in toxic epidermal necrolysis–like acute cutaneous lupus erythematosus (H&E, 100×). |
In all subclasses of LE-specific vesiculobullous disease, immunopathology of lesional skin from direct immunofluorescence (DIF) may show coarse, granular, continuous deposition of multiple immunoglobulins and/or complement, eg, IgG, IgM, IgA (rare), and C3 along the basement-membrane zone. The reported sensitivity of DIF examination is 58%–93%; therefore, negative DIF does not rule out this condition.14,22,30,31 These findings have also been shown in other connective tissue diseases, eg, dermatomyositis, mixed connective-tissue diseases, and systemic sclerosis.30 However, when DIF is positive in both lesional and sun-protected nonlesional skin, especially in the presence of many different immunoreactants (two or more), it is not only highly specific for LE but also helps indicate ACLE over other types of CLE.25,32 In vivo ANA or epidermal nuclear staining can also be observed in SLE, and epidermal cytoplasmic staining can be positive in anti-Ro (SSA)-positive SCLE.30,33 Deposition of immunoreactants in the perivascular, perifollicular, and periadnexal areas can present in the DIF, more so for the chronic subtype of CCLE. Indirect immunofluorescence is negative.
Serological assessment of autoantibodies is essential for SLE. Most patients with ACLE-associated LE have positive ANA (81.8%–100%). Anti-dsDNA is highly specific for SLE, and is present in approximately 40% of reported cases of TEN-like ACLE. Anti-Ro (SSA) has been found to be positive in 50%–90%.20,22 The majority of SCLE-associated vesiculobullous patients have ANA (87.5%) and anti-Ro (62.5%) antibody positivity.22 There were insufficient data regarding anti-La (SSB) and anti-Sm of ACLE- or SCLE-associated LE patients. Data on the serology of cases with bullous DLE are lacking. A relatively large study evaluating the prevalence of autoantibodies in CLE demonstrated that patients with DLE were positive for ANA, anti-ds DNA, anti-Ro, anti-La, and anti-Sm: 47.8%, 14.8%, 25.2%, 5.2% and 4.3% respectively.34 The same results may be anticipated for bullous DLE.
Most patients have associated features of systemic lupus. Barker et al reported hematologic and renal involvement to be 36.3% and 27.2%, respectively, in a case series on SJS/TEN-like LE.32 We recently reported SJS- and TEN-like LE as a marker of internal involvement and active disease at diagnosis in 100% of our patients.35 Cerebral lupus has been reported in several cases.17 Internal lupus in patients with bullous SCLE and DLE has not been reported.
Treatment of CLE-specific diseases includes both topical and systemic therapy. For LE-specific vesiculobullous skin conditions, given their rarity, evidence on treatment options is confined to case reports/series and expert opinions. Indeed, erosive variants of SCLE or CCLE can be treated with antimalarials.22 Patients at the extreme end of the spectrum — TEN-like SCLE and TEN-like ACLE — require hospitalization. High-dose systemic corticosteroids with or without pulse therapy, in conjunction with immunosuppressives (azathioprine, cyclophosphamide, mycophenolate mofetil, methotrexate), intravenous immunoglobulin (IVIG), and plasmapheresis have been reported to be effective.20,36 In review of a relatively large case series on TEN-like ACLE, 56.5% responded well to systemic corticosteroids, while 34.7% were resistant. IVIG alone or in conjunction with systemic corticosteroids offered 75% complete resolution.32 In the era of biologics and targeted therapy, there have been studies on the treatment of SLE patients using TNF inhibitors,37–40 B-cell-depleting anti-CD-20 antibodies (rituximab),41 anti-B-lymphocyte-stimulator antibodies (belimumab),42 anti-IFNα (sifalimumab),43 anti-IFNα receptors (anifrolimumab),44 anti-IFNγ monoclonal antibodies (AMG 811),45 and JAK inhibitors (baricitinib).46–48 TNF inhibitors have been reported to improve the outcome of TEN and TEN-like ACLE; however, due to their known potential to induce lupus or lupus-like syndromes, these drugs are not recommended.37–40 Clinical response to rituximab in CLE is variable among different subtypes. A fairly good response to rituximab has been shown in ACLE patients, while CCLE cases responded poorly.41 Two phase III trials on the effect of belimumab on organ-specific disease activity in SLE revealed that belimumab improved the mucocutaneous domain of SLE.42 In a Phase IIB randomized controlled trial of adult SLE patients, sifalimumab43 and anifrolimumab44 demonstrated promising results, with improvements in both global and organ-specific outcome measures, including the Cutaneous Erythematosus Disease Area and Severity Index (CLASI). A recent trial on baricitimib, a JAK1/2 inhibitor, for SLE showed that although baricitinib improved signs and symptoms of SLE, there was no improvement on the CLASI.48
In a 14-year retrospective study, the prognosis of SJS/TEN-like ACLE and SCLE tended to be better than conventional drug-induced SJS/TEN, with gradual improvement in weeks. Like classic TEN, prognosis depends largely on the amount of body surface–area involvement. Recurrence of SJS/TEN-like ACLE and SCLE was not noted. However, internal lupus, such as lupus nephritis, can occasionally occur.35
LE-aspecific vesiculobullous diseases
Autoimmune
Bullous systemic lupus erythematosus
BSLE is an autoimmune subepidermal blistering eruption occurring rarely in patients with SLE. Patients present with acute onset of vesicles and bullae over inflamed lesional and/or normal skin (Figure 6). The presence of blisters on normal skin without overlying lesional erythema seen in LE-specific vesiculobullous lesions reflects the autoantibody-mediated nature of BSLE. Lesions of acute LE are rarely observed in patients with BSLE.49 Pruritus is usually not present. There is a predilection of lesions to develop on the face, upper trunk, neck, supraclavicular regions, and axillary folds, and tendency for sun-exposed areas; however, generalized distribution is not uncommon.50,51 Mucosal involvement, especially oral mucosa, can occur in 30%–40% of cases.52,53 Blisters often evolve to erosions and crusts without milia formation or scars; however, pigmentary alterations, hypopigmentation, or less commonly hyperpigmentation are common.52 BSLE tends to be strongly associated with SLE, and all patients must satisfy ACR or SLICC criteria for the diagnosis of SLE. BSLE may or may not correspond to exacerbation of systemic disease. Some studies have not shown any clinical or laboratory evidence indicating flaring of disease.3,54 However, some case reports/series have suggested that blistering parallels internal involvement, particularly lupus nephritis and hematologic abnormalities.52,55
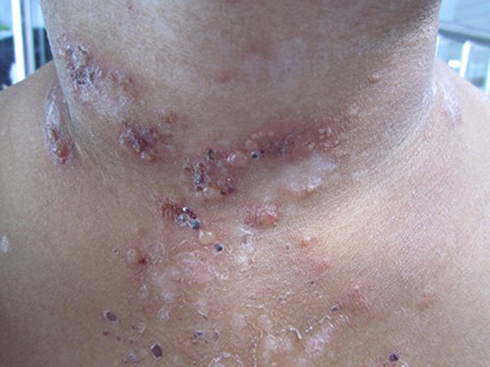 |
Figure 6 A 12-year-old Thai female diagnosed with bullous systemic lupus erythematosus. Multiple tense vesicles, bullae, and crusts on erythematous edematous patches and plaques in a V shape on the neck. Some lesions healed with postinflammatory hypopigmentation. |
The pathogenesis of BSLE is related to circulating antibodies that target type VII collagen, which causes weakening of basement membrane–dermal adhesion, creating subepidermal blistering. Type VII collagen is the major component of the anchoring fibrils connecting the epidermis to the dermis. The major antigenic epitope for autoantibody has been shown to reside within the noncollagenous domain type I of type VII collagen.3,54 Chan et al also identified other autoantibodies, such as BPAg1, laminin 5, laminin 6, and BPAg2, in patients with BSLE.56 Epitope spreading may be the reason for this, as a primary autoimmune insult against collagen type VII could expose epitopes, causing a secondary autoimmune response to the newly exposed targets.49
Classic histopathological findings are subepidermal blistering with predominance of neutrophils in the upper dermis concentrated on the papillary tip, with the association of nuclear dust and fibrin, indistinguishable from DH (Figure 7A). Occasionally, neutrophils may be distributed in a band-like pattern within the entire papillary dermis and into the blistering cavity. Marked dermal edema is associated with perivascular inflammatory infiltrates in the superficial and mid-dermis, with a large amount of mucin in the reticular dermis.54
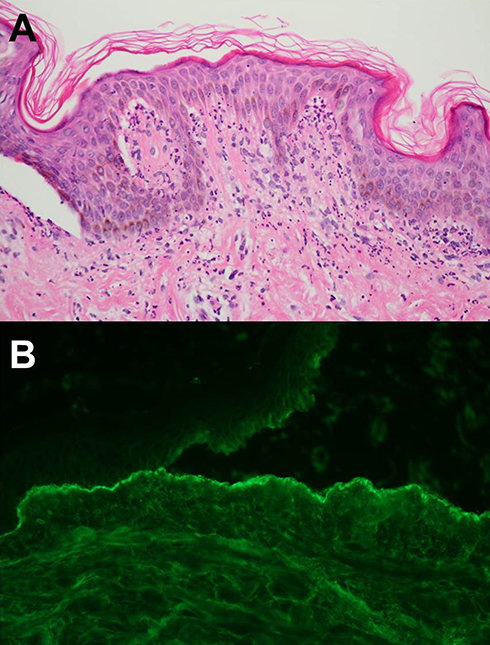 |
Figure 7 (A) Subepidermal blistering with predominance of neutrophils in the upper dermis concentrated on the dermal papillae (H&E, 400×). (B) Direct immunofluorescence on NaCl-spit skin shows immunoglobulins G in a linear and granular pattern along the base of the blister cavity (400×). |
The characteristic immunopathological feature in BSLE is the deposition of immunoreactants along the dermoepidermal junction under DIF staining. DIF staining of perilesional and clinically uninvolved skin often demonstrates all major classes of immunoglobulins — IgG, IgA, and IgM — and complement in a linear, granular, or mixed pattern.57 In BSLE, IgA deposition is positive more than twice as often as other forms of SLE, which explains the neutrophilic predominance of BSLE. DIF studies on NaCl-spit skin have shown immunoreactant along the base of the blister cavity, where type VII collagen is located (Figure 7B). Indirect immunofluorescence staing results of BSLE may show positivity for anti-type VII collagen antibody, the same antibody as EBA.3 Three subsets of BSLE can be classified according to autoantibodies reacting with collagen type VII or others (type I), yet undefined epitope(s) of the basement membrane (type II), or epidermis (type III).51 However, there are no differences between the subtypes from a clinical standpoint.3
All cases of BSLE require the diagnosis of SLE and must meet ACR and/or SLICC diagnostic criteria for SLE.58,59 As the SLICC criteria for SLE classification require at least one immunological criterion, SLE-related serologies are often positive in BSLE patients. ANA (high titer ≥1:320), anti-dsDNA, and anti-Sm are highly positive in BSLE cases, occurring in 90%, 60%, and 30%, respectively.52 It is unclear whether the flare of BSLE parallels disease activity. BSLE in conjunction with internal organ involvement (26%–100%), active lupus nephritis (>500 mg proteinuria/24 hours and/or urine protein and creatinine ratio >0.5), and/or hematologic abnormalities (anemia and/or leukopenia/lymphopenia on two occasions) have been reported.52,60 In such cases, proteinuria, abnormal complete blood count, elevated erythrocyte sedimentation rate, and low complement levels are usually present.52
Dapsone is a good first-line drug for BSLE.61 However, if systemic disease is active or skin disease extensive, then starting with steroids is routine and normally effective.61Immunosuppressives are helpful as steroid-sparing agents, but typically do not work as single-agent treatment. Methotrexate, azathioprine, cyclophosphamide, and mycophenolate mofetil have been used in patients with BSLE, with variable results.52,61 Rituximab, a chimeric monoclonal antibody that reacts with CD20, has shown promising result in refractory cases of BSLE.62
Lesions usually clear up without scarring, although postinflammatory hypo- or hyperpigmentation can occur, especially in patients with darker skin types.52 In a long-term retrospective review, recurrence of BSLE rarely occured. However, a few patients may experience flares and/or uncontrolled internal lupus, reflecting the relapsing–remitting and chronic active pattern of SLE.52
LE-related autoimmune bullous diseases
Autoimmune blistering diseases and SLE have been reported to coexist. Approximately 30% of SLE patients have an additional autoimmune disease.63 Both pemphigus and pemphigoid are reported to occur together with various other autoimmune diseases, eg, connective-tissue diseases, particularly SLE, myasthenia gravis, thymoma, and chronic thyroiditis.6–8 The temporal relationship has not been clearly elucidated through prospective studies. Although definitive explanation for the coexistence of SLE and autoimmune bullous diseases is lacking, specific immunomechanisms and immunogenetics may offer valuable insights. There is a high frequency of human leukocyte antigen in SLE and virtually all autoimmune bullous conditions.9,10
Pemphigus and LE are both B-cell-mediated autoimmune diseases, dependent on autoreactive CD4+ T lymphocytes to modulate autoimmune B-cell response. Sezin et al identified two novel regulatory genes, namely IRF8 and STAT1, as genetic markers that were significantly associated with pemphigus and SLE.64 According to a recent systemic review and meta-analysis, the prevalence of SLE was slightly higher among patients with pemphigus (0.5%) than controls (0.3%), although the association did not exceed the level of statistical significance (OR 1.85, 95% CI 0.89–3.82).65 Simultaneous occurrence of SLE and various forms of pemphigus have been documented, such as PE, PF, PV, PH, and PNP.6,7,11,65–70 PE, also known as Senear–Usher syndrome, is currently recognized as a variant of PF manifesting with overlapping immunological and serological features of SLE. Classic clinical presentation of PE is erythematous, scaly, erosive patches along the seborrheic areas, yet exhibits hints of photodistribution, often involving the malar area (Figure 8). While sharing common histopathology and immunofluorescence findings with PF, PE has positive circulating ANA and positive DIF along the basement-membrane zone. Patients with PE have been documented to have internal lupus (ie, hematologic renal involvement).71 A few reported cases of PF coexisting with LE have been published.7,66 Sawamura et al reported a case of SLE with PF, myasthenia gravis, and chronic thyroiditis after a thymectomy for thymoma, which are rare coexisting autoimmune diseases.7 A recent report by Temel et al demonstrated an unusual case with prominent features of both DLE and PF.66 PV has also been reported to coexist with LE.6,65,67–69 Among those who meet criteria for both SLE and PV, the demographic profile, clinical outcome, and organ-specific involvement (eg, arthritis, hematologic, renal, and neurological involvement) are more typical of SLE.6 Patients with coexisting PV and SLE do not appear to have life-threatening systemic consequences of SLE.69 To date, there has been only one reported case of PNP in association with SLE and polymyositis70 and one case of coexistence of PH and SLE in the literature.11
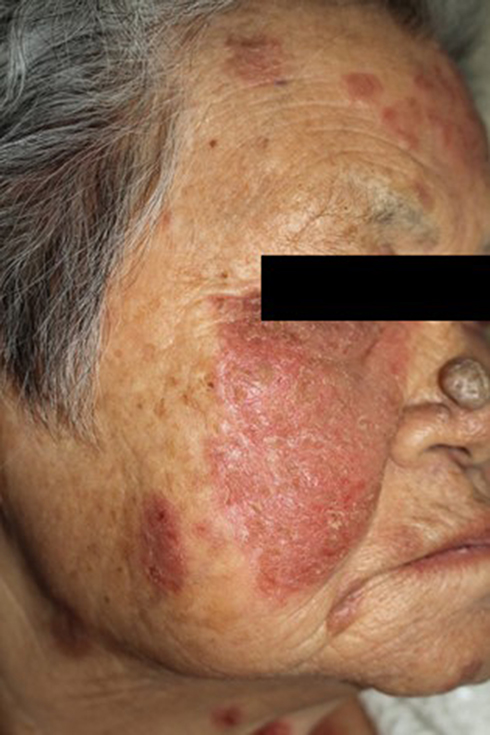 |
Figure 8 An 80-year-old female patient with pemphigus erythematosus presented with a 3-month history of erythematous and scaly, erosive patches along the seborrheic areas, aggravated by sun exposure. |
The co-occurrence of LE and autoimmune subepidermal vesiculobullous disease could be from interface dermatitis of LE-specific disease resulting in exposure to epidermal and dermal antigens, causing sensitization and production of autoantibodies responsible for the development of autoimmune bullous diseases. This hypothesis would be consistent for autoantibodies against basement-membrane antigens, eg, BP, LABD, MMP, p200 pemphigoid, DH, and EBA.22 LE with BP, LABD, MMP, DH, and EBA have all been documented.8–10,12,13 Moreover, specific immunomechanisms and immunogenetics could play a role in the concurrence of these conditions. For example, there seems to be a high frequency of human leukocyte antigens DR4 and DQ B1*0301 in MMP coexisting with connective-tissue disease.10 DH and SLE patients have been shown to share HLA-B8 and DR3 haplotypes.9 However, it is important to note that the simultaneous occurrence of SLE and autoimmune subepidermal dermatosis is exceedingly rare. Some reports date far back before specific diagnostic methods were available. Therefore, these patients could simply represent a specific subset of bullous eruptions of SLE, rather than a separate diagnostic entity.
Nonautoimmune disorders in association with LE
Many nonautoimmune conditions, namely EM, SJS/TEN, and PCT, have been reported in SLE patients. Uncertainty lies as to whether the occurrence of these conditions in SLE patients is related to immunodysregulation, increased incidence of medication or infection, or if there is no relation at all. Differentiation between classic SJS/TEN in patients with underlying SLE and SJS/TEN-like LE may be difficult, and relies on a complete evaluation of clinical findings and histopathology. Moreover, case reports/series suggest that SLE is a risk factor for developing SJS/TEN.28
The association of EM with LE has been documented by Rowell et al.19 EM is mostly precipitated by infections (herpes simplex virus and mycoplasma), and is generally not associated with specific autoimmune abnormalities. In contrast to EM-like LE, infections are usually not the trigger. SJS/TEN has been reported to occur with increased frequency in patients with connective-tissue disease. A relatively large case–control study demonstrated that SJS, SJS/TEN, and TEN occurred in 4%–8% of patients with collagen vascular diseases.72 Several case reports and case series have suggested that SLE is a risk factor for developing SJS/TEN.73,74 Ziemer et al reported a frequency of LE up to 1.2%–2% in the registry on SJS/TEN.28 Codependent risk factors could share mechanisms of acute diffuse epidermal apoptosis with associated production of inflammatory cytokines, eg, genetic predisposition and prior use of immunosuppressive drugs.28 However, the presence of acute onset, widespread mucosal involvement, prominent systemic features, history of culprit drugs, and poor prognosis in SLE patients who developed SJS/TEN did not differ from the classic SJS/TEN. Therefore, LE may act as an etiologic cofactor in TEN, but does not modify its presentation or course. The coexistence of PCT with different variants of LE has been reported in SLE, DLE, and SCLE patients.15,75,76 PCT can occurr before, simultaneously with, or after LE presentation. Because both conditions present clinically with photosensitivity, one may conceal the presence of the other. The coexistence of PCT and LE is thought to be from a common genetic factor. Several genetic susceptibility loci for LE have been identified on chromosome I in closed genetic vicinity to the uroporphyrinogen decarboxylase, the enzyme deficient in PCT. Furthermore, porphyrin deposits in the skin cause increasing photosensitivity and could trigger LE. In PCT, there is ongoing inflammatory reaction in the dermis that may lead to the release of autoantigens with subsequent autoantibody production in genetically and epidemiologically susceptible individuals.15,75
Conclusion
LE-associated vesiculobullous diseases have variable presentation. With this review, we have organized vesiculobullous diseases in relation to LE into four groups consisting of LE-specific and aspecific vesiculobullous diseases, LE-related autoimmune bullous diseases, and LE in association with nonautoimmune conditions. It is important for physicians, dermatologists, and rheumatologists to recognize and fully understand these conditions. A thorough evaluation of the patients’ history, and complete clinical, histopathological, and/or immunopathological information to establish a definitive diagnosis of LE-associated vesiculobullous disease is essential. However, a uniform classification is lacking, which causes diagnostic heterogeneity. Therefore, additional work is needed to fully understand the spectrum of vesiculobullous LE skin diseases, and a consensus is required to improve the classification of these conditions.
Statement of ethics
The patients provided written informed consent to perform all necessary investigations, to take clinical photographs, and to use them for research purposes and publication.
Acknowledgment
We acknowledge the assistance of Dr Poonkiat Suchonwanit in the preparation of illustrations.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hejazi EZ, Werth VP. Cutaneous lupus erythematosus: an update on pathogenesis, diagnosis and treatment. Am J Clin Dermatol. 2016;17(2):135–146. doi:10.1007/s40257-016-0173-9
2. Pons-Estel GJ, Salerni GE, Serrano RM, et al. Therapeutic plasma exchange for the management of refractory systemic autoimmune diseases: report of 31 cases and review of the literature. Autoimmun Rev. 2011;10(11):679–684. doi:10.1016/j.autrev.2011.04.028
3. Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15(6):517–524. doi:10.1007/s40257-014-0098-0
4. Gilliam JN, Sontheimer RD. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J Am Acad Dermatol. 1981;4(4):471–475. doi:10.1016/s0190-9622(81)80261-7
5. Sontheimer RD. The lexicon of cutaneous lupus erythematosus–a review and personal perspective on the nomenclature and classification of the cutaneous manifestations of lupus erythematosus. Lupus. 1997;6(2):84–95. doi:10.1177/096120339700600203
6. Malik M, Ahmed AR. Concurrence of systemic lupus erythematosus and pemphigus: coincidence or correlation? Dermatology. 2007;214(3):231–239. doi:10.1159/000099588
7. Sawamura S, Kajihara I, Makino K, et al. Systemic lupus erythematosus associated with myasthenia gravis, pemphigus foliaceus and chronic thyroiditis after thymectomy. Australas J Dermatol. 2017;58(3):e120–e122. doi:10.1111/ajd.12510
8. Stoll DM, King LE
9. Kurano TL, Lum CA, Izumi AK. The association of dermatitis herpetiformis and systemic lupus erythematosus. J Am Acad Dermatol. 2010;63(5):892–895. doi:10.1016/j.jaad.2009.05.037.
10. Malik M, Gurcan HM, Ahmed AR. Coexistence of mucous membrane pemphigoid and connective-tissue disease. Clin Exp Dermatol. 2010;35(2):156–159. doi:10.1111/j.1365-2230.2009.03222.x
11. Marinovic B, Basta-Juzbasic A, Bukvic-Mokos Z, Leovic R, Loncaric D. Coexistence of pemphigus herpetiformis and systemic lupus erythematosus. J Eur Acad Dermatol Venereol. 2003;17(3):316–319.doi:10.1046/j.1468-3083.2003.00738.
12. McHenry PM, Dagg JH, Tidman MJ, Lever RS. Epidermolysis bullosa acquisita occurring in association with systemic lupus erythematosus. Clin Exp Dermatol. 1993;18(4):378–380.doi:10.1111/j.1365-2230.1993.tb02224.x
13. Tobon GJ, Toro CE, Bravo JC, Canas CA. Linear IgA bullous dermatosis associated with systemic lupus erythematosus: a case report. Clin Rheumatol. 2008;27(3):391–393. doi:10.1007/s10067-007-0752-5
14. Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67(3):417–421. doi:10.1016/j.jaad.2011.10.012
15. van Tuyll van Serooskerken AM, Habets JM, Badeloe S, Poblete-Gutierrez P, Frank J. Porphyria cutanea tarda in pre-existent lupus erythematosus–is there an association? Int J Dermatol. 2007;46(Suppl 3):S50–S52. doi:10.1111/j.1365-4632.2007.03515.x
16. Ting W, MS S, Racila D, Scofield RH, Sontheimer RD. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus and the spectrum of the acute syndrome of apoptotic pan-epidermolysis (ASAP): a case report, concept review and proposal for new classification of lupus erythematosus vesiculobullous skin lesions. Lupus. 2004;13(12):941–950. doi:10.1191/0961203304lu2037sa
17. Lee HY, Tey HL, Pang SM, Thirumoorthy T. Systemic lupus erythematosus presenting as Stevens-Johnson syndrome and toxic epidermal necrolysis: a report of three cases. Lupus. 2011;20(6):647–652. doi:10.1177/0961203310385162
18. Zeitouni NC, Funaro D, Cloutier RA, Gagne E, Claveau J. Redefining Rowell’s syndrome. Br J Dermatol. 2000;142(2):343–346. doi:10.1046/j.1365-2133.2000.03306.x
19. Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions. A syndrome with characterisctic immunological abnormalitities. Arch Dermatol. 1963;88(2):176–180. doi:10.1001/archderm.1963.01590200064012
20. Yildirim Cetin G, Sayar H, Ozkan F, Kurtulus S, Kesici F, Sayarlioglu M. A case of toxic epidermal necrolysis-like skin lesions with systemic lupus erythematosus and review of the literature. Lupus. 2013;22(8):839–846. doi:10.1177/0961203313492242
21. Duarte-Garcia A, Fang H, To CH, Magder LS, Petri M. Seasonal variation in the activity of systemic lupus erythematosus. J Rheumatol. 2012;39(7):1392–1398. doi:10.3899/jrheum.111196
22. Merklen-Djafri C, Bessis D, Frances C, et al. Blisters and loss of epidermis in patients with lupus erythematosus: a clinicopathological study of 22 patients. Medicine (Baltimore). 2015;94(46):e2102. doi:10.1097/MD.0000000000000874
23. Perera GK, Black MM, McGibbon DH. Bullous subacute cutaneous lupus erythematosus. Clin Exp Dermatol. 2004;29(3):265–267. doi:10.1111/j.1365-2230.2004.01498.x
24. Ryan E, Marshman G, Astill D. Toxic epidermal necrolysis-like subacute cutaneous lupus erythematosus. Australas J Dermatol. 2012;53(4):303–306. doi:10.1111/j.1440-0960.2011.00842.x
25. Walling HW, Sontheimer RD. Cutaneous lupus erythematosus: issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10(6):365–381. doi:10.2165/11310780-000000000-00000
26. Achtman JC, Werth VP. Pathophysiology of cutaneous lupus erythematosus. Arthritis Res Ther. 2015;17:182. doi:10.1186/s13075-015-0706-2
27. Wenzel J, Tuting T. An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in “interface dermatitis”. J Invest Dermatol. 2008;128(10):2392–2402. doi:10.1038/jid.2008.96
28. Ziemer M, Kardaun SH, Liss Y, Mockenhaupt M. Stevens-Johnson syndrome and toxic epidermal necrolysis in patients with lupus erythematosus: a descriptive study of 17 cases from a national registry and review of the literature. Br J Dermatol. 2012;166(3):575–600. doi:10.1111/j.1365-2133.2011.10705.x
29. Crowson AN, Magro C. The cutaneous pathology of lupus erythematosus: a review. J Cutan Pathol. 2001;28(1):1–23.doi:10.1034/j.1600-0560.2001.280101.x
30. Chhabra S, Minz RW, Saikia B. Immunofluorescence in dermatology. Indian J Dermatol Venereol Leprol. 2012;78(6):677–691. doi:10.4103/0378-6323.102355
31. Yu J, Brandling-Bennett H, Co DO, Nocton JJ, Stevens AM, Chiu YE. Toxic epidermal necrolysis-like cutaneous lupus in pediatric patients: a case series and review. Pediatrics. 2016;137(6):e20154497. doi:10.1542/peds.2015-4497
32. Baker MG, Cresce ND, Ameri M, Martin AA, Patterson JW, Kimpel DL. Systemic lupus erythematosus presenting as Stevens-Johnson syndrome/toxic epidermal necrolysis. J Clin Rheumatol. 2014;20(3):167–171. doi:10.1097/RHU.0000000000000088
33. Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45(6):803–822. (quiz 822-804). doi:10.1067/mjd.2001.117518
34. Patsinakidis N, Gambichler T, Lahner N, Moellenhoff K, Kreuter A. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30(12):2097–2104. doi:10.1111/jdv.13769
35. Tankunakorn J, Sawatwarakul S, Vachiramon V, Chanprapaph K. Stevens-Johnson syndrome and toxic epidermal necrolysis-like lupus erythematosus. J Clin Rheumatol. 2019;25(5):224–231. doi:10.1097/RHU.0000000000000830
36. Simsek I, Cinar M, Erdem H, Pay S, Meric C, Dinc A. Efficacy of plasmapheresis in the treatment of refractory toxic epidermal necrolysis-like acute cutaneous lupus erythematosus. Lupus. 2008;17(6):605–606. doi:10.1177/0961203308089341
37. Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatology. 2012;224(2):134–139. doi:10.1159/000338202
38. Moulis G, Sommet A, Lapeyre-Mestre M, Montastruc JL. Is the risk of tumour necrosis factor inhibitor-induced lupus or lupus-like syndrome the same with monoclonal antibodies and soluble receptor? A case/non-case study in a nationwide pharmacovigilance database. Rheumatology (Oxford). 2014;53(10):1864–1871. doi:10.1093/rheumatology/keu214
39. Napolitano M, Giampetruzzi AR, Didona D, Papi M, Didona B. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus successfully treated with a single dose of etanercept: report of three cases. J Am Acad Dermatol. 2013;69(6):e303–e305. doi:10.1016/j.jaad.2013.07.036
40. Paradisi A, Abeni D, Bergamo F, Ricci F, Didona D, Didona B. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71(2):278–283. doi:10.1016/j.jaad.2014.04.044
41. Vital EM, Wittmann M, Edward S, et al. Brief report: responses to rituximab suggest B cell-independent inflammation in cutaneous systemic lupus erythematosus. Arthritis Rheumatol. 2015;67(6):1586–1591. doi:10.1002/art.39085
42. Manzi S, Sanchez-Guerrero J, Merrill JT, et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: combined results from two phase III trials. Ann Rheum Dis. 2012;71(11):1833–1838. doi:10.1136/annrheumdis-2011-200831
43. Khamashta M, Merrill JT, Werth VP, et al. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(11):1909–1916. doi:10.1136/annrheumdis-2015-208562
44. Furie R, Khamashta M, Merrill JT, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 2017;69(2):376–386. doi:10.1002/art.39962
45. Werth VP, Fiorentino D, Sullivan BA, et al. Brief report: pharmacodynamics, safety, and clinical efficacy of AMG 811, a human anti-interferon-gamma antibody, in patients with discoid lupus erythematosus. Arthritis Rheumatol. 2017;69(5):1028–1034. doi:10.1002/art.40052
46. Kubo S, Nakayamada S, Tanaka Y. Baricitinib for the treatment of rheumatoid arthritis and systemic lupus erythematosus: a 2019 update. Expert Rev Clin Immunol. 2019;15(7):696–700. doi:10.1080/1744666X.2019.1608821.
47. Shreberk-Hassidim R, Ramot Y, Zlotogorski A. Janus kinase inhibitors in dermatology: A systematic review. J Am Acad Dermatol. 2017;76(4):745–753.e719. doi:10.1016/j.jaad.2016.12.004
48. Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392(10143):222–231. doi:10.1016/S0140-6736(18)31363-1
49. Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29(4):649–653.doi:10.1016/j.det.2011.06.002.
50. Pan M, Tang HD, Zhu HQ, Dong J, Gill J, Zheng J. Partial epilepsy as an initial manifestation in bullous systemic lupus erythematosus. Lupus. 2011;20(8):886–890. doi:10.1177/0961203311398511
51. Yell JA, Allen J, Wojnarowska F, Kirtschig G, Burge SM. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132(6):921–928. doi:10.1111/j.1365-2133.1995.tb16950.x
52. Chanprapaph K, Sawatwarakul S, Vachiramon V. A 12-year retrospective review of bullous systemic lupus erythematosus in cutaneous and systemic lupus erythematosus patients. Lupus. 2017;26(12):1278–1284. doi:10.1177/0961203317699714
53. Nico MM, Lourenco SV. Multiple blisters along the lip vermilion are a clue to bullous lupus erythematosus. Acta Derm Venereol. 2012;92(4):404–405. doi:10.2340/00015555-1276
54. Vassileva S. Bullous systemic lupus erythematosus. Clin Dermatol. 2004;22(2):129–138. doi:10.1016/j.clindermatol.2003.12.020
55. Sirka CS, Padhi T, Mohanty P, Patel DK, Parida PR, Kar CR. Bullous systemic lupus erythematosus: response to dapsone in two patients. Indian J Dermatol Venereol Leprol. 2005;71(1):54–56.doi:10.4103/0378-6323.13795
56. Chan LS, Lapiere JC, Chen M, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol. 1999;135(5):569–573. doi:10.1001/archderm.135.5.569
57. Barbosa WS, Rodarte CM, Guerra JG, Maciel VG, Fleury Junior LF, Costa MB. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 Suppl 1):S92–S95. doi:10.1590/s0365-05962011000700024
58. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi:10.1002/1529-0131(199707)40:7<1267::AID-ART11>3.0.CO;2-L
59. Petri M, Orbai AM, Alarcon GS, et al. Derivation and validation of the systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–2686. doi:10.1002/art.34473
60. Malcangi G, Brandozzi G, Giangiacomi M, Zampetti M, Danieli MG, Bullous SLE. response to methotrexate and relationship with disease activity. Lupus. 2003;12(1):63–66. doi:10.1191/0961203303lu241cr
61. Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
62. Alsanafi S, Kovarik C, Mermelstein AL, Werth VP. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17(3):142–144. doi:10.1097/RHU.0b013e318214f30c
63. McDonagh JE, Isenberg DA. Development of additional autoimmune diseases in a population of patients with systemic lupus erythematosus. Ann Rheum Dis. 2000;59(3):230–232. doi:10.1136/ard.59.3.230
64. Sezin T, Vorobyev A, Sadik CD, Zillikens D, Gupta Y, Ludwig RJ. Gene expression analysis reveals novel shared gene signatures and candidate molecular mechanisms between pemphigus and systemic lupus erythematosus in CD4(+) T cells. Front Immunol. 2017;8:1992. doi:10.3389/fimmu.2017.01992
65. Kridin K, Laufer-Britva R, Kridin M, Comaneshter D, Batat E, Cohen AD. The relationship between pemphigus and systemic lupus erythematosus: a cross-sectional study, systematic review, and meta-analysis. Immunol Res. 2019;67(1):116–122. doi:10.1007/s12026-019-9065-4
66. Bilgic Temel A, Ergun E, Poot AM, et al. A rare case with prominent features of both discoid lupus erythematosus and pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2019;33(1):e5–e7. doi:10.1111/jdv.15099
67. Calebotta A, Cirocco A, Giansante E, Reyes O. Systemic lupus erythematosus and pemphigus vulgaris: association or coincidence. Lupus. 2004;13(12):951–953. doi:10.1191/0961203304lu1073cr
68. Hidalgo-Tenorio C, Sabio-Sanchez JM, Tercedor-Sanchez J, Leon-Ruiz L, Perez-Alvarez F, Jimenez-Alonso J. Pemphigus vulgaris and systemic lupus erythematosus in a 46-y-old man. Lupus. 2001;10(11):824–826. doi:10.1177/096120330101001112
69. Malik M, Ahmed AR. Dual diagnosis of pemphigus vulgaris and connective tissue disease. J Am Acad Dermatol. 2006;55(4):699–704. doi:10.1016/j.jaad.2006.04.052
70. Mascaro JM
71. Vassileva S, Drenovska K, Manuelyan K. Autoimmune blistering dermatoses as systemic diseases. Clin Dermatol. 2014;32(3):364–375. doi:10.1016/j.clindermatol.2013.11.003
72. Auquier-Dunant A, Mockenhaupt M, Naldi L, Correia O, Schroder W, Roujeau JC. Correlations between clinical patterns and causes of erythema multiforme majus, Stevens-Johnson syndrome, and toxic epidermal necrolysis: results of an international prospective study. Arch Dermatol. 2002;138(8):1019–1024. doi:10.1001/archderm.138..8.1019
73. Horne NS, Narayan AR, Young RM, Frieri M. Toxic epidermal necrolysis in systemic lupus erythematosus. Autoimmun Rev. 2006;5(2):160–164.doi:10.1016/j.autrev.2005.10.003
74. Vachvanichsanong P, Dissaneewate P. Childhood systemic lupus erythematosus in songklanagarind hospital: a potential unique subgroup. Clin Rheumatol. 1993;12(3):346–349.
75. Gibson GE, McEvoy MT. Coexistence of lupus erythematosus and porphyria cutanea tarda in fifteen patients. J Am Acad Dermatol. 1998;38(4):569–573. doi:10.1016/s0190-9622(98)70119-7
76. Peitsch WK, Lorentz K, Goebeler M, Goerdt S. Subacute cutaneous lupus erythematosus with bullae associated with porphyria cutanea tarda. J Dtsch Dermatol Ges. 2007;5(3):220–222. doi:10.1111/j.1610-0387.2007.06201.x
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.