Back to Journals » Vascular Health and Risk Management » Volume 21
Vascular Endothelium in Health and Disease: Structure, Function, Assessment and Role in Metabolic Disorders
Authors Pluta W ![]() , Lubkowska A
, Lubkowska A ![]() , Dudzińska W
, Dudzińska W
Received 25 January 2025
Accepted for publication 23 July 2025
Published 3 September 2025 Volume 2025:21 Pages 729—747
DOI https://doi.org/10.2147/VHRM.S519426
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Pietro Scicchitano
Waldemar Pluta,1,* Anna Lubkowska,1,* Wioleta Dudzińska1,2,*
1Department of Functional Diagnostics and Physical Medicine, Pomeranian Medical University in Szczecin, Szczecin, 71-210, Poland; 2Institute of Biology, University of Szczecin, Szczecin, 71-412, Poland
*These authors contributed equally to this work
Correspondence: Anna Lubkowska, Department of Functional Diagnostics and Physical Medicine, Pomeranian Medical University in Szczecin, Żołnierska 54, Szczecin, 71-210, Poland, Email [email protected]
Abstract: The vascular endothelium is responsible for regulating vascular tone, maintaining fluid homeo-stasis, and preventing platelet aggregation, exhibits regulatory properties in vasorelaxation and vasoconstriction – it produces, among others, nitric oxide and endothelin. The imbalance of vasoactive molecules leads to the loss of their function, known as endothelial dysfunction. Impaired endothelial function is observed in people with metabolic disorders, often preceding the onset of the disease by several years. Endothelial function assessment can be performed using invasive (eg coronary angiography) and non-invasive (flow-dependent dilation, biochemical markers) techniques. Among metabolic abnormalities, increasing scientific interest has focused on metabolic obesity in normal-weight individuals. People with this type of obesity, despite having a normal body mass index, exhibit both metabolic abnormalities typical of obesity (including hypertension and type 2 diabetes) and endothelial dysfunction. This comprehensive review summarizes the current knowledge of the structure and function of the vascular endothelium, methodologies for assessment, and pathophysiological mechanisms of endothelial dysfunction in metabolic disorders, with particular emphasis on diagnostic approaches and clinical implications for cardiovascular risk stratification.
Keywords: vascular endothelium, endothelial dysfunction, metabolic disorders, cardiovascular risk
Introduction
The vascular endothelium (VE) is the innermost structure of endothelial cells that covers internal capillaries, veins, arteries and lymph vessels. This heterogeneous monolayer of highly specialized cells is the first barrier to circulating pathogens, cells, and other molecules in the bloodstream. Its action is selective, therefore, it controls the exchange of relationships between the bloodstream and the surrounding tissue in an organ-specific manner. The total area of the vascular endothelium is about 1 km2 - for this reason, VE can be regarded as the largest organ in the human body.1 For many years, endothelial cells were thought to be inactive. It is now known that they have unique properties – they are able to sense changes in hemodynamic forces and the biochemistry of flowing blood.2
VE has many functions that vary by location.3 VE is responsible for regulating vascular tone, maintaining fluid homeostasis, and preventing platelet aggregation. The vascular endothelium also exhibits regulatory properties in vasorelaxation and vasoconstriction – it produces, among others, nitric oxide (NO) and endothelin (ET). The imbalance of vasoactive molecules leads to the loss of their function, known as endothelial dysfunction.4 Impaired endothelial function is observed in people with metabolic disorders, often preceding the onset of the disease by several years.
This comprehensive review summarizes the current knowledge of the structure and function of the vascular endothelium, methodologies for assessment, and pathophysiological mechanisms of endothelial dysfunction in metabolic disorders, with particular emphasis on diagnostic approaches and clinical implications for cardiovascular risk stratification.
The Vascular Endothelium
The vascular endothelium is composed of endothelial cells (ECs), with dimensions of about 10 × 30 µm and a cytoplasm thickness of 0.2 µm. Usually, ECs are flat, however, their appearance depends on their location in the vascular network. In arteries and arterioles, they take on a spindle shape and are oriented according to the direction of blood flow. This shape is a consequence of the shear stress occurring in the vessel and can change depending on the action of these forces. In contrast, in veins and capillaries, ECs take on a more rounded shape.1
ECs have a polar structure - the basal surface of ECs is separated from surrounding tissues by a glycoprotein basement membrane, while the luminal membrane is directly exposed to blood components and circulating cells.5 Basement membrane, which is formed by the extracellular matrix (ECM), is rich in type IV and type V collagen, fibronectin and laminin. Strong adhesion of the endothelium to the basement membrane is possible thanks to numerous integrin receptors that bind it to ECM proteins and are located in the basolateral part of its cells. The luminal membrane forms a smooth layer, on which sparse microvilli are formed from the plasmatic membrane, restricting the sites of intercellular connections.6
Structural integrity is essential for the VE to function properly. Individual ECs are connected to each other by intercellular connections, for whose interactions they are responsible molecules located in the lateral part of them. These include primarily constitutive and induced adhesion molecules: E-selectin (CD62E), P-selectin (CD62P); immunoglobulin-like addressins: intercellular adhesion molecule-1 (ICAM-1, CD54), intercellular adhesion molecule-2 (ICAM-2, CD102), platelet endothelial cell adhesion molecule (PECAM-1, CD31), and vascular cell adhesion molecule (VCAM-1, CD106).7,8
Among the intercellular connections occurring in the endothelium, can be distinguish adherens junction (AJ), tight junction (TJ) and gap junction (GJ). These connections alternate, creating a characteristic system consisting of tight, adherent and gap connections, occurring from the top to the base of the endothelial cell. Connections are responsible for maintaining the integrity of the endothelium and are involved in regulating the flow of signals generated in the course of many biological processes (such as apoptosis or proliferation).9
Adherens junctions connect ECs, providing the structural basis for the mechanical stability of the interendothelium. AJs are formed by integral membrane proteins belonging to the family of Ca2+- dependent cadherins with the subtypes VE-cadherin, P-cadherin, N-cadherin and E-cadherin. In addition, adherens junctions include nectin-2 and PECAM-1. Tight junctions contribute to maintaining cell polarity and seal off neighboring cells, inhibiting uncontrolled intercellular exchange of small molecules, macromolecules and water. TJs are formed mainly by occludins – proteins that, by binding to the actin filaments of the cytoskeleton of the basement membrane, close the intercellular zone, thus restricting the transport of substances. Other proteins include junctional adhesion molecule, endothelial cell-selective adhesion molecule and nectins. TJs are particularly characteristic of mammalian brain blood vessels, where they form the cerebrovascular barrier. Gap junctions are formed by clusters of trans-membrane channels that connect the cytoplasms of neighboring cells. These channels, called connexins, are made up of six transmembrane proteins of the connexin family. GJ occurs between endothelial cells located throughout the vascular system, as well as between ECs and smooth muscle cells surrounding resistance vessels. Through these connections, intercellular transfer of ions and small molecules is possible, allowing for rapid coordination of cellular functions.9,10
Most of the surface of the ECs is covered by a multilayer structure - the endothelial surface layer (ESL). Its role is to limit the access of cellular and macromolecular blood components to the surface of the VE. ESL is several hundred nanometres thick (up to 8 µm in some larger blood vessels) and consists mainly of glycocalyx (eGCX), which is formed by negatively charged glycoproteins, proteoglycans (PGs), and glycosaminoglycans (GAGs). PGs and GAGs anchor the eGCX to the endothelium and create an extensive matrix containing soluble components.11,12
PGs have a protein core to which negatively charged GAG side chains are attached. The most common glycosaminoglycan (50–90% GAGs) is heparan sulfate (HS), while others include hyaluronic acid, chondroitin, dermatan, and keratan sulfates.13,14 Non-covalently bonded hyaluronans of a non-sulphate nature play an important function related to the cytosolic aspect of eGCX, eg, mediating shear stress or vascular permeability.11 Degradation of heparan sulfate proteoglycans (HSPGs) can lead to VE dysfunction. GAGs are being released into the circulation leading to a decrease in the thickness of the eGCX and endothelial cell membranes. There is a change in the permeability of blood vessels and an increase in the flow of fluid, leukocytes and albumin from the lumen to the tissue spaces, causing edema. Reduction of eGCX leads to an increased likelihood of a prothrombotic environment in which exposed adhesion molecules attract platelets, increase thrombin production, and promote fibrinolysis.15
The exact composition and thickness of the layer varies depending on the location. By forming a luminal lattice, eGCX provides endothelial cells with a skeleton to bind plasma proteins and soluble GAGs. The eGCX itself is inactive; however, binding or immersion of plasma components in this structure creates a physiologically active ESL.16
The distinctive structure makes the eGCX perform many physiological functions. On the one hand, it acts as a barrier, on the other hand, the other negatively charged structure makes eGCX act as a macromolecular sieve, repelling negatively charged particles (including platelets and white and red blood cells) and attracting many enzymes and regulatory molecules – for example, extracellular superoxide dismutase (ecSOD) or lipoprotein lipase (LPL).12,15 It is known that macromolecules larger than 70 kDa are excluded from the tight structure of eGCX. Nevertheless, for albumin (67 kDa negatively charged), it is closely related to eGCX due to its amphoteric nature.17 However, this binding leads to a reduction in the hydraulic conductivity across the vascular barrier. Because of this, some proteins in the albumin group leak through the structure.5
Conformational changes in eGCX and excretion of microparticles contribute to the regulation of vascular tone and thus oxygen distribution by triggering the release of nitric oxide. Through this rheological mechanism, eGCX contributes to the maintenance of homeostasis in peripheral tissues.18 The glycocalyx is also an important binding site for blood proteins – including fibroblast growth factor, antithrombin III, and extracellular superoxide dismutase – so it plays an important role in vascular protection by inhibiting coagulation and leukocyte adhesion.12 The integrity of eGCX, which depends on blood chemistry and flow along blood vessels, plays a key role in preventing vascular complications. High amounts of neurotransmitters, hormones, and vasoactive factors in the blood can lead to degradation of eGCX. This results in the exposure of glycoprotein components that facilitate inflammation and thrombus formation. However, the greatest impact on eGCX structure is due to increases in blood flow. A strong, endothelium-protective glycocalyx occurs along vessel walls in which the flow is uniform. In vessels with complex geometry in which blood flow is uneven – eGCX becomes thinner within the resident endothelial cells.2,19
A characteristic structure of ECs is the Weible-Palade bodies (WPBs) located in the cytoplasm, which contain a pro-coagulation glycoprotein that carries von Willebrand factor (VWF). This factor, representing 90% of the WPB, enables platelets to adhere to the vascular endothelium and aggregate at the site of injury. WPBs also contain P-selectin (adhesion of leukocytes), angiopoetin-2 (wound healing), interleukin-8 (Il-8; boost inflammation), tissue plasminogen activator (tPA; preventing excessive formation of fibrin) and ET-1 (vasoconstriction). Activation and exocytosis of WPB can be triggered by several stimuli including vascular endothelial growth factor (VEGF), thrombin, or epinephrine either through Ca2+/calmodulin-dependent pathways or in response to cyclic adenosine monophosphate-raising agonists distinctly influencing cytoskeletal function. Exocytosis of WPBs is considered the first vascular response to injury.20–22
ECs exhibit structural heterogeneity including differences in cell morphology (thickness, size and position of the nucleus), extracellular matrix production (basal lamina components), gene expression profiles and cell surface properties (varying compositions of the GCX and the amounts of structural components of the endocytic and transcytosis pathway).5 Particularly important is the heterogonicity of ECs depending on their location and the role they play in the body. Endothelium can be classified based on their intercellular connections (continuous, fenestrated, discontinuous).
Continuous junctions are characterized by luminal and abluminal plasma membranes joining only in tight junctions, which are the main pathway for the exchange of water, glucose, urea and other hydrophilic molecules. Such a tight barrier is found in the vessels of the skin, lungs, heart and brain (blood–brain barrier). The windowed monolayer is characterized by pores 50–60 nm in diameter, closed by a membrane. They are more permeable to low-molecular hydrophilic molecules and water. It is located in places that are characterized by increased filtration or increased transendothelial transport – vessels of endocrine glands, intestinal and gastric mucosa, glomeruli, and a subpopulation of renal tubules. In contrast, discontinuous monolayers have large inter- and intracellular gaps with a diameter of 100–200 nm. In these gaps, there is no or a basement membrane, which is not a solid structure, but can undergo dynamic changes. This structure is characteristic of liver vessels.9,23,24
ECs also show variation depending on the vessel they are lining. Lymphatic ECs are surrounded by a thin, discontinuous basement membrane; do not have perivascular cells and entail discontinuous “button-like” cell junctions. Between these connections are “flap valves” that allow fluid and immune cells to enter. Fiber-anchored ECs, which are capable of detecting changes in interstitial pressure, are responsible for modulating the opening and closing. Capillary ECs are mainly involved in the exchange of oxygen and nutrients in the circulatory system. To ensure the most efficient exchange of substances, the lining of ECs is very thin (less than 0.1 µm). Capillary ECs have a much higher density of caveolae compared to other vessels (up to 10000 per cell). ECs located in veins promote transfer of deoxygenated blood back to heart, form valves and do not have smooth muscle cell investment. The wall thickness of ECs is small (as in capillaries less than 0.1 µm), allows them to act as volume reserves. ECs lining the arteries do not develop valves and are invested with smooth muscle cells. They form thick vessel walls, which allows them to function as “pressure reserves”, which propel blood rapidly with each heartbeat. Due to the high blood flow rate in the arteries, ECs have an elongated or ellipsoidal shape.24,25
A special type of endothelium is the so-called high endothelial venules (HEV), which allow leukocyte migration. HEV cells are more cuboidal in shape than other ECs and are connected by discontinuous, “spot-welded” tight junctions. HEVs are found primarily in the extravillous venules of nonlymphatic organs and the peripheral vessels of peripheral organs (lymph nodes, Peyer’s tufts, tonsils). Unlike normal venules, where leukocyte migration occurs only under the influence of inflammation, transmigration by HEV allows continuous circulation of leukocytes between the blood and lymphoid organs.23,26
Such an extensive and complicated structure plays an important role in physiological processes. VE primarily maintains hemostasis of blood vessels, thus conditioning the proper blood flow in the circulatory system. This is the effect of the ECs synthesizing and secreting the entire spectrum of factors maintaining the state of equilibrium.27
By synthesizing and releasing anticoagulant factors (which under physiological conditions predominate over procoagulant factors), VE prevents excessive blood clotting. VE secretes numerous anticoagulant factors, the most important of which is antithrombin III (ATIII). It is a glycoprotein whose task is to inhibit active blood coagulation factors, in particular, factor II (thrombin), IXa, Xa, XIa, XIIa, plasmin and kallikrenin.28 Its action, leading to the inactivation of enzymes, is supported by the glycosaminoglycans found on the surface of the endothelium, which have heparin groups in their structure, because the heparin contained in them is a cofactor for antithrombin. The anticoagulant factors secreted by the endothelium also include heparin cofactor II, proteins S and C, and protease nexins I and II.29 These factors participate in the blood coagulation cascade. Heparin cofactor II is primarily involved in the inactivation of thrombin, but also factor Xa.30,31 The presence of thrombin causes the activation of protein C, and the cofactor of this process is thrombomodulin. The active form of protein C is capable of proteolysis, and thus inactivation of factors Va and VIIIa, but only in the presence of the cofactor, which is protein S.32 In turn, protein S is also involved in antigen presenting cells (APC)-independent activation of prothrombin by factors Va and Xa. ECs also secrete another factor involved in the blood coagulation cascade, the tissue factor pathway inhibitor (TFPI). TFPI is a protein expressed mainly on endothelial cells and to a lesser extent on active platelets. It has the ability to bind to blood plasma lipoproteins and is primarily responsible for the inhibition of factor Xa. TFPI located on thrombocytes is involved in regulating the activity of tissue factor (TF), secreted into the blood from activated endothelial cells and leukocytes. In addition, TFPI participates in the inhibition of the formation of a complex of tissue factor with factor VII (TF-fVIIa). The transport of TF to the clot takes place in microbubbles and is a necessary condition for the proper repair of damaged endothelium. If this process is not properly regulated, it can lead to vascular thrombosis.33,34
VEs secrete appropriate substances that are involved in maintaining normal fibrinolytic potential. These compounds primarily include plasminogen activators, which convert plasminogen into active plasmin in the presence of fibrin. One such factor is tPA, which is considered a marker of endothelial activation as well as damage. tPA secreted continuously by endothelium has the ability to combine with annexin II, also found in endothelium, so that plasminogen activation can occur even in the absence of fibrin. tPA is found in large amounts in the thyroid gland, which is the most circulating organ. The endothelium also secretes the urokinase plasminogen activator (uPA), but in lower amounts than tPA. Serine proteases are another factor involved in plasmin formation. To ensure proper balance in fibrinolysis, fibrinolysis inhibitors are secreted in addition to plasminogen activators. In physio-logical states, the amount of plasminogen activators dominates over fibrinolysis inhibitors. The well-known inhibitors of tPA and uPA are plasminogen activator inhibitors (PAI), among which are PAI-1, PAI2, PAI-3 and PAI-4 also called protease annexin I. PAI-1 is the most important in the inhibition of tPA, uPA and thrombin.35,36
ECs also secrete ectonucleases, which are responsible for degrading adenosine diphosphate (ADP) and adenosine triphosphate (ATP), resulting in the formation of inosine and adenosine, which act to inactivate platelet stimulating factors. Ectonucleases primarily include adenosine ectodiphosphatase and ectotriphosphatase.22
VE plays an important role in regulating blood flow and maintaining proper tone of the vessel wall. This is possible thanks to the vasodilating and vasoconstrictive factors secreted by it, which are released in response to chemical and biomechanical stimuli. These substances are produced by the normal endothelium in very small amounts. Vascular homeostasis is maintained because these factors are balanced, marginally in favor of vasodilation.5 Vasodilating factors cause relaxation of the smooth muscles of the vessel, which leads to dilatation of the blood vessel lumen, and thus slowing down the blood flow. The main compound with a vasodilating effect is NO. It is produced by nitric oxide synthases (NOS) in a stepwise redox reaction with L-arginine. The VE isoform of nitric oxide, eNOS, supplies NO under physiological conditions to maintain vascular tone under changing pressure and blood flow. NO, thanks to its lipophilic properties, has the ability to penetrate into the cells of the vascular muscles, where, by activating guanylate cyclase, it contributes to the relaxation of the blood vessel. NO is an unstable compound, relatively quickly captured and bound by hemoglobin, therefore, to maintain the state of vasodilation, it is necessary to continuously secrete this factor by endothelial cells. In addition to nitric oxide, the endothelium secretes prostacyclin (PGl2), acting synergistically with NO. This substance is classified as eicosanoids, and it is secreted in response to factors such as hypoxia, abrasion forces and the presence of acetylcholine or serotonin. PGl2 is involved in the relaxation of the vascular muscles, leading to its contraction by activating potassium channels and increasing the concentration of cyclic adenosine monophosphate (cAMP) in smooth muscle cells. An important vasodilating compound released by ECs is the endothelium derived hyperpolarizing factor (EDHF). This factor directly activates the ATP-dependent potassium channel, thus contributing to the hyperpolarization of the plasma membrane of endothelial cells. Hyperpolarization and reduced influx of potassium ions into the cell cause the weakening of contractile proteins.37 VE also secretes vasodilator-stimulated phosphoprotein (VASP) and leukotrienes C3 and C4, which are also vasodilators. Secretion of vasodilating factors is usually under the influence of bradykinin after its binding to the B2 receptor located in endothelial cells. This leads to many changes that cause the dilation of blood vessels due to the relaxation of their smooth muscles. Chemical stimuli that stimulate the endothelium to release vasodilating factors include acetylcholine, bradykinin, ADP, cytokines (endotoxin, interleukin 1b, tumor necrosis factor-α (TNF-α)), insulin, substance P, hormones, eg estrogens, while the biomechanical stimulus is primarily abrasion forces caused by the flow of blood cells.5
Vasoconstrictive factors are responsible for the contraction of vascular smooth muscle cells (VSMCs), which causes vasoconstriction. The strongest vasoconstrictor is ET-1. Its action is possible due to the existence of endothelin type B receptors located on endothelial cells and endothelin type A receptors present on smooth muscle cells. Serum ET-1 levels are elevated by pro-inflammatory EC activating signaling. Due to the vasoconstriction of the inflamed area, an encapsulating effect is obtained for pathogens and delays the leukocyte transmigration. In addition, ET-1 signaling leads to increased expression of adhesion molecules (eg VCAM) in the EC and promotes neutrophil aggregation. The synthesis and secretion of ET-1 are inhibited by NO. Among the compounds with a vasoconstrictive effect, we also distinguish thromboxane A2 (TXA2), angiotensin II (AII), platelet-activating factor (PAF), prostaglandin H, prostaglandin F and leukotrienes (except C4 and D4).5,22
eGCX modulates local blood viscosity and hematocrit in microcirculation – pushes red blood cells away from VE and shields it from platelet and leukocyte interactions. Cell adhesion molecules (selectins and integrins) protruding into the lumen of the vessel are surrounded by a more expansive ESL, as a result of which damaged eGCX causes increased adhesion of leukocytes and platelets.11
eGCX is considered a key transducer that translates fluid shear stress into a cellular signal (ie, mechanotransduction). The basis of action is the “bush-like” clusters of proteoglycans that protrude from anchor points in the cytoskeleton of endothelial cells (first described in 2003 by Weinbaum et al38). As a result of the mechanical distortion of the entire bush, a force is generated which deforms the cytoskeleton underneath. The expression of eNOS is increased, which catalyzes the production of NO and increases the hydraulic conductivity.11
Through the release of pro-angiogenic and anti-angiogenic factors, as well as the expression of surface proteins, the endothelium is involved in the formation of blood vessels, both by vasculogenesis and angiogenesis.39 An important factor regulating the formation of new blood vessels is the rate of proliferation of endothelial cells. Unactivated ECs are able to survive for about 1000 days, while during angiogenesis their rate of proliferation increases significantly and divisions may occur every 5 days.40 A significant role for the course of the angiogenesis process is also played by various adhesion molecules presented on the surface of endothelial cells, involved in cell migration during the formation of new vessels, and enzyme proteins responsible for the proteolysis of the extracellular matrix surrounding the vessels.41 The process of angiogenesis is also regulated by a number of factors, the so-called pro-angiogenic cells, many of which are secreted by the endothelium itself. Among them, the following stand out: VEGF, basic fibroblast growth factor, acidic fibroblast growth factor, placental factor growth factor, TNF-α, transforming growth factor α (TGF-α), platelet-derived growth factor derived growth factor, PDGF, adenosine, and prostaglandins E1 and E2.42 During the course of the angiogenesis process, endothelial cells show migration, proliferative and division abilities, thanks to which they can form “branches” from an existing vessel.41 The movement of these cells to the place of formation of a new vessel depends on proangiogenic factors, as well as on the interaction of endothelial cells with ECM proteins. An important role here is played by integrins, which mediate the interaction of the endothelium with ECM proteins, such as vitronectin, laminin or fibronectin.39
Techniques for Assessing Endothelial Function
Determining the function of the vascular endothelium is difficult due to its diversity and heterogeneity. The basic principle underlying techniques to assess endothelial function is that healthy arteries dilate in response to reactive hypertension or as a response after pharmacological stimuli. In pathological states, endothelium-dependent vasodilation is attenuated or even absent.43 Techniques for the assessment of endothelial function include physical methods and biochemical methods. Physical methods can be divided into invasive and non-invasive techniques. For preclinical studies, the technique used should bear the characteristics of being noninvasive while being reliable, reproducible, inexpensive, and methodologically simple.44
Physical Methods
Invasive Techniques
The gold standard in invasive techniques for evaluating vascular endothelium is the quantitative coronary angiography (QCA) with intracoronary infusion of vasoactive agents. The technique allows both the assessment of the basic function of VE (by infusion of nitrogen oxygen synthase inhibitors) and the assessment of dose-response relationship of VE agonists and antagonists. In a properly functioning endothelium, a coronary infusion of vasoactive substances (acetylcholine, bradykinin, serotonin, or substance P) leads to the stimulation of NO production by VE cells, resulting in the expansion of the coronary arteries. In the case of VE dysfunction, vasoactive substance directly affects the underlying smooth muscles leading to vasoconstriction. The advantage of the technique is the possivantages including the cost-consumption, invasiveness and the inability to use the technique as a screening test for the general population. In addition, the examination is associated with the risk of coronary catheterization.45
Invasive methods for assessing VE function also include venous occlusion plethysmography, which has been used for more than a century to assess blood flow. The method involves inflating the cuff on the arm to a pressure below the systolic pressure – usually about 40 mmHg at intervals of about 10 seconds, followed by 5 seconds of deflation over 2–3 minutes. The pressure used in the test is so optimal that, despite stopping venous return, it does not impair arterial flow toward the forearm, leading to a linear increase in blood flow to the limb. The test uses a strain gauge, which allows for any change in tissue volume.46
The demonstration of the relationship between the functioning of the VE in the coronary arteries and the function in the peripheral arteries47 made it possible to adapt the technique to the much more easily accessible brachial artery. The advantages of the technique are reliability, repeatability and methodological ease. Nevertheless, as an invasive technique, it may risk damaging the median nerve or the brachial artery.45
Non-Invasive Techniques
Non-invasive techniques take advantage of the fact that peripheral arteries respond to physical and chemical stimuli by regulating the vascular tone and blood flow expected endothelium-dependent dilation.
The most common method uses imaging of the arteries (primarily the brachial artery) in response to reactive congestion. An artery in the distal part of the hand (or part of the forearm) is closed by inflating a closure cuff, followed by evacuation enabling vasodilation in response to reactive congestion in the distal and proximal vascular beds, mediated by the release of endothelial factors. One of these is NO, whose activity induces increased shear stress and dilation, resulting in increased flow in the proximal artery. Such a response is known as flow-mediated dilation (FMD).46 Introduced in 1992 by Celermajer et al,48 FMD is a technique in which ultrasound imaging is performed in stages, at baseline (before occlusion) and during reactive hyperemia (5 min after arterial occlusion). Brachial FMD is used to indirectly measure the release of NO by VE following a transient flow stimulus. Ghiadoni et al49 demonstrated the dependence of FMD on NO by intra-arterial infusion of the specific eNOS inhibitor NG-monomethyl-L-arginine. As a result, arterial dilation decreased by about 66% in response to shear stress. FMD measurement can stratify both individuals at low, moderate and high risk of future cardiometabolic events.50 FMD is not without its drawbacks and limitations, however. First of all, the lack of standardization and differences in cuff placement make it difficult to compare results. In some situations, the degree of reactive congestion can vary even after being exposed to the same stimulus. Additional factors limiting FMD assessment are changes in blood vessel structure and impaired vasodilation. FMD is among the methods that are strongly observer-dependent, so this can lead to receiving different values.46,51
Idei et al52 proposed using an oscillometric method to quantify FMD in the enclosed zone – enclosed zone flow-mediated dilation (ezFMD). Compared to FMD, which is based on changes in the diameter of the vessel imaged by USG, ezFMD uses changes in vessel volume determined indirectly by the amplitude of the oscillations. Oscillometric measurement of blood pressure is carried out using an automatic device while assessing the amplitude of oscillations. It has been demonstrated that there is a proportional relationship between the amplitude of the oscillation and the volumetric change with the brachial pulse pressure. A 5-minute occlusion leads to a change in peak oscillation amplitude and reflects the volume change induced by reactive congestion. The advantage of the method is automation and independence from the operator.
VE function can be reflected using reactive hyperemia pulse artery tonometry (RH-PAT). This technique combines FMD with disposable pneumatic finger-tip probes to measure arterial pulse wave amplitude and obtain the reactive hyperaemia index. The test requires the patient to be in a supine position with hands at heart level, with pneumatic probes placed on the index fingers to record pulse wave amplitudes. A baseline measurement is carried out, followed by closure of the arterial flow to the arm with the help of a pressure gauge administered to a pressure of 40 mmHg above the systolic pressure. After a period of 5 minutes, the pressure gauge is rapidly deflated and there is a transient increase in blood flow. The RH-PAT ratio is calculated from the ratio between the pulse wave amplitude values after and before the closure. The test is also performed on the opposite finger to correct for changes in systemic vascular tone, allowing the values to be normalized. The advantages of RH-PAT certainly include safety, speed and operator independence. On the other hand, the method is sensitive to autonomic tone, shows low reproducibility and lacks correlation with FMD. In addition, structural vascular aspects may affect the outcome.53,54
Vascular endothelial dysfunction is mediated by changes in the glycocalyx structure. Assessment of ultrastructural modifications to the glycalis can be performed using imaging techniques. The most reliable tool for studying both the structure of the glycocalyx and its cellular environment is electron microscopy (EM). The technique combines high spatial resolution and analytical tools. Initially, EM imaging was based on the reactivity of sugar with the dye Alciana blue, ruthenium red or the interaction of the negatively charged surface with cationic colloids (eg dysprosium). Improvements in scanning electron microscopy (SEM) and transmission electron microscopy (TEM) made it possible to achieve multi-scale and chemical information, offering a new way to study the structure of the glycocalyx.55
Biochemical Methods
Cell Adhesion Molecules
Endothelial cells play key roles in regulating permeability and transport of molecules between the blood and the interstitial space. In addition, they are responsible for controlling signaling pathways associated with innate immunity. Activation of endothelial cells can occur both as a result of pro-inflammatory stimuli, such as C-reactive protein (CRP), interleukin 1B (Il-1b), TNF-α, and through hemodynamic forces associated with blood flow. This results in the expression of adhesion molecules (ICAM-1, VCAM-1 and E-selectin). Therefore, adhesion molecules are considered early systemic inflammation and endothelial cell activation markers.56
ICAM-1 mediates many cell–cell interactions, including adhesion and transmigra-tion of leukocytes to the vascular endothelial wall. Ridker et al demonstrated that ICAM-1 expression activates VE cells and leads to inflammation, which in turn is an important step in the initiation and progression of atherosclerosis.57
In the case of VCAM-1, its expression is restricted to VE and occasionally to spindle cells. Moreover, it is not expressed on healthy vascular endothelial cells. It is suspected that the expression of these adhesion molecules provokes VE activation due to increased monocyte recruitment and improved monocyte-endothelial interactions during the initial stages of myoepithelial lesion formation.58
In contrast, E-selectin, which belongs to the C-type lectin family, is probably the most specific marker of endothelial activation. Its expression is limited to VE, induction occurs via inflammatory cytokines. In addition, it is involved in the recruitment of leukocytes to inflammatory sites and in the slow rolling of leukocytes in inflamed venules.59
C Reactive Protein (CRP)
C-reactive protein, which is considered a classic biomarker of inflammation, exhibits proatherogenic properties by reducing NO bioavailability, increasing the expression of adhesion molecules or promoting vasoconstriction and VE dysfunction. Endothelial activation is promoted through the expression of ICAM-1, VCAM-1, E-selectins and CCL2 and activation of macrophages expressing cytokines and tissue factors.60 CRP can also increase the production of the potent endothelin-dependent vasoconstrictor endothelin 1- and the key pro-inflammatory cytokine interleukin 6.61
CC Chemokine Ligand 2 (CCL2)
CCL2 belongs to the CC chemokine family and is highly involved in the initiation and progression of atherosclerosis. Moreover, studies have shown that CCL2 might mediate the pro-atherogenic effects of dyslipidemia.62,63 The increase in CCL2 expression leads to the attraction of monocytes from the vessel lumen to the subendothelial space and, in combination with adhesion molecules, facilitates the binding of monocytes to endothelial cells and their subsequent transmigration to the inner membrane layer. There, monocytes differentiate into macrophages, which begin to express receptors such as CD36, lecitin-like oxidized LDL receptor 1 (LOX-1) or macrophage scavenger receptor (SR-A) that internalize modified LDL beginning the atherosclerotic process. Increased CCL2 expression is observed in VE damage, so measuring systemic CCL2 levels is considered a biomarker of VE dysfunction.64
Asymmetric Dimethylarginine (ADMA)
ADMA, which is an endogenous inhibitor of NO synthase, can be regarded as a biomarker of NO impairment and atherosclerosis. ADMA levels have been shown to independently correlate with VE function measurements,65,66 and its elevated plasma levels are associated with, among others, hypertension, hyperlipidemia, diabetes, and myocardial infarction.67–69
Endothelial Cell-Specific Molecule 1
ESM-1 is an endothelial cell-specific proteoglycan which is expressed by pulmonary endothelial cells. The synthesis and secretion of ESM-1 is increased by pro-inflammatory mediators, including interleukin 1β (Il-1β), TNF-α; bacterial components, including lipopolysaccharide (LPS); and angiogenic factors such as vascular endothelial growth factor (VEGF) or fibroblast growth factor-2 (FGF-2).70 Elevated levels of ESM-1 are associated with cardiometabolic disorders including coronary artery disease,71 hypertension,72 type 2 diabetes73 or atherosclerosis.74 In atherosclerosis, ESM-1 interferes with VE function by promoting cell adhesion, oxidative stress, and inflammation.75
Myeloperoxidase (MPO)
MPO is an enzyme of the heme peroxidase superfamily that is produced by activated monocytes, neutrophils and tissue macrophages. Activated MPOs catalyze the formation of reactive oxygen species (ROS) which results in oxidative damage to proteins and lipids in the body.56 In the vascular system, MPO attaches to glycosaminoglycans, which interferes with NO release, resulting in VE dysfunction.76
Matrix Metalloproteinases (MMPs) and Tissue Inhibitor of Metalloproteinases (TIMPs)
MMPs are a family of zinc-dependent endoproteases that have a variety of functions in tissue remodeling.77 Through ECM degradation and remodeling, they are involved in both physiological and pathological complications of obesity and MetS.78 MMPs may be involved in cell apoptosis, tissue repair, angiogenesis, immune response, and induce cell proliferation, migration and differentiation.77 The regulation of MMP activity is responsible for endogenous inhibitors tissue inhibitor of metalloproteinases (TIMPs).79
A study by Boumiza et al79 that correlated the levels of MMPs and TIMPs and with endothelial function showed that MMP-9 levels and the MMP-9/TIMP-1 ratio can predict VE dysfunction. The authors suggested that MMPs and TIMPs may be used as clinical biomarkers in obesity-associated CDV such as MetS and hypertension.
Angiopoietin-Related Protein 2 (ANGPTL2)
ANGPTL2, which belongs to the glycoprotein family, is a pro-inflammatory mediator that promotes vascular inflammation and atherosclerosis. Their expression is observed in many cell types – muscle, fat, some endocrine organs and VE.80 Increased expression in ECs and macrophages infiltrating atherosclerotic plaques was also demonstrated. The promotion of vascular inflammation is thought to be mediated through the NF-κB signaling pathway in ECs and increased chemotaxis of monocytes/macrophages.81
Homocysteine (Hcy)
The nonproteinogenic methionine-derived amino acid, homocysteine (Hcy), is considered an independent risk factor for many diseases, including neuropsychiatric and neurodegenerative diseases, as well as cerebral ischemia.82 The association of elevated homocysteine levels with MetS and diabetes has been confirmed.83
Elevated homocysteine levels may promote angiotensin conversion enzyme (ACE) activity and act synergistically with angiotensin II to cause damage to the vasculature and lead to a deterioration in the function of the endothelium.84 Studies in rats have confirmed that an increase in local/vascular Hcy production leads to VE dysfunction.83
Vascular Endothelial Dysfunction in Metabolic Disorders
Diabetes and Insulin Resistance
Endothelial damage has long been suspected as one of the components of diabetes pathophysiology. Relatively recently it has been confirmed that damage to eGCX is observed in both type 1 and type 2 diabetes. Nieuwdorp et al85 using lateral jet dark field imaging imaged the gap between erythrocytes and endothelium in the sublingual microcirculation before and after leukocyte passage. Patients with type 1 diabetes (T1DM) have been shown to have eGCX damage that is exacerbated in the presence of microalbuminuria. In addition, a correlation has been shown between a decrease in systemic eGCX volume and increased levels of circulating hyaluronan (HA) and hyaluronidase (an enzyme that degrades, cleaves HA in plasma). In type 2 diabetes (T2DM), a reduction in eGCX volume was observed in the sublingual and retinal pole.86 Studies in rat and mouse models of retinopathy with T1DM have also shown a significant reduction in eGCX in retinal vessels.87,88 In patients with type 1 diabetes, a significant increase in circulating MMP2 and MMP9 was also observed. These compounds have properties that cleave the components of eGCX.89
The enzyme capable of cleaving GAGs in diabetes is heparanase (HPSE). It has been shown that during diabetes – both in humans and in animal models – there are elevated levels of the active form of HPSE, which degrades HS.90–92
An important mediator of the diabetic environment in both T1DM and T2DM, affecting eGCX status, is hyperglycemia. Nieuwdorp et al in their study showed that acute induction of hyperglycemia in healthy subjects leads to a decrease in eGCX volume within 6 hours.93 Studies on the immortalized GEnC line showed an inhibitory effect of hyperglycemia on the synthesis of sulfated and non-sulfated GAGs, with no apparent changes in proteoglycan expression.94
VE dysfunction not always present at time of T2DM diagnosis. Zaharia et al95 demonstrated that people with T2DM experience an approximate 14% decline in VE function within 5 years of disease diagnosis.
A role in endothelial dysfunction in diabetic patients has been attributed to existing insulin resistance and hyperglycemia.96 Insulin resistance (IR) is defined as the reduced ability of insulin to stimulate glucose uptake in skeletal muscle adipose tissue and to inhibit glucose secretion by the liver. IR often precedes the onset of hyperglycemia and diabetes by many years.97 Insulin has a dual function in maintaining homeostasis of blood vessels.98 On the one hand, insulin stimulates endothelial production of NO (which has vasodilatory effects), and on the other, it mediates the release of the potent vasoconstrictive factor ET-1.
VE cells express the insulin receptor, which belongs to a family of membrane-bound receptors that have intrinsic tyrosine kinase activity, whose ligands include growth factors such as vascular endothelial growth factor, insulin-like growth factor-1, epidermal growth factor and platelet-derived growth factor.99
Under physiological conditions, the vasoprotective phosphoribositide-3kinase (PI3-K)/Akt pathway dominates the action of insulin. The biochemical insulin signaling pathway in VE involves phosphorylation of insulin receptor substrate 1 (IRS-1), which then binds and activates phosphatidylinositol (PI) 3-kinase, leading to phosphoinositide-dependent kinase-1 (PDK-1) phosphorylation and activation, which in turn phosphorylates and activates Akt. Akt directly phosphorylates eNOS at Ser1177, resulting in increased eNOS activity and NO production.100–102 Stimulation of ET-1 production by insulin is via MAPK-dependent (but not PI3K-dependent) signaling pathways. In addition, this pathway stimulates increased expression of PAI-1, VCAM-1 and E-selectin on the endothelium.103
The occurrence of IR leads to a shift in the balance towards mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), which mediates inflammation, vasoconstriction and proliferation of vascular smooth muscle cells.100–102 IR leads to an increase in the level of prothrombotic factors, pro-inflammatory markers and ROS, which lead to an increase in intracellular levels of adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM-1).100–102 Insulin resistance also affects VE indirectly by stimulating proliferation and migration of vascular smooth muscle cells (VSMCs), and in adipose tissue, in turn, is associated with excessive secretion of free fatty acids (FFA), which provokes pathogenic gene expression through activation of protein kinase C and increased oxidative stress.104 Excessive release of FFAs leads to the development of a proatherogenic lipid profile and dyslipidemia. Studies, both in vivo and in humans, have shown that acute infusion of FFAs leads to a reduction in VE-dependent vasodilation.105,106 FFA lipotoxicity can lead to VE dysfunction through multiple related mechanisms, including increased ROS production, increased AGE formation and activation of PKC, the hexosamine pathway and pro-inflammatory signaling. Induction of ROS production in the vascular system occurs by uncoupling mitochondria and increasing the expression and protein content of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. ROS activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which stimulates the production of further pro-inflammatory cytokines.107 FFAs activating protein kinase C (PKC) lead to increased phosphorylation of IRS-1 and consequently reduced activation of phosphorylated kinase-1, PI-3K, Akt and eNOS, which culminates in impaired NO production by VE. Ultimately, increased FFAs lead to decreased NO bioavailability, increased vascular oxidative stress, VE apoptosis and increased inflammation.97
In the case of hyperglycemia, numerous studies in both animal models and humans have shown that prolonged hyperglycemia impairs normal VE function at both the micro- and macrovascular levels.108,109 Conducted by Loader et al,110 a systematic review and meta-analysis found that in 9 of 11 studies in healthy young adults, acute hyperglycemia reduced FMD; in the remaining 2, there was no significant change. VE dysfunction caused by acute hyperglycemia can be further compounded by the presence of cardiovascular risk factors, such as elevated blood pressure or LDL.110
Arterial Hypertension
According to the guidelines of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC) from 2018,111 arterial hypertension (AH) is diagnosed when systolic blood pressure (SBP) ≥140 mmHg and/or diastolic blood pressure (DBP) ≥90 mmHg. Optimal blood pressure is defined as <120/80 mmHg, while the normal range is 120–129 mmHg for systolic pressure and/or 80–84 mmHg for diastolic pressure. There are 3 degrees of AH, respectively: 1st degree: 140–159 mmHg systolic and/or 90–99 mmHg diastolic; 2nd degree: 160–179 mmHg systolic and/or 100–109 mmHg; 3rd degree: >180 mmHg systolic and >100 mmHg diastolic. Hypertension is a worldwide problem, in 2015 it was responsible for 10 million deaths,111 while, according to 2022 data, 1 billion adults worldwide suffer from AH.112 The presence of arterial hypertension shows an independent and consistent association with the incidence of cardiovascular events, including heart failure, ischaemic stroke, haemorrhagic stroke, sudden death, myocardial infarction and peripheral artery disease.111
Symptoms of VE dysfunction may precede the development of hypertension.113 The study showed that a 0.62% reduction in endothelial function, as quantified by FMD, is associated with a 20 mmHg increase in SBP.114 AH itself causes functional and structural changes in the resistance arteries, which consequently leads to an increase in peripheral vascular resistance.113 To fully understand the mechanisms that affect hypertension, it is important to understand the difference between constant laminar flow and oscillatory flow.
Physiologically, the endothelium is subjected to uniform, laminar shear stress. Laminar flow activates mechanosensory and mechanosensory channels, resulting in increased production of vasodilating factors. Laminar shear leads to mechanosensing activation of the glycocalyx, which is transferred through the cytoskeleton to integrins. These, in turn, distribute force through microtubules, actin microfilaments and intermediate filaments through focal adhesion of c-Src kinases, which consequently leads to the maintenance of endothelial integrity.115 The consequence of increasing shear stress is an increase in the concentration of calcium in the VE cytoplasm, subsequently leading to the activation of eNOS and an increase in NO production. Moreover, an increase in the concentration of cytosolic calcium results in the opening of calcium-activated potassium channels, leading to hyperpolarization of ECs and thus to vasorelaxation. In addition, PECAM-1 together with caveolin, tyrosine-specific phosphotransferase Fyn, VEGF receptor 2 and VE cadherin forms a mechanosensory complex conferring appropriate sensitivity to the beneficial effects of shear stress in ECs.116 PECAM-1 exerts a force so strong that it triggers activation of the VEGF receptor 2 in the absence of its ligand, thereby inducing integrin-mediated signaling and ultimately leading to inhibition of inflammatory pathways.117 In vivo studies on a mouse model118 showed that increased shear stress leads to AMP-activated protein kinase (AMPK)-induced phosphorylation and sirtuin-1-mediated deacetylation, with the combined effect of promoting eNOS compartmentalization and activation with atherosclerotic protective effects.
In turn, the oscillatory flow leads to a decrease in eNOS expression and promotes smooth muscle proliferation, leukocyte infiltration and the secretion of pro-inflammatory molecules, including monocyte chemotactic protein (MCP1), PDGF, ET-1, whose activity leads to vasoconstriction, increased blood pressure and the development of atherosclerosis in the larger arteries. There is also activation of genes sensitive to mechanisms in ECs, leading to the induction of ROS growth and the activation of several transcription factors, including NF-κB, Kruppel-like factor (KLF2/4), activator protein 1 (AP-1), early growth response 1 (EGR1), c-Jun, c-fos, c-myc proteins, as well as kinase activation mitogen-activated protein (MAPK) and small ubiquitin-like modifier (SUMO) signaling. It is noteworthy that SUMOylation may reduce the expression of the protective transcription factor p53, which may lead to the development of cardiovascular complications in AH. In addition to the above-mentioned, the oscillatory flow induces the activation of the PI3Kinase-Akt pathway, leading to the assembly of the NADPH oxidase-2 (NOX-2) and to the production of ROS, finally inducing inflammation and vascular remodeling.119,120
Oxidative Stress and Inflammation
Oxidative stress is defined as an imbalance between antioxidants and reactive oxygen species due to excessive production of the latter.121 ROS is reactive intermediates of molecular oxygen, physiologically formed in cells as by-products of cellular metabolism. When ROS are at physiological levels, their function is the transmission of intracellular signals.122 Overproduction of ROS, exceeding the capacity of antioxidant systems (or damage of antioxidant enzymes) are involved in the pathogenesis of many diseases, such a cancer, kidney disease, lung disease, neurodegenerative disorders or metabolic diseases (including obesity and diabetes).123 In addition, toxic oxygen affects all organic molecules, including proteins, nucleic acids, free amino acids, carbohydrates and lipids.4
It has been shown that ROS can lead to a decrease in NO bioavailability and thus cause VE dysfunction. The probable mechanism of induction of VE dysfunction is as follows:
A significant reduction in NO bioavailability occurs due to eNOS uncoupling. This process involves the dissociation of the active dimeric form of eNOS (which produces NO) into eNOS monomers capable of producing O2−•. During uncoupling, there is an electron transfer from NADPH to molecular oxygen (instead of NO) by eNOS, resulting in an increase in O2−• with a decrease in NO generation.124 In addition, the production of O2−• by uncoupled eNOS contributes to a vicious circle of ONOO– production and NO consumption, where the O2−• produced by eNOS can further reduce the levels of available NO by reacting with it and converting it to ONOO-. It is noteworthy that ONOO– by itself can also cause eNOS uncoupling.122
ROS can induce inflammation, thereby leading to increased expression of pro-inflammatory factors and adhesion molecules by ECs. Many of these compounds, such as TNF-α and IL-6, can cause chronic inflammation and change the activity of ECs to a prothrombotic, proinflammatory and proliferative state. In inflammatory conditions, ECs are first activated Type I and then Type II, which has a number of consequences: Increased expression and presentation of adhesion molecules, increased permeability of plasma proteins to the vasculature, increased expression of chemokines and pro-inflammatory cytokines. Most of these processes are mediated by NF-κB.125 ROS inducing oxidation of the IκB kinase (IKK) activates NF-κB and translocation to the nucleus.126 NF-κB’s function in the nucleus includes mediating the activation of its target genes, including the VE dysfunction-inducing VCAM-1, ICAM-1 or inflammatory mediators.127
Hypertriglyceridemia
Hypertriglyceridemia is defined as a fasting serum triglyceride level ≥150 mg/dL and has been associated with an increased risk of cardiovascular disease.128 The relationship between triglyceride levels and VE function has been demonstrated in both healthy subjects,129 those with hypertriglyceridemia130 and those with MetS.131 Despite many studies underlying this association are not fully understood.132
Transport of triglycerides (TGs) takes place along with cholesterol in triglyceride-rich lipoproteins triglycerides (TRLs); thus, elevated serum TG levels are associated with elevated TRLs. These, in turn, participating in various mechanisms plays an important role in VE dysfunction. Residual TRLs are small enough to penetrate the intima of the artery; moreover, they can be directly taken up by macrophages and cause inflammation and foam cell formation. Another mechanism involves lipoprotein lipase (LPL). This enzyme is located on the surface of the VE or within the intima of the artery and hydrolyzes TGs to free fatty acids and monoacylglycerols, which generate local inflammation.133 Experimental studies have shown that lipolysis of TRL products leads to damage to ECs by increasing the deposition of very low-density lipoprotein (VLDL) particles in the arterial wall, increasing the permeability of the VE monolayer, increasing the expression of ROS and inducing apoptosis.134,135 In addition, TRL residuals increase the generation of nicotinamide adenine dinucleotide phosphate oxidasedependent superoxide and induction of tumor necrosis factor-a and interleukin-1b via activated lectin-like oxidized LDL receptor-1 in endothelial cell.132
Increased levels of TGs in the blood are associated with the formation of small dense LDL (sdLDL), which exhibit high atherogenicity.136 An important role is played by cholesterol ester transfer protein (CETP), which facilitates the exchange of TGs and cholesterol esters between TRLs and LDL, resulting in more LDL-rich triglycerides. These in turn are hydrolyzed by hepatic lipase to sdLDL. sdLDL is easily oxidized and can penetrate the subendothelial space of the vascular wall. Oxidative LDL stimulates oxidative stress, endothelial cell inflammation and apoptosis, leading to VE dysfunction.137
HDL can also interact with the endothelium. HDL has an atheroprotective function, promotes NO bioavailability, exhibits anti-inflammatory, anti-thrombotic, antioxidant effects and reduces apoptosis of ECs.138 HDL concentration in blood is dependent on TRL. Increasing TRLs leads to an increase in the CETP-mediated exchange of cholesterol esters from HDL to TRLs and simultaneous transfer of triglycerides into HDL. HDL, rich in triglycerides, can be further hydrolysed by hepatic lipase into smaller, denser HDL. TRLs, by blocking sterol efflux from monocytes and macrophages, suppress the anti-inflammatory and atheroprotective effects of HDL, which can lead to VE dysfunction.132
Hyperglycemia causes an imbalance between oxidant production and antioxidant cellular defense systems. There is an increase in the generation of ROS – especially superoxide anion (O2•–) – which, in turn, reduces the bioavailability of NO, resulting in a reduction in the biological activity of this compound. O2•– reacting with NO forms a highly unstable and reactive nitrogen species – peroxynitrite (ONOO−) – which, by damaging proteins, lipids and nucleic acids, leads to the predominant state of vasospasm. Loader et al in their study showed that microvascular and macrovascular VE dysfunction is partly caused by oxidative stress-induced impairment of NO bioavailability.139 Moreover, hyperglycemia through specific cellular mechanisms leads to vascular bed damage. These mechanisms include increased production of intracellular advanced glycation end products (AGEs), increased expression of AGE receptors (RAGEs) and their ligands, increased polyol and hexosamine flux, activation of PKC and overactivation of the hexosamine pathway. AGEs inducing a decrease in NO synthesis and eNOS expression, and an increase in ET-1 expression, lead to VE dysfunction.97,140
The principal mechanisms underlying vascular endothelial dysfunction in metabolic disorders are summarized in Figure 1.
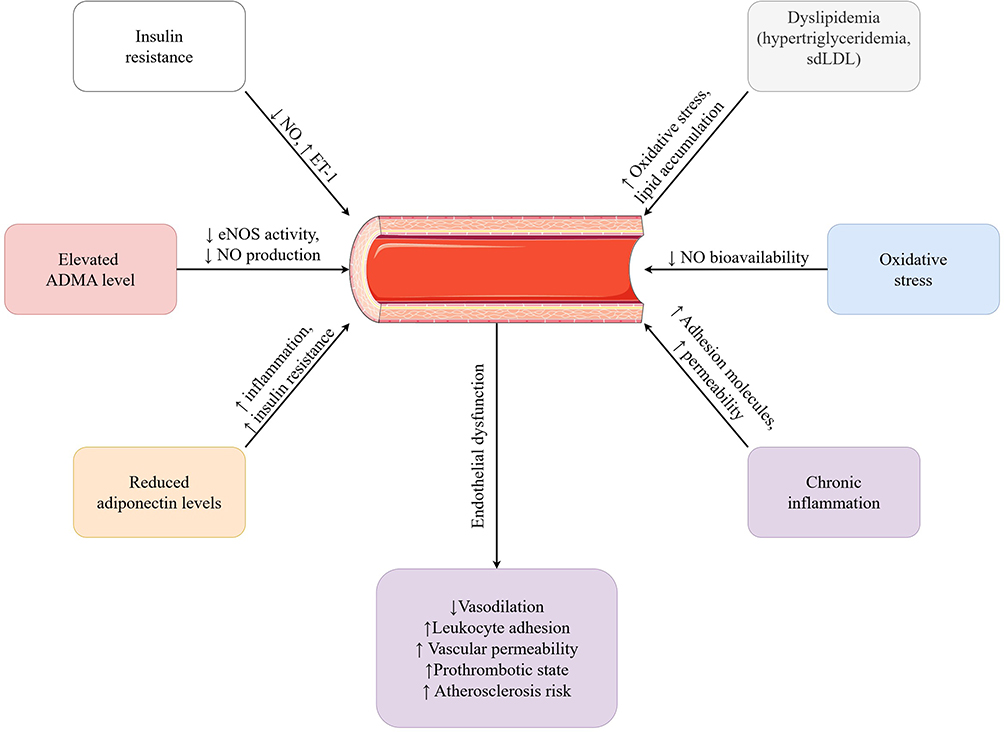 |
Figure 1 Pathophysiological mechanisms of endothelial dysfunction in metabolic disorders. Abbreviations: ADMA, asymmetric dimethylarginine; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; sdLDL, small, dense low-density lipoprotein; ET-1, endothelin-1; ↑, increase; ↓, decrease. |
Metabolic Obesity Normal Body Weight (MONW)
Among metabolic abnormalities, increasing scientific interest has focused on metabolic obesity in normal-weight individuals (MONW). Individuals with MONW show symptoms typical of metabolic syndrome (MetS) – reduced insulin sensitivity, hypertension, type 2 diabetes and hypertriglyceridemia – despite a normal body mass index (BMI). This relatively recently discovered phenomenon is still controversial due to the lack of consensus on diagnostic criteria. Therefore, there is a need to develop predictors that can quickly and accurately identify MONW.141
Keirns et al142 examined both the aforementioned types of obesity and subjects without metabolic disorders and obesity (the control group) for VE function using the FMD method. The study showed that subjects with MetS had less vasodilation compared to the control group. In contrast, those with MONW showed no statistically significant difference from either group in these respects. Similar conclusions were reached by Sprung et al.143 The study showed that people with MetS exhibit endothelial dysfunction (manifested by reduced vasodilation), regardless of the degree of obesity. Of particular note is the demonstrated association of increased cardiovascular disease (CVD) risk in individuals with metabolic disorders, regardless of their obesity status. This may suggest that preserved metabolic health may, to some extent, have a protective function for the cardiovascular system. Also, studies by Rakhmat et al showed endothelial damage in both normal weight individuals and obese individuals suffering from metabolic syndrome. MetS components were shown to correlate positively with VCAM-1 as markers of endothelial dysfunction.144
A study145 involving 3 cohorts of geographically diverse backgrounds confirmed the association of impaired PAT measurements with both metabolic risk factors and obesity. Comparisons of individuals within the same BMI category showed that vascular indices were more impaired in the presence of metabolic abnormalities. This suggests that not only increased body weight but also the presence of metabolic abnormalities is associated with progressive deterioration of microvascular function. Nurver Turfaner et al146 examined the relationship between FMD in patients with MetS and controls without MetS. There were no statistically significant differences in FMD between the study groups. In a study by Iwańczyk et al,147 the degree of VE dysfunction in MetS patients was assessed by determining the serum concentration of endocan. They found higher concentrations of the endocan compound in the patient group compared to the control group. This confirms that VE dysfunction is one of the initial stages of MetS development.
Conclusion
The vascular endothelium is a highly specialized structure of the human organism. It performs a number of functions in the body, forming the border between metabolic health and cardiovascular disease. It performs many critical functions, from regulating vascular tone and maintaining hemostatic balance to modulating inflammatory responses. Its amazing structure acts not only as a protective barrier but also as a mechanical sensor that translates hemodynamic forces into cellular responses. Dysfunction of such a specialized organ can lead to a number of disorders, both at the microvascular and macrovascular levels. The prothrombotic and proatherogenic effects of endothelial dysfunction can contribute to the development of cardio-metabolic abnormalities at an early age.
Modern techniques for detecting endothelial dysfunction are at a very high level and include both invasive (including the “gold standard” - coronary angiography) and non-invasive methods (including flow-dependent dilation (FMD) and biochemical biomarkers). The development of automated techniques such as closed zone FMD (ezFMD) and the identification of new biomarkers, including ADMA, VCAM-1 and endocan, provide physicians with increasingly advanced tools for early detection of vascular dysfunction. These methods enable risk stratification many years before clinical cardiovascular events occur.
The pathophysiological mechanisms underlying endothelial dysfunction in metabolic disorders converge on several key pathways. These include insulin resistance with impaired PI3K/Akt signalling, increased oxidative stress with eNOS uncoupling, chronic low-grade inflammation with NF-κB activation and dyslipidaemia with the formation of atherogenic small, dense LDL particles. These interconnected mechanisms often perpetuate each other, creating a vicious cycle of progressive endothelial impairment. The recognition that endothelial dysfunction precedes clinical CVD by years underscores its potential as both a therapeutic target and a biomarker for early intervention.
As demonstrated in the identified literature, endothelial dysfunction can also occur in individuals with a seemingly normal metabolic profile (metabolic obesity in normal-weight individuals). It seems reasonable to use predictors of vascular endothelial dysfunction, which have been shown to be effective in obesity, diabetes and insulin resistance – all components of MONW.
Understanding the molecular mechanisms of endothelial dysfunction opens avenues for targeted therapeutic interventions. Strategies aimed at improving NO bioavailability, reducing oxidative stress, modulating inflammatory pathways, and optimizing metabolic parameters may help preserve or restore endothelial function. The development of personalized approaches based on individual risk profiles and endothelial function assessment represents a promising direction for precision medicine in cardiovascular disease prevention.
Future research should focus on: (1) standardizing the methodology for assessing endothelial function to enable comparisons across studies, (2) developing biomarkers for widespread clinical use, (3) longitudinal studies examining the temporal relationship between metabolic changes and endothelial dysfunction, (4) exploring novel therapeutic targets within endothelial signaling pathways, and (5) establishing evidence-based diagnostic criteria for emerging metabolic phenotypes such as MONW.
In summary, the vascular endothelium represents a critical therapeutic frontier in metabolic medicine. As our understanding of endothelial biology deepens and assessment techniques become more widely available, the potential for early intervention and personalized cardiovascular risk management continues to expand. Integrating endothelial function assessment into routine clinical practice has the potential to ultimately transform our approach to preventing cardiovascular disease in patients with metabolic disorders.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hennigs JK, Matuszcak C, Trepel M, Körbelin J. Vascular endothelial cells: heterogeneity and targeting approaches. Cells. 2021;10(10). doi:10.3390/CELLS10102712
2. Mitra R, Leland O’neil G, Harding IC, Cheng MJ, Mensah SA, Ebong EE. Glycocalyx in atherosclerosis-relevant endothelium function and as a therapeutic target. Curr Atherosclerosis Rep. 2017;19(12). doi:10.1007/s11883-017-0691-9
3. Di Dedda U. Endothelial function and microcirculation. In: Ranucci M editor. The Coagulation Labyrinth of Covid-19. Springer International Publishing; 2022:103–142. doi:10.1007/978-3-030-82938-4_8
4. Medina-Leyte DJ, Zepeda-García O, Domínguez-Pérez M, González-Garrido A, Villarreal-Molina T, Jacobo-Albavera L. Endothelial dysfunction, inflammation and coronary artery disease: potential biomarkers and promising therapeutical approaches. Int J Mol Sci. 2021;22(8):3850. doi:10.3390/IJMS22083850
5. Krüger-Genge A, Blocki A, Franke R-P, Jung F. Vascular endothelial cell biology: an update. Int J Mol Sci. 2019;20(18):4411. doi:10.3390/ijms20184411
6. McKay TB, Schlötzer-Schrehardt U, Pal-Ghosh S, Stepp MA. Integrin: basement membrane adhesion by corneal epithelial and endothelial cells. Exp Eye Res. 2020;198:108138. doi:10.1016/j.exer.2020.108138
7. Zhang J, Huang S, Zhu Z, Gatt A, Liu J. E-selectin in vascular pathophysiology. Front Immunol. 2024;15:1401399. doi:10.3389/fimmu.2024.1401399
8. Milošević N, Rütter M, David A. Endothelial cell adhesion molecules- (un)attainable targets for nanomedicines. Front Med Technol. 2022;4:846065. doi:10.3389/fmedt.2022.846065
9. Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci. 2013;126(Pt 12):2545–2549. doi:10.1242/jcs.124529
10. Schnittler HJ. Structural and functional aspects of intercellular junctions in vascular endothelium. Basic Res Cardiol. 1998;93(Suppl 3):30–39. doi:10.1007/s003950050205
11. Pillinger NL, Kam PCA. Endothelial glycocalyx: basic science and clinical implications. Anaesth Intensive Care. 2017;45(3):295–307. doi:10.1177/0310057X1704500305
12. Ushiyama A, Kataoka H, Iijima T. Glycocalyx and its involvement in clinical pathophysiologies. J Intensive Care. 2016;4(1):1–11. doi:10.1186/S40560-016-0182-Z/TABLES/3
13. Reitsma S, Slaaf DW, Vink H, van Zandvoort MAMJ, Oude Egbrink MGA. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345–359. doi:10.1007/s00424-007-0212-8
14. Moore KH, Murphy HA, George EM. The glycocalyx: a central regulator of vascular function. Am J Physiol Regul Integr Comp Physiol. 2021;320(4):R508–R518. doi:10.1152/ajpregu.00340.2020
15. Hiebert LM. Heparan sulfate proteoglycans in diabetes. Semin Thromb Hemost. 2021;47(03):261–273. doi:10.1055/s-0041-1724118
16. Suzuki K, Miura T, Okada H. The endothelial glycocalyx—all the same? No, it is not. Acute Med Surg. 2023;10(1):e896. doi:10.1002/ams2.896
17. Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflügers Archiv. 2000;440(5):653–666. doi:10.1007/S004240000307
18. Dragovich MA, Chester D, Fu BM, et al. Mechanotransduction of the endothelial glycocalyx mediates nitric oxide production through activation of TRP channels. Am J Physiol Cell Physiol. 2016;311(6):C846–C853. doi:10.1152/ajpcell.00288.2015
19. Dogné S, Flamion B. Endothelial glycocalyx impairment in disease. Am J Pathol. 2020;190(4):768–780. doi:10.1016/j.ajpath.2019.11.016
20. McCormack JJ, Lopes da Silva M, Ferraro F, Patella F, Cutler DF. Weibel-palade bodies at a glance. J Cell Sci. 2017;130(21):3611–3617. doi:10.1242/jcs.208033
21. El-Mansi S, Nightingale TD. Emerging mechanisms to modulate VWF release from endothelial cells. Int J Biochem Cell Biol. 2021;131:105900. doi:10.1016/j.biocel.2020.105900
22. Sturtzel C. Endothelial Cells. Adv Exp Med Biol. 2017;1003:71–91. doi:10.1007/978-3-319-57613-8_4
23. Pries AR, Kuebler WM. Normal endothelium. Handb Exp Pharmacol. 2006;(176 Pt 1):1–40. doi:10.1007/3-540-32967-6_1
24. Aird WC. Phenotypic heterogeneity of the endothelium: i. Structure, function, and mechanisms. Circ Res. 2007;100(2):158–173. doi:10.1161/01.RES.0000255691.76142.4a
25. Przysinda A, Feng W, Li G. Diversity of organism-wide and organ-specific endothelial cells. Curr Cardiol Rep. 2020;22(4):19. doi:10.1007/s11886-020-1275-9
26. Blanchard L, Girard JP. High endothelial venules (HEVs) in immunity, inflammation and cancer. Angiogenesis. 2021;24(4):719–753. doi:10.1007/s10456-021-09792-8
27. Pablo-Moreno JAD, Serrano LJ, Revuelta L, Sánchez MJ, Liras A. The vascular endothelium and coagulation: homeostasis, disease, and treatment, with a focus on the von willebrand factor and factors VIII and V. Int J Mol Sci. 2022;23(15):8283. doi:10.3390/ijms23158283
28. Wulftange WJ, Kucukal E, Man Y, et al. Antithrombin-III mitigates thrombin-mediated endothelial cell contraction and sickle red blood cell adhesion in microscale flow. Br J Haematol. 2022;198(5):893–902. doi:10.1111/bjh.18328
29. Madjene C, Boutigny A, Bouton MC, Arocas V, Richard B. Protease nexin-1 in the cardiovascular system: wherefore art thou? Front Cardiovasc Med. 2021;8:652852. doi:10.3389/fcvm.2021.652852
30. He L, Vicente CP, Westrick RJ, Eitzman DT, Tollefsen DM. Heparin cofactor II inhibits arterial thrombosis after endothelial injury. J Clin Invest. 2002;109(2):213–219. doi:10.1172/JCI13432
31. Tollefsen DM. Does heparin cofactor ii modulate atherosclerosis and restenosis? Circulation. 2004;109(22):2682–2684. doi:10.1161/01.CIR.0000130436.14464.FC
32. Gandrille S. Endothelial cell protein C receptor and the risk of venous thrombosis. Haematologica. 2008;93(6):812–816. doi:10.3324/haematol.13243
33. White TA, Johnson T, Zarzhevsky N, et al. Endothelial-derived tissue factor pathway inhibitor regulates arterial thrombosis but is not required for development or hemostasis. Blood. 2010;116(10):1787–1794. doi:10.1182/blood-2009-10-250910
34. Wittig J, Drekolia MK, Kyselova A, et al. Endothelial-dependent S-Sulfhydration of tissue factor pathway inhibitor regulates blood coagulation. Redox Biol. 2023;62:102694. doi:10.1016/j.redox.2023.102694
35. Oliver JJ, Webb DJ, Newby DE. Stimulated tissue plasminogen activator release as a marker of endothelial function in humans. Arteriosclerosis Thrombosis Vasc Biol. 2005;25(12):2470–2479. doi:10.1161/01.ATV.0000189309.05924.88
36. Valls MD, Soldado M, Arasa J, et al. Annexin A2-mediated plasminogen activation in endothelial cells contributes to the proangiogenic effect of adenosine A2A receptors. Front Pharmacol. 2021;12:654104. doi:10.3389/fphar.2021.654104
37. Garland CJ, Hiley CR, Dora KA. EDHF: spreading the influence of the endothelium. Br. J. Pharmacol. 2011;164(3):839–852. doi:10.1111/j.1476-5381.2010.01148.x
38. Mechanotransduction and flow across the endothelial glycocalyx | PNAS. Available from: https://www.pnas.org/doi/10.1073/pnas.1332808100.
39. Patan S. Vasculogenesis and angiogenesis. Cancer Treat Res. 2004;117:3–32. doi:10.1007/978-1-4419-8871-3_1
40. Ezaki T, Baluk P, Thurston G, La Barbara A, Woo C, McDonald DM. Time course of endothelial cell proliferation and microvascular remodeling in chronic inflammation. Am J Pathol. 2001;158(6):2043–2055. doi:10.1016/S0002-9440(10)64676-7
41. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100(6):782–794. doi:10.1161/01.RES.0000259593.07661.1e
42. Ribatti D, Crivellato E. Immune cells and angiogenesis. J Cell Mol Med. 2009;13(9a):2822–2833. doi:10.1111/j.1582-4934.2009.00810.x
43. Flammer AJ, Anderson T, Celermajer DS, et al. The assessment of endothelial function: from research into clinical practice. Circulation. 2012;126(6):753–767. doi:10.1161/CIRCULATIONAHA.112.093245
44. Al-Qaisi M. Measurement of endothelial function and its clinical utility for cardiovascular risk. VHRM. 2008;4:647–652. doi:10.2147/VHRM.S2769
45. Tousoulis D, Antoniades C, Stefanadis C. Evaluating endothelial function in humans: a guide to invasive and non-invasive techniques. Heart. 2005;91(4):553–558. doi:10.1136/hrt.2003.032847
46. Storch AS, de Mattos JD, Alves R, Dos Galdino IS, Rocha HNM. Methods of endothelial function assessment: description and applications. Int J Cardiovasc Sci. 2017;30:262–273. doi:10.5935/2359-4802.20170034
47. Anderson TJ, Uehata A, Gerhard MD, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26(5):1235–1241. doi:10.1016/0735-1097(95)00327-4
48. Celermajer DS, Sorensen KE, Gooch VM, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340(8828):1111–1115. doi:10.1016/0140-6736(92)93147-F
49. Ghiadoni L, Versari D, Magagna A, et al. Ramipril dose-dependently increases nitric oxide availability in the radial artery of essential hypertension patients. J Hypertens. 2007;25(2):361–366. doi:10.1097/HJH.0b013e3280115901
50. Yeboah J, Folsom AR, Burke GL, et al. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation. 2009;120(6):502–509. doi:10.1161/CIRCULATIONAHA.109.864801
51. Areas GPT, Mazzuco A, Caruso FR, et al. Flow-mediated dilation and heart failure: a review with implications to physical rehabilitation. Heart Fail Rev. 2019;24(1):69–80. doi:10.1007/s10741-018-9719-7
52. Idei N, Ukawa T, Kajikawa M, et al. A novel noninvasive and simple method for assessment of endothelial function: enclosed zone flow-mediated vasodilation (ezFMD) using an oscillation amplitude measurement. Atherosclerosis. 2013;229(2):324–330. doi:10.1016/j.atherosclerosis.2013.05.016
53. Bonetti PO, Barsness GW, Keelan PC, et al. Enhanced external counterpulsation improves endothelial function in patients with symptomatic coronary artery disease. J Am Coll Cardiol. 2003;41(10):1761–1768. doi:10.1016/S0735-1097(03)00329-2
54. Weisrock F, Fritschka M, Beckmann S, et al. Reliability of peripheral arterial tonometry in patients with heart failure, diabetic nephropathy and arterial hypertension. Vasc Med. 2017;22(4):292–300. doi:10.1177/1358863X17706752
55. Chevalier L, Selim J, Castro C, et al. Combined electron microscopy approaches for arterial glycocalyx visualization. Front Cardiovasc Med. 2022;9:840689. doi:10.3389/fcvm.2022.840689
56. Badimon L, Romero JC, Cubedo J, Borrell-Pagès M. Circulating biomarkers. Thromb Res. 2012;130:S12–S15. doi:10.1016/j.thromres.2012.08.262
57. Ridker PM, Hennekens CH, Roitman-Johnson B, Stampfer MJ, Allen J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet. 1998;351(9096):88–92. doi:10.1016/S0140-6736(97)09032-6
58. Li H, Cybulsky MI, Gimbrone MA, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb. 1993;13(2):197–204. doi:10.1161/01.atv.13.2.197
59. Hidalgo A, Peired AJ, Wild MK, Vestweber D, Frenette PS. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity. 2007;26(4):477–489. doi:10.1016/j.immuni.2007.03.011
60. Yeh ETH, Willerson JT. Coming of Age of C-Reactive Protein. Circulation. 2003;107(3):370–371. doi:10.1161/01.CIR.0000053731.05365.5A
61. Verma S, Li SH, Badiwala MV, et al. Endothelin antagonism and interleukin-6 inhibition attenuate the proatherogenic effects of C-reactive protein. Circulation. 2002;105(16):1890–1896. doi:10.1161/01.CIR.0000015126.83143.B4
62. Association between plasma monocyte chemoattractant protein-1 levels and the extent of atherosclerotic peripheral artery disease. Available from: https://www.jstage.jst.go.jp/article/tjem/224/4/224_4_301/_article/-char/en.
63. Deo R, Khera A, McGuire DK, et al. Association among plasma levels of monocyte chemoattractant protein-1, traditional cardiovascular risk factors, and subclinical atherosclerosis. J Am Coll Cardiol. 2004;44(9):1812–1818. doi:10.1016/j.jacc.2004.07.047
64. New markers of inflammation and endothelial cell activation | circulation. Available from: https://www.ahajournals.org/doi/10.1161/01.CIR.0000089190.95415.9F.
65. Wang F, Xiong R, Feng S, Lu X, Li H, Wang S. Association of circulating levels of ADMA with carotid intima-media thickness in patients with CKD: a systematic review and meta-analysis. Kidney Blood Press Res. 2018;43(1):25–33. doi:10.1159/000486743
66. Yilmaz MI, Sonmez A, Saglam M, et al. ADMA levels correlate with proteinuria, secondary amyloidosis, and endothelial dysfunction. J Am Soc Nephrol. 2008;19(2):388–395. doi:10.1681/ASN.2007040461
67. Böger RH, Sullivan LM, Schwedhelm E, et al. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation. 2009;119(12):1592–1600. doi:10.1161/CIRCULATIONAHA.108.838268
68. Caglar K, Yilmaz MI, Sonmez A, et al. ADMA, proteinuria, and insulin resistance in non-diabetic stage I chronic kidney disease. Kidney Int. 2006;70(4):781–787. doi:10.1038/sj.ki.5001632
69. Kobayashi S, Oka M, Maesato K, et al. Coronary artery calcification, ADMA, and insulin resistance in CKD patients. Clin J Am Soc Nephrol. 2008;3(5):1289–1295. doi:10.2215/CJN.00010108
70. Chenevier-Gobeaux C, Ducastel M, Meritet JF, et al. Plasma endocan as a biomarker of thrombotic events in COVID-19 patients. J Clin Med. 2022;11(19):5560. doi:10.3390/jcm11195560
71. Ziaee M, Mashayekhi S, Ghaffari S, Mahmoudi J, Sarbakhsh P, Garjani A. Predictive value of endocan based on TIMI risk score on major adverse cardiovascular events after acute coronary syndrome. Angiology. 2019;70(10):952–959. doi:10.1177/0003319718815241
72. Klisic A, Kavaric N, Vujcic S, Spasojevic-Kalimanovska V, Ninic A, Kotur-Stevuljevic J. Endocan and advanced oxidation protein products in adult population with hypertension. Eur Rev Med Pharmacol Sci. 2020;24(12):7131–7137. doi:10.26355/eurrev_202006_21707
73. Klisic A, Kavaric N, Stanisic V, et al. Endocan and a novel score for dyslipidemia, oxidative stress and inflammation (DOI score) are independently correlated with glycated hemoglobin (HbA1c) in patients with prediabetes and type 2 diabetes. Arch Med Sci. 2020;16(1):42–50. doi:10.5114/aoms.2019.87541
74. He XW, Ke SF, Bao YY, et al. Serum levels of endocan and endoglin are associated with large-artery atherosclerotic stroke. Clin Chim Acta. 2018;478:157–161. doi:10.1016/j.cca.2017.12.040
75. Bar A, Targosz‐Korecka M, Suraj J, et al. Degradation of glycocalyx and multiple manifestations of endothelial dysfunction coincide in the early phase of endothelial dysfunction before atherosclerotic plaque development in apolipoprotein E/low‐density lipoprotein receptor‐deficient mice. J Am Heart Assoc. 2019;8(6):e011171. doi:10.1161/JAHA.118.011171
76. Pennathur S, Heinecke JW. Oxidative stress and endothelial dysfunction in vascular disease. Curr Diab Rep. 2007;7(4):257–264. doi:10.1007/s11892-007-0041-3
77. Cui N, Hu M, Khalil RA. Biochemical and biological attributes of matrix metalloproteinases. Prog Mol Biol Transl Sci. 2017;147:1–73. doi:10.1016/bs.pmbts.2017.02.005
78. Tinahones FJ, Coín-Aragüez L, Mayas MD, et al. Obesity-associated insulin resistance is correlated to adipose tissue vascular endothelial growth factors and metalloproteinase levels. BMC Physiology. 2012;12(1):4. doi:10.1186/1472-6793-12-4
79. Boumiza S, Chahed K, Tabka Z, Jacob MP, Norel X, Ozen G. MMPs and TIMPs levels are correlated with anthropometric parameters, blood pressure, and endothelial function in obesity. Sci Rep. 2021;11(1):20052. doi:10.1038/s41598-021-99577-2
80. Yu Z, Yang W, He X, et al. Endothelial cell-derived angiopoietin-like protein 2 supports hematopoietic stem cell activities in bone marrow niches. Blood. 2022;139(10):1529–1540. doi:10.1182/blood.2021011644
81. Horio E, Kadomatsu T, Miyata K, et al. Role of endothelial cell–derived Angptl2 in vascular inflammation leading to endothelial dysfunction and atherosclerosis progression. Arteriosclerosis Thrombosis Vasc Biol. 2014;34(4):790–800. doi:10.1161/ATVBAHA.113.303116
82. Wyse ATS, Bobermin LD, Dos Santos TM, Quincozes-Santos A. Homocysteine and Gliotoxicity. Neurotox Res. 2021;39(3):966–974. doi:10.1007/s12640-021-00359-5
83. Role of homocysteinylation of ACE in endothelial dysfunction of arteries - PMC. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4340149/.
84. Qin X, Li Y, Sun N, et al. Elevated homocysteine concentrations decrease the antihypertensive effect of angiotensin-converting enzyme inhibitors in hypertensive patients. Arteriosclerosis Thrombosis Vasc Biol. 2017;37(1):166–172. doi:10.1161/ATVBAHA.116.308515
85. Nieuwdorp M, Mooij HL, Kroon J, et al. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes. 2006;55(4):1127–1132. doi:10.2337/diabetes.55.04.06.db05-1619
86. Broekhuizen LN, Lemkes BA, Mooij HL, et al. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia. 2010;53(12):2646–2655. doi:10.1007/s00125-010-1910-x
87. Kim YH, Kim YS, Roh GS, Choi WS, Cho GJ. Resveratrol blocks diabetes-induced early vascular lesions and vascular endothelial growth factor induction in mouse retinas. Acta Ophthalmol. 2012;90(1):e31–37. doi:10.1111/j.1755-3768.2011.02243.x
88. Kumase F, Morizane Y, Mohri S, Takasu I, Ohtsuka A, Ohtsuki H. Glycocalyx degradation in retinal and choroidal capillary endothelium in rats with diabetes and hypertension. Acta Med Okayama. 2010;64(5):277–283. doi:10.18926/AMO/40502
89. Lipowsky HH. Protease activity and the role of the endothelial glycocalyx in inflammation. Drug Discov Today. 2011;8(1):57–62. doi:10.1016/j.ddmod.2011.05.004
90. van den Hoven MJ, Rops AL, Bakker MA, et al. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. 2006;70(12):2100–2108. doi:10.1038/sj.ki.5001985
91. Shafat I, Ilan N, Zoabi S, Vlodavsky I, Nakhoul F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS One. 2011;6(2):e17312. doi:10.1371/journal.pone.0017312
92. Wang F, Wan A, Rodrigues B. The function of heparanase in diabetes and its complications. Can J Diabetes. 2013;37(5):332–338. doi:10.1016/j.jcjd.2013.05.008
93. Nieuwdorp M, van Haeften TW, Gouverneur MCLG, et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 2006;55(2):480–486. doi:10.2337/diabetes.55.02.06.db05-1103
94. Singh A, Fridén V, Dasgupta I, et al. High glucose causes dysfunction of the human glomerular endothelial glycocalyx. Am J Physiol Renal Physiol. 2011;300(1):F40–48. doi:10.1152/ajprenal.00103.2010
95. Zaharia OP, Schön M, Löffler L, et al. Metabolic factors predict changes in endothelial function during the early course of type 1 and type 2 diabetes. J Clin Endocrinol Metab. 2022;107(10):e4167–e4176. doi:10.1210/clinem/dgac480
96. Grismaldo A, Sobrevia L, Morales L. Role of platelet-derived growth factor c on endothelial dysfunction in cardiovascular diseases. BBA. 2022;1866(10):130188. doi:10.1016/j.bbagen.2022.130188
97. Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: molecular insights and therapeutic strategies. Cardiovasc Diabetol. 2018;17(1):121. doi:10.1186/s12933-018-0763-3
98. Honka MJ, Latva-Rasku A, Bucci M, et al. Insulin-stimulated glucose uptake in skeletal muscle, adipose tissue and liver: a positron emission tomography study. Eur J Endocrinol. 2018;178(5):523–531. doi:10.1530/EJE-17-0882
99. Rajendran P, Rengarajan T, Thangavel J, et al. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9(10):1057–1069. doi:10.7150/ijbs.7502
100. Janus A, Szahidewicz-Krupska E, Mazur G, Doroszko A. Insulin resistance and endothelial dysfunction constitute a common therapeutic target in cardiometabolic disorders. Mediators Inflamm. 2016;2016:3634948. doi:10.1155/2016/3634948
101. Kim J, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction. Circulation. 2006;113(15):1888–1904. doi:10.1161/CIRCULATIONAHA.105.563213
102. Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuñiga FA. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol. 2018;17(1):122. doi:10.1186/s12933-018-0762-4
103. Muniyappa R, Sowers JR. Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord. 2013;14(1):5–12. doi:10.1007/s11154-012-9229-1
104. Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi:10.2337/diabetes.49.11.1939
105. Steinberg HO, Tarshoby M, Monestel R, et al. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J Clin Invest. 1997;100(5):1230–1239. doi:10.1172/JCI119636
106. Diabetes and vascular disease | circulation. Available from: https://www.ahajournals.org/doi/full/10.1161/01.CIR.0000091257.27563.32.
107. Sena C, Pereira AM, Seiça R. Endothelial dysfunction — a major mediator of diabetic vascular disease. BBA. 2013;1832(12):2216–2231. doi:10.1016/J.BBADIS.2013.08.006
108. Grassi D, Desideri G, Necozione S, et al. Protective effects of flavanol-rich dark chocolate on endothelial function and wave reflection during acute hyperglycemia. Hypertension. 2012;60(3):827–832. doi:10.1161/HYPERTENSIONAHA.112.193995
109. Candido R, Bernardi S, Allen TJ. Linking diabetes and atherosclerosis. Expert Rev Endocrinol Metab. 2009;4(6):603–624. doi:10.1586/eem.09.46
110. Loader J, Montero D, Lorenzen C, et al. Acute hyperglycemia impairs vascular function in healthy and cardiometabolic diseased subjects. Arteriosclerosis Thrombosis Vasc Biol. 2015;35(9):2060–2072. doi:10.1161/ATVBAHA.115.305530
111. Williams B, Mancia G, Spiering W, et al. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur Heart J. 2018;39(33):3021–3104. doi:10.1093/eurheartj/ehy339
112. Carey RM, Moran AE, Whelton PK. Treatment of hypertension: a review. JAMA. 2022;328(18):1849–1861. doi:10.1001/jama.2022.19590
113. S C, D M, L F, V M. Angiotensin type 2 receptor in hypertensive cardiovascular disease. Curr Opin Nephrol Hypertens. 2011;20(2). doi:10.1097/MNH.0b013e3283437fcd
114. Clinical correlates and heritability of flow-mediated dilation in the community: the framingham heart study - pubmed. Available from: https://pubmed.ncbi.nlm.nih.gov/14769683/.
115. Chatterjee S. Endothelial mechanotransduction, redox signaling and the regulation of vascular inflammatory pathways. Front Physiol. 2018;9:524. doi:10.3389/fphys.2018.00524
116. He M, Martin M, Marin T, Chen Z, Gongol B. Endothelial mechanobiology. APL Bioeng. 2020;4(1):010904. doi:10.1063/1.5129563
117. Coon BG, Baeyens N, Han J, et al. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J Cell Biol. 2015;208(7):975–986. doi:10.1083/jcb.201408103
118. Shentu TP, He M, Sun X, et al. AMP-activated protein kinase and sirtuin 1 coregulation of cortactin contributes to endothelial function. Arterioscler Thromb Vasc Biol. 2016;36(12):2358–2368. doi:10.1161/ATVBAHA.116.307871
119. Abe J, Berk BC. Novel mechanisms of endothelial mechanotransduction. Arterioscler Thromb Vasc Biol. 2014;34(11):2378–2386. doi:10.1161/ATVBAHA.114.303428
120. Gallo G, Volpe M, Savoia C. Endothelial dysfunction in hypertension: current concepts and clinical implications. Front Med. 2022;8:798958. doi:10.3389/fmed.2021.798958
121. Grassi D, Desideri G, Ferri L, Aggio A, Tiberti S, Ferri C. Oxidative stress and endothelial dysfunction: say NO to cigarette smoking! CPD. 2010;16(23):2539–2550. doi:10.2174/138161210792062867
122. Shaito A, Aramouni K, Assaf R, et al. Oxidative stress-induced endothelial dysfunction in cardiovascular diseases. Front Biosci. 2022;27(3):105. doi:10.31083/j.fbl2703105
123. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. doi:10.1016/j.vph.2017.05.005
124. Förstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010;459(6):923–939. doi:10.1007/s00424-010-0808-2
125. Yang X, Chang Y, Wei W. Endothelial dysfunction and inflammation: immunity in rheumatoid arthritis. Mediators Inflamm. 2016;2016:6813016. doi:10.1155/2016/6813016
126. Tenório MB, Ferreira RC, Moura FA, Bueno NB, de Oliveira ACM, Goulart MOF. Cross-talk between oxidative stress and inflammation in preeclampsia. Oxid Med Cell Longev. 2019;2019:8238727. doi:10.1155/2019/8238727
127. Chen CJ, Fu YC, Yu W, Wang W. SIRT3 protects cardiomyocytes from oxidative stress-mediated cell death by activating NF-κB. Biochem Biophys Res Commun. 2013;430(2):798–803. doi:10.1016/j.bbrc.2012.11.066
128. Oh RC, Trivette ET, Westerfield KL. Management of hypertriglyceridemia: common questions and answers. Am Fam Physician. 2020;102(6):347–354.
129. Lundman P, Eriksson MJ, Stühlinger M, Cooke JP, Hamsten A, Tornvall P. Mild-to-moderate hypertriglyceridemia in young men is associated with endothelial dysfunction and increased plasma concentrations of asymmetric dimethylarginine. J Am Coll Cardiol. 2001;38(1):111–116. doi:10.1016/s0735-1097(01)01318-3
130. Kajikawa M, Maruhashi T, Matsumoto T, et al. Relationship between serum triglyceride levels and endothelial function in a large community-based study. Atherosclerosis. 2016;249:70–75. doi:10.1016/j.atherosclerosis.2016.03.035
131. Nakamura T, Takano H, Umetani K, et al. Remnant lipoproteinemia is a risk factor for endothelial vasomotor dysfunction and coronary artery disease in metabolic syndrome. Atherosclerosis. 2005;181(2):321–327. doi:10.1016/j.atherosclerosis.2005.01.012
132. Kajikawa M, Higashi Y. Triglycerides and endothelial function: molecular biology to clinical perspective. Curr Opin Lipidol. 2019;30(5):364–369. doi:10.1097/MOL.0000000000000630
133. Schwartz EA, Reaven PD. Lipolysis of triglyceride-rich lipoproteins, vascular inflammation, and atherosclerosis. Biochim Biophys Acta. 2012;1821(5):858–866. doi:10.1016/j.bbalip.2011.09.021
134. Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res. 2009;50(2):204–213. doi:10.1194/jlr.M700505-JLR200
135. Aung HH, Lamé MW, Gohil K, An CI, Wilson DW, Rutledge JC. Induction of ATF3 gene network by triglyceride-rich lipoprotein lipolysis products increases vascular apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2013;33(9):2088–2096. doi:10.1161/ATVBAHA.113.301375
136. Ivanova EA, Myasoedova VA, Melnichenko AA, Grechko AV, Orekhov AN. Small dense low-density lipoprotein as biomarker for atherosclerotic diseases. Oxid Med Cell Longev. 2017;2017:1273042. doi:10.1155/2017/1273042
137. Suciu CF, Prete M, Ruscitti P, Favoino E, Giacomelli R, Perosa F. Oxidized low density lipoproteins: the bridge between atherosclerosis and autoimmunity. Possible implications in accelerated atherosclerosis and for immune intervention in autoimmune rheumatic disorders. Autoimmun Rev. 2018;17(4):366–375. doi:10.1016/j.autrev.2017.11.028
138. Pirillo A, Catapano AL, Norata GD. Biological consequences of dysfunctional HDL. Curr Med Chem. 2019;26(9):1644–1664. doi:10.2174/0929867325666180530110543
139. Loader J, Meziat C, Watts R, et al. Effects of sugar-sweetened beverage consumption on microvascular and macrovascular function in a healthy population. Arteriosclerosis Thrombosis Vasc Biol. 2017;37(6):1250–1260. doi:10.1161/ATVBAHA.116.308010
140. Idris-Khodja N, Ouerd S, Mian MOR, et al. Endothelin-1 overexpression exaggerates diabetes-induced endothelial dysfunction by altering oxidative stress. Am J Hypertens. 2016;29(11):1245–1251. doi:10.1093/ajh/hpw078
141. Pluta W, Dudzińska W, Lubkowska A. Metabolic Obesity in People with Normal Body Weight (MONW)—review of diagnostic criteria. Int J Environ Res Public Health. 2022;19(2):624. doi:10.3390/ijerph19020624
142. Keirns BH, Hart SM, Sciarrillo CM, Poindexter KL, Clarke SL, Emerson SR. Postprandial triglycerides, endothelial function, and inflammatory cytokines as potential candidates for early risk detection in normal-weight obesity. Obes Res Clin Pract. 2022;16(5):386–392. doi:10.1016/j.orcp.2022.08.008
143. Sprung VS, Bowden Davies KA, Norman JA, et al. Metabolic syndrome is associated with reduced flow mediated dilation independent of obesity status. Eur J Endocrinol. 2020;183(2):211–220. doi:10.1530/EJE-20-0098
144. Rakhmat II, Nugraha GI, Ariyanto EF, et al. Strong association of metabolic parameters with ADMA and VCAM-1 in normo-weight subjects with metabolic syndrome. DMSO. 2024;17:833–839. doi:10.2147/DMSO.S448650
145. Brant LCC, Wang N, Ojeda FM, et al. Relations of metabolically healthy and unhealthy obesity to digital vascular function in three community‐based cohorts: a meta‐analysis. JAHA. 2017;6(3):e004199. doi:10.1161/JAHA.116.004199
146. Sipahioglu NT, Ilerigelen B, Gungor ZB, et al. Relation of biochemical parameters with flow-mediated dilatation in patients with metabolic syndrome. Chinese Med J. 2017;130(13):1564–1569. doi:10.4103/0366-6999.208231
147. Iwańczyk S, Smukowska-Gorynia A, Woźniak P, Grygier M, Lesiak M, Araszkiewicz A. Increased endocan expression as a biomarker of endothelial dysfunction in patients with metabolic syndrome. Pol Arch Intern Med. 2022;132(7–8):16292. doi:10.20452/pamw.16292
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.