Back to Journals » Vascular Health and Risk Management » Volume 16
Vascular Calcification: An Important Understanding in Nephrology
Authors Zununi Vahed S ![]() , Mostafavi S
, Mostafavi S ![]() , Hosseiniyan Khatibi SM, Shoja MM, Ardalan M
, Hosseiniyan Khatibi SM, Shoja MM, Ardalan M
Received 21 December 2019
Accepted for publication 17 April 2020
Published 12 May 2020 Volume 2020:16 Pages 167—180
DOI https://doi.org/10.2147/VHRM.S242685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Harry Struijker-Boudier
Sepideh Zununi Vahed,1 Soroush Mostafavi,1 Seyed Mahdi Hosseiniyan Khatibi,1 Mohammadali M Shoja,2 Mohammadreza Ardalan1
1Kidney Research Center, Tabriz University of Medical Sciences, Tabriz, Iran; 2Department of Surgery, University of Texas Medical Branch, Galveston, TX, USA
Correspondence: Mohammadreza Ardalan
Kidney Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
Tel +98 914 116 8518
Fax +98 41 3336 65 79
Email [email protected]
Abstract: Vascular calcification (VC) is a life-threatening state in chronic kidney disease (CKD). High cardiovascular mortality and morbidity of CKD cases may root from medial VC promoted by hyperphosphatemia. Vascular calcification is an active, highly regulated, and complex biological process that is mediated by genetics, epigenetics, dysregulated form of matrix mineral metabolism, hormones, and the activation of cellular signaling pathways. Moreover, gut microbiome as a source of uremic toxins (eg, phosphate, advanced glycation end products and indoxyl-sulfate) can be regarded as a potential contributor to VC in CKD. Here, an update on different cellular and molecular processes involved in VC in CKD is discussed to elucidate the probable therapeutic pathways in the future.
Keywords: chronic kidney disease, CKD, uremic toxins, hyperphosphatemia, uremia, calcification
Introduction
The growing burden of cardiovascular disease (CVD) in chronic kidney disease (CKD) patients and general population is presumably, at least in part, due to vascular calcification (VC).1 VC is an active, complex, and extremely regulated procedure that involves cell-mediated processes and a complex interaction between the inhibitor and promoter factors.2,3 VC is a result of the pathological deposition of calcium phosphate mineral in soft tissues that decreases the blood vessels’ elasticity and elevates blood pressure.
Disturbed mineral homeostasis due to an impaired renal function, the uremic milieu and CKD provides a storm of risk factors for VC and the development of CVD. Dysregulated mineral metabolism and the elevated levels of circulating calcium (Ca) and phosphate (Pi) are key factors for the initiation and progression of VC in CKD since major resident cells in the media layer of blood vessels, vascular smooth muscle cells (VSMCs), are sensitive to these factors.4,5 VSMCs can undergo trans-differentiation to osteoblast-like cells and extrude matrix vesicles (MVs) that contain proteins similar to osteoblastic vesicles. When these proteins are secreted by VSMCs, the osteogenic environment is created and resulted in VC.6,7
Numerous pathological landscapes are associated with the development of VC. The impaired homeostasis of Ca/Pi and high levels of parathyroid hormone (PTH) cause Ca/Pi release by bone under severe hyperphosphatemia. The endothelial dysfunction, oxidative stress, chronic inflammation, VSMCs trans-differentiation, proliferation and apoptosis, loss of mineralization inhibitors, increased remodeling of extracellular matrix (ECM), and release of calcifying extracellular vesicles (cEVs) are the most important contributors to VC.8 Moreover, the calciprotein particles (CPPs), the complexes of Pi, Ca, and proteins, are recognized to drive the calcification process.9 It is reported that in CKD, uremic EVs and CPPs can modulate VSMCs' responses through an inflammation stress and trans-differentiation, causing an increase in mineral deposition. Hence, these circulating particles play an important role in the mechanisms of widespread calcification.10 Additionally, several uremia-related factors contribute to the development of VC among patients with CKD. In addition to disrupted metabolism and other pathologies that promote VC, genetics, hereditary predisposition, and epigenetics are involved in VC development.7,11 In this review, we will review the recently updated state of knowledge on cellular and molecular mechanisms of VC in CKD. Unraveling the signaling pathways involved in VC in CKD patients will eventually offer novel therapies to limit the vicious effects of VC.
Vascular Calcification in CKD
Under physiological circumstances, the active mineralization inhibitors including matrix Gla protein (MGP), pyrophosphate (PPi), fetuin-A, osteoprotegerin (OPG), adenosine, bone morphogenetic protein 7 (BMP-7) and osteopontin (OPN) protect blood vessels from the formation of stable hydroxyapatite crystals.12,13 A decline in these inhibitors along with elevated levels of active inducers of VC lead to a high prevalence of VC in the CKD population (Figure 1A). Although CKD patients can develop calcification in both media and intima layers of vessel wall (Figure 1B and C), the media calcification is more common in these patients, especially in pediatrics.14 It has been shown that all types of VC increase the mortality and morbidity rates in CKD patients.15
 |
Figure 1 Schematic view of vascular calcification in CKD. (A) As renal function continues to fall, normal defense mechanisms for Pi and Ca homeostasis (PTH, FGF-23, and klotho) become overwhelmed and the endocrine system of FGF23-klotho-VitaminD and RAAS is disturbed. As a result of nephron loss and higher levels of FGF-23, 1α-hydroxylase activity is diminished in the kidney, leading to elevated levels of inhibitor of this enzyme (FGF-23) and a decrease in 1,25(OH)2-vitamin D (calcitriol) production43 that, in turn, upregulates the production of renin in the kidney. Subsequently, the elevated levels of angiotensin II lead to kidney klotho loss, disruption of FGF-23 signaling, and the impairment of phosphaturia. Elevated levels of FGF-23 may activate the RAAS either by suppressing ACE-2 directly94 or decreasing calcitriol levels indirectly.107 (B) Ca and Pi deposition in the VSMCs of medial layers may cause VC. (C) In the intimal calcification process, more diverse cells are involved including osteoclast-like cells, Gli1+-MSCs of the adventitia, and CCCs. The interaction of different factors and these cells causes atherosclerosis. Uremic toxins cause VSMCs trans-differentiation into osteoblast-like cells. In the process of calcification, macrophage differentiation into osteoclast-like cells is inhibited. In turn, macrophages increase apoptosis and accumulation of apoptotic bodies through transition into foam cells. A pro-inflammatory form of circulating monocytes (M1 macrophages) promotes the initial calcium deposition within the necrotic core of the lesions. All the above factors together cause atherosclerosis. For more details, see the full text. Abbreviations: CKD, chronic kidney disease; FGF-23, fibroblast growth factor-23; PTH, parathyroid hormone; VC, vascular calcification; MMPs, matrix metalloproteinases; DH-VitD, 1, 25-dihydroxyvitamin D. Gli1+-MSCs, Gli1+ mesenchymal stem cells; CCCs, calcifying circulating cells; ACE-2, angiotensin-converting enzyme-2; RAAS, renin-angiotensin-aldosterone system; HA, hydroxyapatite crystal; ECs, endothelial cells; MQ, macrophage; IS, indoxyl-sulfate; VSMC, vascular smooth muscle cell; OS, oxidative stress. |
Beyond a high incidence of the traditional risk factors in CKD patients, for instance, age, family history, sex, diabetes, hypertension, dyslipidemia, and tobacco use, VC in this population is connected with numerous other factors. Oxidative stress, inflammation, the CKD-related disorder of mineral metabolism, and bone are the most important non-traditional risk factors that accelerate VC in CKD patients.16 The accumulation of uremic toxins [ie, Pi, advanced glycation end products (AGEs), and indoxyl-sulfate (IS)] and uremia-related factors (ie, malnutrition, hyperhomocysteinemia, and anemia) may also directly enhance the VC in CKD patients. As a source of uremic toxins, the gut microbiome is a potential contributor to CVD in CKD. In CKD, p-cresyl sulfate (PCS) and IS stimulate toxin-induced VC directly through the activation of coagulation and inflammation pathways in the arterial wall.17 Furthermore, CKD risk factors including the history of dialysis, phosphate retention, high doses of vitamin D therapy, extra calcium, and uremic hyperparathyroidism can promote the VC development in patients with CKD.18 It is also reported that the dysregulated mineral metabolism derives oxidative DNA injury and premature senescence to stimulate inflammation and VC in children on dialysis.19 Different cells and factors can regulate the VC process in CKD patients. Epigenetics (microRNAs), the formation and release of extracellular vesicles, elastin degradation, and CPPs continue to disclose the involved mechanisms in the initiation and development of VC in CKD. Autophagy, mitochondrial dysfunction, microtubule destabilization, and endoplasmic reticulum stress are also involved in the pathogenesis of VC in CKD and restoring their functions can be effective therapeutic targets.20–25 Hortells et al (2017) clarified the expression patterns of factors contributed to the pathogenesis of VC in uremic rats in detail.26 In the following sections, we highlight the most common factors in the development of VC.
Cells Involved in Vascular Calcification
In addition to the fact that osteogenic transition of VSMCs is the main cause of VC, other cells are involved in this process. Among them, osteoclast-like cells, endothelial progenitor cells (EPCs), Gli1+ mesenchymal stem cells (Gli1+-MSCs) of the adventitia, and calcifying circulating cells (CCCs) can be mentioned.
Vascular Smooth Muscle Cells
In CKD, the oxidative stress, chronic inflammation, and uremic toxins may influence the VSMCs’ physiological functions directly. Under these circumstances, the cellular environment is capable to stimulate a VSMC trans-differentiation from a contractile cell to an osteoblastic/chondroblastic-like cell and undergoes irregular senescence, proliferation, apoptosis, migration, and calcification (Figure 2).27
 |
Figure 2 The impact of uremic toxins on CKD-induced VSMC dysfunction and VC. Due to hyperphosphatemia, hypercalcemia, elevated oxidative stress, and inflammation,132 VSMCs manifest dysregulated functions and phenotype. Uremic toxins including Pi, IS, AGEs, IL-1β, IL-6, and TNF-α are involved in CV. (A) IL-1β, IL-6, and TNF-α induce osteoblast-like trans-differentiation of VSMCs through different mechanisms.16 Interaction of AGEs with their receptor (RAGE) induces the expression of Pit-1 via ROS production49 and leads to osteogenic transition. It also causes apoptosis through NAD(P)H oxidase-derived oxidative stress.133 (B) In CKD, normal Ca homeostasis is also dysregulated. This homeostasis is mediated by klotho, PTH, active vitamin D metabolites, and calcitonin. In VSMCs, Ca signaling is mediated by Ca channels, CaR, and pumps that maintain Ca concentrations in these cells.134 Higher level of extracellular Ca is associated with the release of MVs and cell death promotion and release of apoptotic bodies.43 (C) Extracellular Pi, as a signaling molecule, can trigger numerous changes in VSMCs through regulating different molecular pathways. NPP1 is responsible for extracellular ATP degradation to AMP and PPi, CD73 degrades AMP to adenosine and Pi and TNAP breaks PPi into phosphate and adenosine.15 Higher Pi level simultaneously upregulates the expression of osteo/chondrogenic genes (Runx2, ALP, OPN, and osterix) and downregulates VSMCs genes (SM22α and αSMA). ALP controls vascular matrix mineralization by degradation and inactivation of the VC inhibitors (PPi and P-OPN) to allow uncontrolled tissue mineralization and simultaneously releasing free Pi.43 These osteo-/chondroblast-like cells actively induce apoptosis and vesicle release, a reduction in calcification inhibitors, elastin degradation, increased ECM remodeling, and a pro-inflammatory state. Moreover, under high levels of Pi, VSMCs synthesize collagen at high amount and provide a collagen-enriched ECM. Downregulation of Gas6 and Bcl2 may be the basic mechanism of VSMCs apoptosis. The released apoptotic bodies provide a further nidus for deposition of Pi and Ca. For more details, see the full text. Abbreviations: Ca, calcium; Pi, phosphate; PPi, pyrophosphate; ECM, extracellular matrix; MMP, matrix metalloproteinases; Gas6, growth arrest-specific gene 6; ALP, alkaline phosphatase; ROS, reactive oxygen species; SM, α-smooth muscle actin; CPPs, calciprotein particles; CaR, Ca sensing receptor; MVs, matrix vesicles; AGEs, advanced glycation end products; RAGE, receptor for advanced glycation end products. |
A switch to the osteogenic- and/or chondrogenic-like phenotype is characterized by the expression of Runx2 also known as core-binding factor a-1 (CBFA1), SRY-Box 9 (SOX9), Msh Homeobox 2 (MSX2), and Osterix that are important transcription factors for both intimal and medial VC.28 The VSMCs trans-differentiation into osteo-/chondrogenic-like cells is coordinated by a complex network of intracellular signaling pathways like nuclear factor kappa beta (NF-κB), receptor activator of NF-κB (RANK)/RANK ligand (RANKL), Wnt-β-catenin, mitogen-activated protein kinase (MAPK) p38, calcium-induced signaling, and nitric oxide–plasminogen activator inhibitor-1 (PAI-1) pathways.29–32 Moreover, trans-differentiation can be mediated by extracellular signal-regulated kinases 1/2 (ERK1/2) pathways.31 The downstream impacts of ERK1/2 during VC are not still completely understood.3
Primary CPPs are amorphic and solid-phase Ca-Pi that bound to serum fetuin-A protein and calcium-regulatory proteins and disperse as colloids in the circulation to eliminate mineral crystal formation and guard against the ectopic calcification. This defense mechanism maintains blood mineral homeostasis and inhibits calcification. In the pathology of VC, the balance between the primary CPP formation and absorption is dysregulated and these CPPs may undergo a transition to the crystalline (secondary CPPs) phase. Secondary CPPs are stretched particles that contain predominantly hydroxyapatite surrounding proteins. Clinical studies have showed that the level of serum CPP was elevated with the decline of kidney function and connected with inflammation and VC in CKD patients.13,33 The CPPs of CKD patients had the features of secondary CPPs; a reduced level of fetuin-A and GRP (Gla-rich protein).10 The secondary CPPs may directly stimulate VC through the induction of VSMCs trans-differentiation.10 This event is mediated by uptake of the CPPs, increasing the intracellular levels of Ca2+, initiation of oxidative stress and inflammatory responses (TNF-α) in VSMCs to promote mineralization via its receptor (TNFR1).34 This event may be involved in VC but no sufficient proof is available to underline that this is the only pathway.
In CKD patients, the elevated levels of Ca and Pi induce the discharge of membrane-bound MVs from VSMCs. These MVs contain lipids, microRNAs, and proteins that are essential to induce the calcification cascade. Proteins for import of Pi and Ca into the MVs and proteins-related to cellular stress, cytoskeleton, and extracellular mineralization along with other intracellular proteins can be found in MVs. These MVs also contain tissue nonspecific alkaline phosphatase (TNAP) to degrade PPi for making a local calcifying microenvironment.35 The apoptotic bodies and Ca/Pi-loaded MVs (released from VSMCs and macrophages) are different types of extracellular vesicles that eventually form hydroxyapatite crystals and deposit on a collagen matrix in the vessel wall and provide a bed for nucleation and VC in the media layer.8,27
Additionally, macrophage-released calcifying MVs are directly associated with arterial medial early intimal and calcification in the CKD patients.36 A pathological calcification-inflammation cycle exists between VSMCs and macrophages. The presence of Ca/Pi mineral in ECM, cEVs, and secondary CPPs of blood vessels can trigger pro-inflammatory responses in VSMCs and immune cells. Osteogenic-like VSMCs cause ECM calcification by releasing cEVs and increasing pro-inflammatory responses in macrophage. This inflammatory response in macrophages contributes to the elevated VC via releasing cEVs and inducing VSMCs osteogenic trans-differentiation.37
Elastin degradation is facilitated by proteases such as Cathepsin-S and matrix metalloproteinases (MMP-2 and −9) that are upregulated in CKD. Elastin disruption in the aortic wall causes an increase in the expression level of transforming growth factor (TGF-β) involved in osteoblast differentiation and increases arterial stiffness in CKD.38,39 After the VSMCs phenotypical alteration toward osteoblast-like phenotype, the deposition of mineral crystals (biomineralization) happens. This regulated process requires MVs release to concentrate the Ca and Pi and support the nucleation of mineral crystals via the matrix proteins.18 Kapustin et al proposed a model for depicting the possible mechanism of MVs calcification. Phosphate is taken up by Pi transporters, while Ca passes through the MV membrane by voltage-dependent anion-selective channel protein 1 (VDAC1). The nucleation complexes are formed via the binding of mineral ions (Pi and Ca) with phosphatidylserine and annexin A6 on the inner and outer MVs surfaces. The formation of these complexes stimulates the growth of crystal apatite. Moreover, MMP-2 degrades elastin and stimulates calcification on the MV surface.40
It should be noted that the VSMCs are believed to take up chondrogenic properties. The VSMCs chondrogenic-like transformation underlies the cartilaginous metaplasia formation that is associated with VC in animal models and humans. TGFβ-Wnt16-Notch signaling is involved in this process.41
Sensing and Transduction of Osteogenic Signals in CKD
Sensing and the transduction of osteogenic stimuli in CKD are mediated through different signaling molecules, receptors, and channels that modulate the osteogenic response in the VSMC. Some harmful effects of Pi are triggered by its excessive entry into the VSMCs through sodium-dependent phosphate transporters (PiT-1 and −2). Moreover, in response to elevated Pi levels, PiT-1 (but not Pi that is taken through PiT-1) is necessary for ERK1/2 phosphorylation. Therefore, for the calcification process of VSMC, both Pi transport-dependent and -independent effects of PiT-1 are important.42 The phosphate-induced calcification is mediated by reactive oxygen species (ROS) production and oxidative stress, osteochondrogenic differentiation, apoptosis of VSMCs and the release and instability of extracellular vesicles.43–45 Under uremic conditions, PiT-2 is up-regulated along with PiT-1 in the vasculature; however, it defends against VC by unidentified mechanisms.46 Toll-like receptors (TLRs) may be also involved in Pi-sensing; the activation of TLR4/NF-κB signaling in VSMCs directly induces VC in CKD.47 The NF-κB signaling activation, at least, through the serum- and glucocorticoid-inducible kinase 1 (SGK1) can also promote VC in CKD.48
The endocytosis of Ca/Pi particles in lysosomes causes calcium release and apoptosis in VSMCs. It is also reported that VSMCs can be stimulated through AGE products and their receptors (RAGE). RAGE ligands mediate ROS production in VSMC that is involved in the up-regulation of Pit-1 and Runx2.49 Different channels, pumps, and exchangers are involved in the sensing, entrance of Ca into the VSMCs, and preserving of Ca concentrations in the cytosol and sarcoplasmic reticulum. Changes in intra- and extracellular pools of Ca affect VSMC function and phenotype and the regulation of Ca is dependent on the phenotypic state of VSMC.
Circulating Calcifying Cells
Circulating calcifying cells (CCCs), which originate from the bone marrow (BM), play a role in the intima calcification processes. CCCs have an osteogenic phenotype and express bone alkaline phosphatases and osteocalcin (OCN).50 Recent studies have demonstrated that the pool of CCCs contains calcifying endothelial progenitor cells (EPCs), MSC–derived circulating osteoprogenitors and myeloid calcifying cells (a group of circulating monocytes) in CKD patients.51
The endothelium integrity presents a crucial role in the establishment of VC and EPCs support it during endothelial injury. In response to cellular apoptosis or activation, mature endothelial cells secret soluble microparticles (MPs) to regulate procalcificant activity in VSMCs and to differentiate EPCs. In CKD cases with VC, higher endothelial MPs are connected with a lower percentage of EPCs. This data proposesan inequality between the repair procedures and endothelial in patients who suffer from CKD. By the expression of osteogenic factor, OCN, EPCs could contribute to the VC procedure directly. Moreover, the OCN gene is expressed in the fibroblast and VSMCs of CKD patient as a result of MPs action.52,53 Moreover, EPCs undertake an endothelial to procalcific shift in CKD-MBD and trigger VC.54
Bone morphogenetic protein 2 (BMP-2), which increases in uremic patients, promotes the migration of MSCs from BM to other tissues.55–57 Cianciolo et al have proven the relationship between a particular type of EPC subset (CD34+/KDR+/CD133–/CD45– cells) and an increased VC in CKD patients.58 Calcifying myeloid cells in the bloodstream can cause VC but their exact role in CKD patients has not been established.59 Altogether, the issue of CCCs is a new topic in recognizing the pathophysiology of calcification in CKD patients and needs further investigation.2,50
Gli1+ Mesenchymal Stem Cells
Gli1+ MSC-like cells are located in the vascular adventitious layer and play a role in the process of vascular repair and neointima formation.60 These cells are important in maintaining kidney homeostasis, angiogenesis, and vascular stability.61 Gli1+ cells affect arterio- and athero-sclerosis in ApoE−/− mice by migrating to the media and neointima layers.62 These cells are a key source of osteoblast-like cells during VC in the intima and media.62 The interference of Gli1+ in osteogenic differentiation is controlled by the Sonic Hedgehog (SHH) pathway.2 It can be concluded that during the uremic calcification, Gli1+-MSCs are a chief reservoir of osteoblast-like cells that can be therapeutically targeted to inhibit CV in CKD.2
Microbiota
The human intestinal tract homes to a collection of symbiotic, commensal, and pathogenic micro-organisms in a local ecologic community called microbiome.63 The gut microbiome as a “second human genome” has a significant role in both human health and the pathogenesis of kidney diseases.64,65 Recent studies demonstrate dysbiosis, a shift in the bacterial populations, in patients with CKD and end-stage renal disease (ESRD).66,67 The administration of antibiotics and phosphate binders, dietary restriction, and CKD itself may contribute to dysbiosis in kidney disease.68–71 Gut dysbiosis may elevate the production of microbial byproducts that are absorbed from the intestinal lumen. The increased absorption along with a decreased kidney clearance lead to a rise in gut-derived toxin levels in circulation.63 In CKD, the influx of urea and other toxins causes an alteration in the gut microbiome. A diminished number of beneficial bacteria are associated with an increase in uremic toxin-producing bacteria. Because of the degradation of cellular tight junctions and inflammation in intestinal, gut-derived uremic toxins including phenylacetylglutamine, indole-3 acetic acid, IS, trimethylamine-N-oxide (TMAO) and p-cresyl sulfate (PCS), translocate into the bloodstream and cause an extensive oxidative stress damage to the kidney, cardiovascular system, bone-mineral, erythropoiesis, and endocrine systems.72
Recent evidence indicates that different gut-derived byproducts are associated with VC, CVD, and adverse cardiovascular outcomes and mortality in CKD.63 In patients with CKD, the serum levels of IS have an inverse association with renal function and a direct correlation with aortic calcification and cardiovascular mortality.73,74 It is also reported that both PCS and IS can directly stimulate VC in the peripheral arteries and aorta of CKD rat through the stimulation of insulin resistance and hyperglycemia that activate the coagulation pathways and the acute-phase response signaling in the arterial wall.17 In patients on hemodialysis, the serum levels of IS were connected with coronary artery calcium, an independent predictor of cardiovascular events.75
Uremic toxins endorse the proliferation and transformation of VSMCs into osteoblast-like cells, leading to vascular wall thickening and calcification. The effect of IS on VSMCs is mediated by organic anion transporter 3 (OAT3). Moreover, the stimulation of VSMC proliferation is mediated by MAPK activation in vitro. This may be one of the mechanisms which leads to the development of atherosclerotic lesions in ESRD patients.76 Furthermore, IS stimulates the expression of (Pro) renin receptor (PRR) and renin/prorenin in aorta by ROS production, OAT3-mediated uptake as well as aryl hydrocarbon receptor (AhR) and NF-κB p65 activation in VSMCs. The activation of PRR by IS stimulates the proliferation and expression of tissue factor in VSMCs.77
Epigenetics
During the hyperphosphatemia, different epigenetic modifications including DNA methylation, histone modifications, and microRNAs (miRNAs, miRs) dysregulation contribute to the osteo-induced cellular signaling.78,79 It is indicated that through the hypermethylation of Klotho gene, IS can suppress vascular Klotho gene expression and contribute to pathological mechanism of CV in CKD.80 Likewise, the methylation of the SM22α promoter region induces VC at a higher level of Pi.81 It is also reported that through a reduction in the ALP promoter region methylation and an increase in the ALP expression, DNA methyltransferases inhibitors ease the Pi-induced VC.78 Changes in the chromatin conformation, histone modification (histone tail methylation), hypermethylation of calcification inhibitory genes, activation of osteoblast-differentiation genes, or the deregulation of histone deacetylase members may predispose VSMCs to calcification. Furthermore, there is a cross-talk between different epigenetic mechanisms in VC; microRNAs may be upstream regulators of histone deacetylase that can modulate the severity of the calcification.82
Over the last decade, the roles of microRNAs have been identified in the course of CKD, pathogenesis of VC, and atherosclerosis.83,84 microRNAs are small non-coding RNAs that negatively regulate the gene expression at both transcription and translation levels. In a systematic review, the agonistic and antagonistic miRNAs that positively and negatively regulate VC are reviewed comprehensively.82 The protective effect of miR-30b against VC is mediated by stimulating autophagy and mitochondrial membrane potential.85 Increased levels of miR-29b and decreased levels of miR-133b and miR-211 that are correlated with lower and higher expression of inhibitors and RUNX2 of osteoblastic differentiation, respectively, are reported in uremic rats.86 It is also reported that the down-regulation of miR-29b and activation of Wnt/β-catenin signaling may be involved in IS-induced VC in CKD.87 Likewise, down-regulated levels of miR-125-b accelerate trans-differentiation of VSMCs and calcification by targeting Ets1.88 Moreover, miR-142-3p prevents VC in both humans and mice with ESRD.89 It has been proven that miR-223 and miR-126 that are expressed in VSMCs interfere with the trans-differentiation of these cells to an osteoblastic phenotype that increases the vascular wall stiffness.90 There were higher levels of miR-29a/b and miR-223 expression in hemodialysis patients with VC and the calcification intensity was associated with the miR-29a level.91
Inducers and Inhibitors of VC
An imbalance between the inducers and inhibitors of VC happens in CKD and chronic hemodialysis patients.92 In this section, we summarize some of these factors (Table 1).
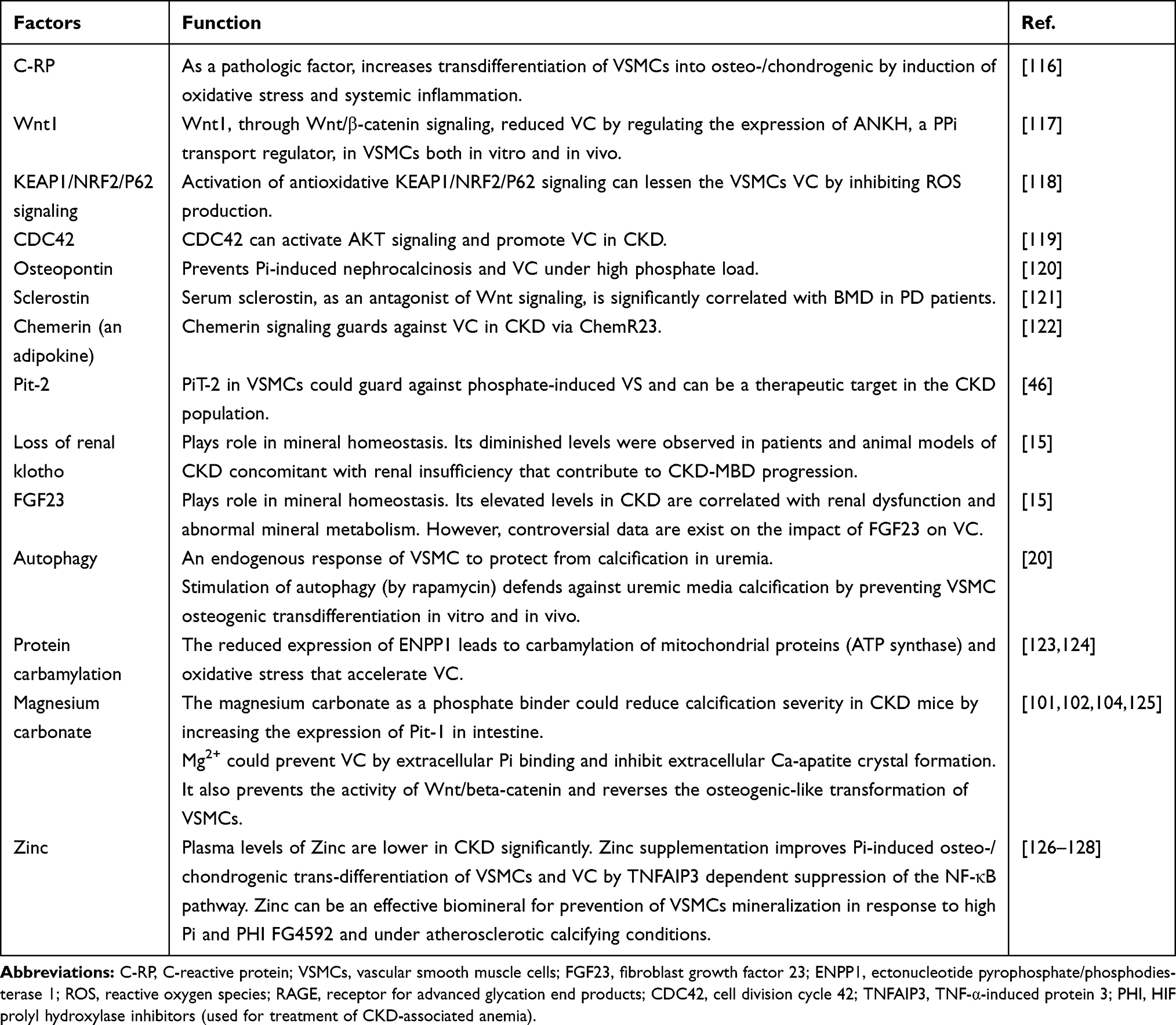 |
Table 1 The Impact of Other Factors on VC in CKD |
Fetuin-A
Fetuin-A (α2 Heremans-Schmid glycoprotein) is a glycoprotein that is secreted into the circulatory system by adipose tissue and liver.93,94 It has been revealed that fetuin-A acts as a prominent protective factor in preventing VC in patients with CKD and ESRD.93 Decreased fetuin-A increases the morbidity and mortality rate in ESRD cases.95 It has been shown that fetuin-A is involved in preventing aortic calcification.95 This is explained by the fact that despite the general similarity of risk factors for aortic and coronary calcification, these factors are specific to each site in the general population.95 Therefore, the pathophysiology involved in the calcification of these two sites is somewhat different.95 Some studies have shown that fetuin-A is also effective in preventing the calcification of the heart valves, some others have not shown a link between them.96 The ability of fetuin-A to prevent the mineralization is mediated by the CPPs formation.97,98
Magnesium
Higher levels of Pi, amorphous Ca2+-Pi particle (ACP) formation, and reduced levels of VC inhibitors in the circulation start the VSMC trans-differentiation that is enhanced by the osteogenic genes expression and amplified by the release of apoptotic bodies and exosomes.99 At different levels, magnesium hinders these processes of VC. Osteogenic differentiation and VC are negatively regulated by magnesium via increasing the activity of its transporter, transient receptor potential melastatin 7 (TRPM7) and its entry into the cell that leads to the expression of anti-calcifying proteins (BMP-7, OPN, and MGP).100 Moreover, by reduction in microRNAs expression (miR-133a, miR-30b, and miR-143a), magnesium influences the expression of osteogenesis (Runx2, Smad1, and Osterix).101 Anti-calcifying impact of magnesium is also mediated by the inhibition of Wnt/β-catenin pathway.102 As an antagonist of Ca-channel, magnesium hinders the Ca entry into the VSMCs, loss of inhibitors, and osteogenic differentiation.103 Moreover, it prevents hydroxyapatite formation in the extracellular space, thereby, avoids VSMCs calcification.104 Although several recent studies propose that the advantageous role of magnesium on VC can be elucidated via a delayed formation of secondary CPP,9 the definitive proof to support this hypothesis is missing.
Hormones
Vascular autocrine and paracrine factors participate in keeping circulatory homeostasis. However, under pathological circumstances, these factors may modulate the pathogenesis of VC and CVD. The constant activation of the RAAS (renin-angiotensin-aldosterone system) has a foremost role in cardiovascular remodeling and CKD progression.105 Angiotensin II (Ang II), an active factor of the RAS, is participated in the control of cardiovascular function and kidney homeostasis through acting on different cells, mainly VSMCs.106 As renal function continues to fall, normal defense mechanisms, as an endocrine axis for Pi and Ca metabolism, become overwhelmed and a disruption happens in this axis. Low kidney klotho, high levels of fibroblast growth factor 23 (FGF-23), vitamin D deficiency, and RAAS activation are associated with adverse kidney outcome in CKD.107 Klotho is a transmembrane protein that acts as a co-receptor of FGF23. Klotho is expressed in kidney, choroid plexus, and parathyroid glands and mediates the functional role of the FGF-23 in regulating the Pi and Ca levels; hence, Klotho deficiency causes hyperphosphatemia. In klotho-hypomorphic mice, deficiency of klotho leads to an uncontrolled formation of calcitriol that increases the reabsorption of Pi in kidney and intestine, increasing phosphate levels.28
In CKD, the impaired kidney elimination of Pi leads to hyperphosphatemia that promotes the FGF-23 secretion, a regulator of serum Pi level, from bone osteocytes. Due to nephron loss and higher levels of FGF-23, the 1α-hydroxylase activity, an enzyme for the production of active vitamin D (1,25(OH)2 vitamin D, calcitriol) is reduced; leading to a reduction in calcitriol level that in turn increases renin production in kidney. The activation of RAAS reduces the expression of renal klotho, a critical factor for accurate FGF-23 signaling.
Phosphate may directly induce aldosterone synthase expression in adrenal glands and vascular tissue that may have an important effect on how FGF-23 mediates the activation of RAAS. On the whole, an increased level of FGF-23 activates the RAAS by two possible mechanisms: (a) indirectly by reducing calcitriol levels and (b) directly by suppressing the activity of angiotensin-converting enzyme 2 (ACE-2) that inhibits the conversion of Ang II into Ang (1–7).108 As a result, the increased levels of Ang II increase the aldosterone production that activates Pit-1; resulting in Pi entrance into VSMCs. Moreover, aldosterone fosters the inflammatory processes by induction of TNF-α.109 It is also reported that Ang II is able to prevent Pi-induced VSMCs calcification by increasing the influx of Mg that is mediated by stimulating the TRPM7 activity as well as prohibition of the canonical Wnt/β-catenin and the activation of the ERK1/2 intracellular signaling pathways.106 Moreover, the activation of angiotensin II type 2 (AT2) receptor could mediate an endogenous protective pathway for VC in CKD since it may decrease the adverse cardiovascular events.110 The secreted FGF-23 from osteogenic cells in the calcified vessel may further increase the serum levels FGF-23 (reviewed in Ref. 109).
The deregulated levels of Ca and vitamin D ease the osteogenic differentiation and mineralization of VSMC; leading to deleterious VC. As discussed, there is a complex relationship between vitamin D, Klotho, and FGF-23 on the vasculature (Figure 1A). Collectively, the perturbation of vitamin D activity, including its turnover and systemic levels along with vitamin D receptor signaling activity are contributed in VC that may ultimately be anti- or pro-calcifying. On the other hand, evidence suggests that vitamin D exerts biphasic impact on the vasculature; both hypo- and hypervitaminosis D can contribute to the VC development through several mechanisms [reviewed comprehensibly in Ref111].
Diagnosis and Treatment of VC
Non-invasive imaging techniques plain X-rays, two-dimensional ultrasound, echocardiography, and computed tomography (CT) are accessible to screen the existence of VC. The multi-detector CT (MDCT) is a highly sensitive method for accurately and quantitatively assessing VC, especially coronary artery calcification (CAC).112 Currently, no definite therapy can reverse VC and available therapeutic modalities can reduce the progression of VC. Most candidate drugs such as phosphate-binders, bisphosphonates, magnesium, and vitamin K are currently under clinical investigation that can correct the imbalance of inhibitors and promoters (ie, hyperphosphatemia) of calcification in VC-affected patients.113,114 The therapeutic potential of antioxidant compounds that can target different pathways in VC pathology is also reported115 (Table 2).
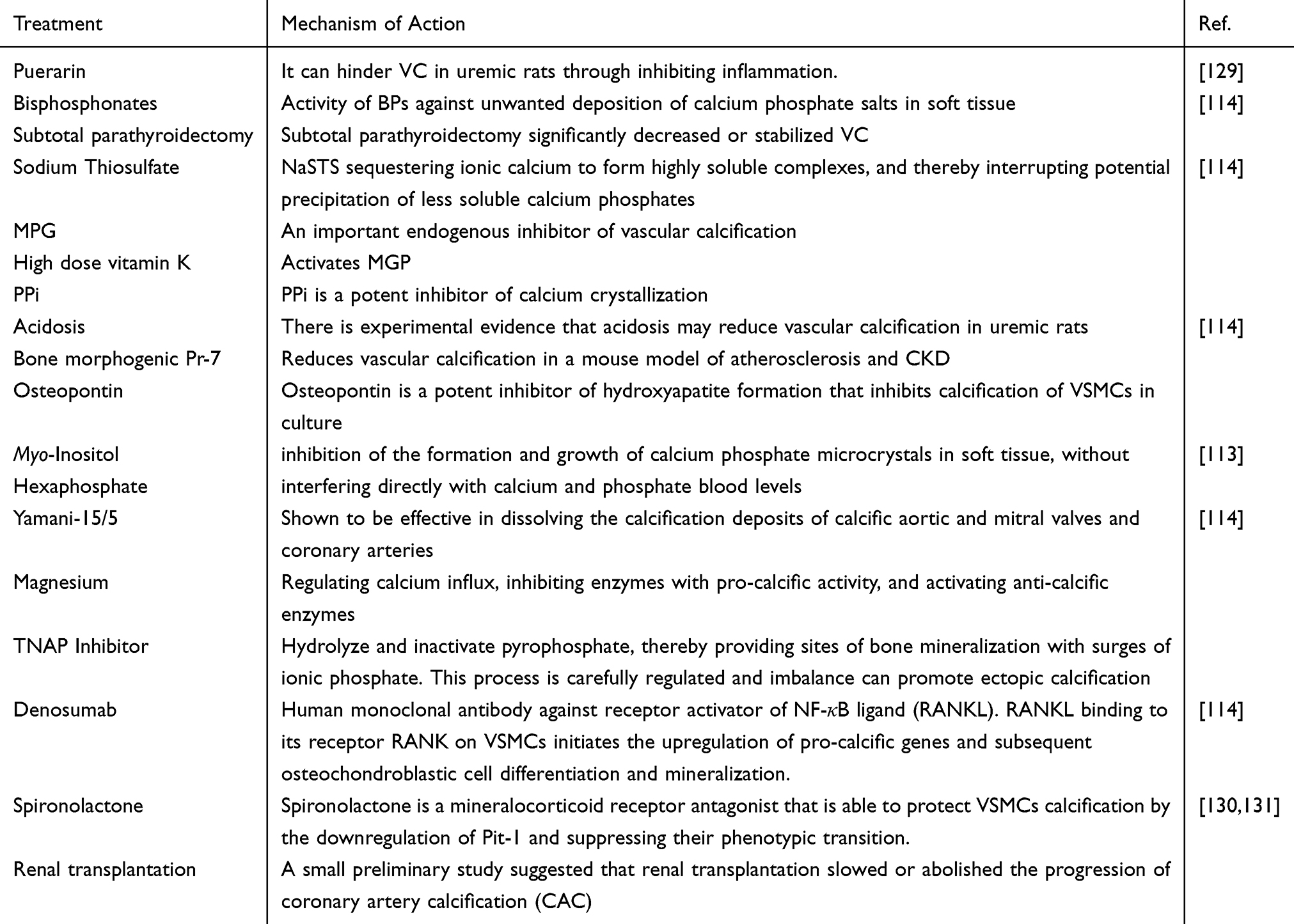 |
Table 2 Therapeutic Strategies for Vascular Calcification |
Conclusion
Overall, a plethora of several contributing factors are associated with VC in CKD. Different conditions (uremia, hyperglycemia, hyperphosphatemia, hyperlipidemia, inflammation, oxidative stress, and hypertension) might coincide with VC pathogenesis. The histological location of VC, the anatomical site of the calcified artery, and many other factors affect the commencement and progression of VC. VSMCs along with CCCs are active members of the calcification process. A range of pathogenic mechanisms are involved in the intracellular molecular mechanisms of VC. Although different therapeutically opportunities have been studied in VC, no study has strongly established that these modulations alter the patient outcome. Thus, future research might focus on the exact demonstration of VC in CKD to develop and assess tailored interventions in the organized clinical trials.
Data Sharing Statement
Research data were not shared. Data sharing is not applicable to this article as no new data were created or analyzed in this study and this is a review article.
Acknowledgment
The authors gratefully acknowledge Fatemeh Zununi Vahed for her kind support.
Funding
No financial support was provided.
Disclosure
The authors declare that they have no conflict of interest regarding this work.
References
1. Shroff R, Long DA, Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol. 2013;24(2):179–189. doi:10.1681/ASN.2011121191
2. Hénaut L, Chillon J-M, Kamel S, Massy ZA,. Updates on the mechanisms and the care of cardiovascular calcification in chronic kidney disease. Semin Nephrol. 2018;38(3):233–250. doi:10.1016/j.semnephrol.2018.02.004
3. Voelkl J, Lang F, Eckardt KU, et al. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci. 2019;76(11):2077–2091. doi:10.1007/s00018-019-03054-z
4. Azpiazu D, Gonzalo S, Gonzalez-Parra E, Egido J, Villa-Bellosta R. Role of pyrophosphate in vascular calcification in chronic kidney disease. Nefrologia. 2018;38(3):250–257. doi:10.1016/j.nefro.2017.07.005
5. Shroff R. Phosphate is a vascular toxin. Pediatr Nephrol. 2013;28(4):583–593. doi:10.1007/s00467-012-2347-x
6. Pérez-Hernández N, Aptilon-Duque G, Blachman-Braun R, et al. Vascular calcification: current genetics underlying this complex phenomenon. Chin Med J (Engl). 2017;130(9):1113–1121. doi:10.4103/0366-6999.204931
7. Bowman MAH, McNally EM. Genetic pathways of vascular calcification. Trends Cardiovasc Med. 2012;22(4):93–98. doi:10.1016/j.tcm.2012.07.002
8. Leopold JA. Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. 2015;25(4):267–274. doi:10.1016/j.tcm.2014.10.021
9. Zeper LW, de Baaij JHF. Magnesium and calciprotein particles in vascular calcification: the good cop and the bad cop. Curr Opin Nephrol Hypertens. 2019;28(4):368–374. doi:10.1097/MNH.0000000000000509
10. Viegas CSB, Santos L, Macedo AL, et al. Chronic kidney disease circulating calciprotein particles and extracellular vesicles promote vascular calcification: a role for GRP (Gla-Rich Protein). Arterioscler Thromb Vasc Biol. 2018;38(3):575–587. doi:10.1161/ATVBAHA.117.310578
11. Rutsch F, Nitschke Y, Terkeltaub R. Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ Res. 2011;109(5):578–592. doi:10.1161/CIRCRESAHA.111.247965
12. Schurgers LJ, Barreto DV, Barreto FC, et al. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: a preliminary report. Clin J Am Soc Nephrol. 2010;5(4):568–575. doi:10.2215/CJN.07081009
13. Smith ER, Ford ML, Tomlinson LA, Rajkumar C, McMahon LP, Holt SG. Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol Dial Transplant. 2012;27(5):1957–1966. doi:10.1093/ndt/gfr609
14. Shroff RC, McNair R, Figg N, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118(17):1748–1757. doi:10.1161/CIRCULATIONAHA.108.783738
15. Yamada S, Giachelli CM. Vascular calcification in CKD-MBD: roles for phosphate, FGF23, and Klotho. Bone. 2017;100:87–93. doi:10.1016/j.bone.2016.11.012
16. Henaut L, Mary A, Chillon JM, Kamel S, Massy ZA. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins (Basel). 2018;10(6):218. doi:10.3390/toxins10060218
17. Opdebeeck B, Maudsley S, Azmi A, et al. Indoxyl sulfate and p-Cresyl sulfate promote vascular calcification and associate with glucose intolerance. J Am Soc Nephrol. 2019;30(5):751–766. doi:10.1681/ASN.2018060609
18. Disthabanchong S. Vascular calcification in chronic kidney disease: pathogenesis and clinical implication. World J Nephrol. 2012;1(2):43–53. doi:10.5527/wjn.v1.i2.43
19. Sanchis P, Ho CY, Liu Y, et al. Arterial “inflammaging” drives vascular calcification in children on dialysis. Kidney Int. 2019;95(4):958–972. doi:10.1016/j.kint.2018.12.014
20. Frauscher B, Kirsch AH, Schabhuttl C, et al. Autophagy protects from uremic vascular media calcification. Front Immunol. 2018;9:1866. doi:10.3389/fimmu.2018.01866
21. Lee K, Kim H, Jeong D. Microtubule stabilization attenuates vascular calcification through the inhibition of osteogenic signaling and matrix vesicle release. Biochem Biophys Res Commun. 2014;451(3):436–441. doi:10.1016/j.bbrc.2014.08.007
22. Ma WQ, Sun XJ, Wang Y, Zhu Y, Han XQ, Liu NF. Restoring mitochondrial biogenesis with metformin attenuates beta-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis. Mol Cell Endocrinol. 2019;479:39–53. doi:10.1016/j.mce.2018.08.012
23. Yao L, Wang J, Tian BY, Xu TH, Sheng ZT. Activation of the Nrf2-ARE signaling pathway prevents hyperphosphatemia-induced vascular calcification by inducing autophagy in renal vascular smooth muscle cells. J Cell Biochem. 2017;118(12):4708–4715. doi:10.1002/jcb.26137
24. Panda DK, Bai X, Sabbagh Y, et al. Defective interplay between mTORC1 activity and endoplasmic reticulum stress-unfolded protein response in uremic vascular calcification. Am J Physiol Renal Physiol. 2018;314(6):F1046–f61. doi:10.1152/ajprenal.00350.2017
25. Lee K, Kim H, Jeong D. Protein kinase C regulates vascular calcification via cytoskeleton reorganization and osteogenic signaling. Biochem Biophys Res Commun. 2014;453(4):793–797. doi:10.1016/j.bbrc.2014.10.026
26. Hortells L, Sosa C, Guillen N, Lucea S, Millan A, Sorribas V. Identifying early pathogenic events during vascular calcification in uremic rats. Kidney Int. 2017;92(6):1384–1394. doi:10.1016/j.kint.2017.06.019
27. Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol. 2014;307(8):F891–F900. doi:10.1152/ajprenal.00163.2014
28. Lang F, Leibrock C, Pelzl L, et al. Therapeutic interference with vascular calcification-lessons from klotho-hypomorphic mice and beyond. Front Endocrinol (Lausanne). 2018;9:207. doi:10.3389/fendo.2018.00207
29. Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc Res. 2008;77(1):221–230. doi:10.1093/cvr/cvm049
30. Cai T, Sun D, Duan Y, et al. WNT/beta-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. 2016;345(2):206–217. doi:10.1016/j.yexcr.2016.06.007
31. Lee GL, Yeh CC, Wu JY, et al. TLR2 promotes vascular smooth muscle cell chondrogenic differentiation and consequent calcification via the concerted actions of osteoprotegerin suppression and IL-6-Mediated RANKL induction. Arterioscler Thromb Vasc Biol. 2019;39(3):432–445. doi:10.1161/ATVBAHA.118.311874
32. Osako MK, Nakagami H, Shimamura M, et al. Cross-talk of receptor activator of nuclear Factor-κB ligand signaling with renin–angiotensin system in vascular calcification. Arterioscler Thromb Vasc Biol. 2013;33(6):1287–1296. doi:10.1161/ATVBAHA.112.301099
33. Hamano T, Matsui I, Mikami S, et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol. 2010;21(11):1998–2007. doi:10.1681/ASN.2009090944
34. Aghagolzadeh P, Bachtler M, Bijarnia R, et al. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis. 2016;251:404–414. doi:10.1016/j.atherosclerosis.2016.05.044
35. New SE, Aikawa E. Role of extracellular vesicles in de novo mineralization: an additional novel mechanism of cardiovascular calcification. Arterioscler Thromb Vasc Biol. 2013;33(8):1753–1758. doi:10.1161/ATVBAHA.112.300128
36. New SE, Goettsch C, Aikawa M, et al. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. 2013;113(1):72–77. doi:10.1161/CIRCRESAHA.113.301036
37. Viegas C, Araujo N, Marreiros C, Simes D. The interplay between mineral metabolism, vascular calcification and inflammation in Chronic Kidney Disease (CKD): challenging old concepts with new facts. Aging (Albany NY). 2019;11(12):4274–4299. doi:10.18632/aging.102046
38. Gauthier-Bastien A, Ung RV, Lariviere R, Mac-Way F, Lebel M, Agharazii M. Vascular remodeling and media calcification increases arterial stiffness in chronic kidney disease. Clin Exp Hypertens. 2014;36(3):173–180. doi:10.3109/10641963.2013.804541
39. Pai AS, Giachelli CM. Matrix remodeling in vascular calcification associated with chronic kidney disease. J Am Soc Nephrol. 2010;21(10):1637–1640. doi:10.1681/ASN.2010040349
40. Kapustin AN, Davies JD, Reynolds JL, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109(1):e1–e12. doi:10.1161/CIRCRESAHA.110.238808
41. Beazley KE, Nurminsky D, Lima F, Gandhi C, Nurminskaya MV. Wnt16 attenuates TGFbeta-induced chondrogenic transformation in vascular smooth muscle. Arterioscler Thromb Vasc Biol. 2015;35(3):573–579. doi:10.1161/ATVBAHA.114.304393
42. Chavkin NW, Chia JJ, Crouthamel MH, Giachelli CM. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp Cell Res. 2015;333(1):39–48. doi:10.1016/j.yexcr.2015.02.002
43. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109(6):697–711. doi:10.1161/CIRCRESAHA.110.234914
44. Ciceri P, Galassi A, Alfieri C, Messa P, Cozzolino M. Uremic patients with increased vascular calcification score have serum with high calcific potential: role of vascular smooth muscle cell osteoblastic differentiation and apoptosis. Blood Purif. 2019;48(2):142–149. doi:10.1159/000497229
45. Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52(10):843–850. doi:10.1016/j.jacc.2008.05.043
46. Yamada S, Leaf EM, Chia JJ, Cox TC, Speer MY, Giachelli CM. PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int. 2018;94(4):716–727. doi:10.1016/j.kint.2018.05.015
47. Zhang D, Bi X, Liu Y, et al. High phosphate-induced calcification of vascular smooth muscle cells is associated with the TLR4/NF-kappab signaling pathway. Kidney Blood Press Res. 2017;42(6):1205–1215. doi:10.1159/000485874
48. Voelkl J, Luong TT, Tuffaha R, et al. SGK1 induces vascular smooth muscle cell calcification through NF-kappaB signaling. J Clin Invest. 2018;128(7):3024–3040. doi:10.1172/JCI96477
49. Belmokhtar K, Ortillon J, Jaisson S, et al. Receptor for advanced glycation end products: a key molecule in the genesis of chronic kidney disease vascular calcification and a potential modulator of sodium phosphate co-transporter PIT-1 expression. Nephrol Dial Transplant. 2019;34(12):2018–2030. doi:10.1093/ndt/gfz012
50. Cianciolo G, Capelli I, Cappuccilli M, Schillaci R, Cozzolino M, La Manna G. Calcifying circulating cells: an uncharted area in the setting of vascular calcification in CKD patients. Clin Kidney J. 2016;9(2):280–286. doi:10.1093/ckj/sfv145
51. Fadini GP, Rattazzi M, Matsumoto T, Asahara T, Khosla S. Emerging role of circulating calcifying cells in the bone-vascular axis. Circulation. 2012;125(22):2772–2781. doi:10.1161/CIRCULATIONAHA.112.090860
52. Soriano S, Carmona A, Trivino F, et al. Endothelial damage and vascular calcification in patients with chronic kidney disease. Am J Physiol Renal Physiol. 2014;307(11):F1302–11. doi:10.1152/ajprenal.00114.2014
53. Shin V, Zebboudj AF, Bostrom K. Endothelial cells modulate osteogenesis in calcifying vascular cells. J Vasc Res. 2004;41(2):193–201. doi:10.1159/000077394
54. Cianciolo G, Capelli I, Cappuccilli M, et al. Is chronic kidney disease-mineral and bone disorder associated with the presence of endothelial progenitor cells with a calcifying phenotype? Clin Kidney J. 2017;10(3):389–396. doi:10.1093/ckj/sfw145
55. Zvaifler NJ, Marinova-Mutafchieva L, Adams G, et al. Mesenchymal precursor cells in the blood of normal individuals. Arthritis Res Ther. 2000;2(6):477–488. doi:10.1186/ar130
56. Otsuru S, Tamai K, Yamazaki T, Yoshikawa H, Kaneda Y. Bone marrow-derived osteoblast progenitor cells in circulating blood contribute to ectopic bone formation in mice. Biochem Biophys Res Commun. 2007;354(2):453–458. doi:10.1016/j.bbrc.2006.12.226
57. Otsuru S, Tamai K, Yamazaki T, Yoshikawa H, Kaneda Y. Circulating bone marrow‐derived osteoblast progenitor cells are recruited to the bone‐forming site by the CXCR4/stromal cell‐derived factor‐1 pathway. Stem Cells. 2008;26(1):223–234. doi:10.1634/stemcells.2007-0515
58. Cianciolo G, La Manna G, Della Bella E, et al. Effect of vitamin D receptor activator therapy on vitamin D receptor and osteocalcin expression in circulating endothelial progenitor cells of hemodialysis patients. Blood Purif. 2013;35(1–3):187–195. doi:10.1159/000347102
59. Fadini GP, Albiero M, Menegazzo L, et al. Widespread increase in myeloid calcifying cells contributes to ectopic vascular calcification in type 2 diabetes. Circ Res. 2011;108(9):1112–1121. doi:10.1161/CIRCRESAHA.110.234088
60. Bardeesi ASA, Gao J, Zhang K, et al. A novel role of cellular interactions in vascular calcification. J Transl Med. 2017;15(1):95. doi:10.1186/s12967-017-1190-z
61. El Agha E, Kramann R, Schneider RK, et al. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. 2017;21(2):166–177. doi:10.1016/j.stem.2017.07.011
62. Kramann R, Goettsch C, Wongboonsin J, et al. Adventitial MSC-like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell. 2016;19(5):628–642. doi:10.1016/j.stem.2016.08.001
63. Jovanovich A, Isakova T, Stubbs J. Microbiome and Cardiovascular Disease in CKD. Clin J Am Soc Nephrol. 2018;13(10):1598–1604. doi:10.2215/CJN.12691117
64. Ramezani A, Massy ZA, Meijers B, Evenepoel P, Vanholder R, Raj DS. Role of the Gut Microbiome in Uremia: a Potential Therapeutic Target. Am J Kidney Dis. 2016;67(3):483–498. doi:10.1053/j.ajkd.2015.09.027
65. Mahmoodpoor F, Rahbar Saadat Y, Barzegari A, Ardalan M, Zununi Vahed S. The impact of gut microbiota on kidney function and pathogenesis. Biomed Pharmacother. 2017;93:412–419. doi:10.1016/j.biopha.2017.06.066
66. Xu KY, Xia GH, Lu JQ, et al. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci Rep. 2017;7(1):1445. doi:10.1038/s41598-017-01387-y
67. Stadlbauer V, Horvath A, Ribitsch W, et al. Author Correction: structural and functional differences in gut microbiome composition in patients undergoing haemodialysis or peritoneal dialysis. Sci Rep. 2019;9(1):8522. doi:10.1038/s41598-019-43263-x
68. Nazzal L, Roberts J, Singh P, et al. Microbiome perturbation by oral vancomycin reduces plasma concentration of two gut-derived uremic solutes, indoxyl sulfate and p-cresyl sulfate, in end-stage renal disease. Nephrol Dial Transplant. 2017;32(11):1809–1817. doi:10.1093/ndt/gfx029
69. Ramezani A, Raj DS. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol. 2014;25(4):657–670. doi:10.1681/ASN.2013080905
70. Riccio E, Sabbatini M, Bruzzese D, et al. Plasma p-cresol lowering effect of sevelamer in non-dialysis CKD patients: evidence from a randomized controlled trial. Clin Exp Nephrol. 2018;22(3):529–538. doi:10.1007/s10157-017-1504-8
71. Andersen K, Kesper MS, Marschner JA, et al. Intestinal dysbiosis, barrier dysfunction, and bacterial translocation account for CKD-related systemic inflammation. J Am Soc Nephrol. 2017;28(1):76–83. doi:10.1681/ASN.2015111285
72. Lau WL, Savoj J, Nakata MB, Vaziri ND. Altered microbiome in chronic kidney disease: systemic effects of gut-derived uremic toxins. Clin Sci (Lond). 2018;132(5):509–522. doi:10.1042/CS20171107
73. Barreto FC, Barreto DV, Liabeuf S, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4(10):1551–1558. doi:10.2215/CJN.03980609
74. Lin CJ, Liu HL, Pan CF, et al. Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch Med Res. 2012;43(6):451–456. doi:10.1016/j.arcmed.2012.08.002
75. Asami M, Tanabe K, Ito S, et al. Impact of indoxyl sulfate on coronary plaques in patients on hemodialysis. Int Heart J. 2018;59(3):489–496. doi:10.1536/ihj.17-351
76. Yamamoto H, Tsuruoka S, Ioka T, et al. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006;69(10):1780–1785. doi:10.1038/sj.ki.5000340
77. Yisireyili M, Saito S, Abudureyimu S, et al. Indoxyl sulfate-induced activation of (pro)renin receptor promotes cell proliferation and tissue factor expression in vascular smooth muscle cells. PLoS One. 2014;9(10):e109268. doi:10.1371/journal.pone.0109268
78. Azechi T, Sato F, Sudo R, Wachi H. 5-aza-2ʹ-Deoxycytidine, a DNA methyltransferase inhibitor, facilitates the inorganic phosphorus-induced mineralization of vascular smooth muscle cells. J Atheroscler Thromb. 2014;21(5):463–476. doi:10.5551/jat.20818
79. Xie SA, Zhang T, Wang J, et al. Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: role of DNA methyltransferase 1. Biomaterials. 2018;155:203–216. doi:10.1016/j.biomaterials.2017.11.033
80. Chen J, Zhang X, Zhang H, et al. Indoxyl sulfate enhance the hypermethylation of klotho and promote the process of vascular calcification in chronic kidney disease. Int J Biol Sci. 2016;12(10):1236–1246. doi:10.7150/ijbs.15195
81. Montes de Oca A, Madueno JA, Martinez-Moreno JM, et al. High-phosphate-induced calcification is related to SM22alpha promoter methylation in vascular smooth muscle cells. J Bone Miner Res. 2010;25(9):1996–2005. doi:10.1002/jbmr.93
82. Hou YC, Lu CL, Yuan TH, Liao MT, Chao CT, Lu KC. The epigenetic landscape of vascular calcification: an integrative perspective. Int J Mol Sci. 2020;21(3):980. doi:10.3390/ijms21030980
83. Fakhry M, Skafi N, Fayyad-Kazan M, et al. Characterization and assessment of potential microRNAs involved in phosphate-induced aortic calcification. J Cell Physiol. 2018;233(5):4056–4067. doi:10.1002/jcp.26121
84. Massy ZA, Metzinger-le Meuth V, Metzinger L. MicroRNAs are associated with uremic toxicity, cardiovascular calcification, and disease. Contrib Nephrol. 2017;189:160–168.
85. Xu TH, Qiu XB, Sheng ZT, et al. Restoration of microRNA-30b expression alleviates vascular calcification through the mTOR signaling pathway and autophagy. J Cell Physiol. 2019;234(8):14306–14318. doi:10.1002/jcp.28130
86. Panizo S, Naves-Diaz M, Carrillo-Lopez N, et al. MicroRNAs 29b, 133b, and 211 regulate vascular smooth muscle calcification mediated by high phosphorus. J Am Soc Nephrol. 2016;27(3):824–834. doi:10.1681/ASN.2014050520
87. Zhang H, Chen J, Shen Z, et al. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/beta-catenin signaling. Toxicol Lett. 2018;284:29–36. doi:10.1016/j.toxlet.2017.11.033
88. Wen P, Cao H, Fang L, et al. miR-125b/Ets1 axis regulates transdifferentiation and calcification of vascular smooth muscle cells in a high-phosphate environment. Exp Cell Res. 2014;322(2):302–312. doi:10.1016/j.yexcr.2014.01.025
89. Kétszeri M, Kirsch A, Frauscher B, et al. MicroRNA-142-3p improves vascular relaxation in uremia. Atherosclerosis. 2019;280:28–36. doi:10.1016/j.atherosclerosis.2018.11.024
90. Fourdinier O, Schepers E, Metzinger-Le Meuth V, et al. Serum levels of miR-126 and miR-223 and outcomes in chronic kidney disease patients. Sci Rep. 2019;9(1):4477. doi:10.1038/s41598-019-41101-8
91. Lee CT, Lee YT, Tain YL, Ng HY, Kuo WH. Circulating microRNAs and vascular calcification in hemodialysis patients. J Int Med Res. 2019;47(7):2929–2939. doi:10.1177/0300060519848949
92. Wang CL, Lin KP, Hsu GW, Liu KL, Guo CH. Altered mineral metabolism and disequilibrium between calcification promoters and inhibitors in chronic hemodialysis patients. Biol Trace Elem Res. 2019.
93. Akbari M, Nayeri H, Nasri H. Association of fetuin-A with kidney disease; a review on current concepts and new data. J Nephropharmacol. 2019;8(2).
94. Dai L, Qureshi AR, Witasp A, Lindholm B, Stenvinkel P. Early vascular ageing and cellular senescence in chronic kidney disease. Comput Struct Biotechnol J. 2019;17:721–729. doi:10.1016/j.csbj.2019.06.015
95. Maréchal C, Schlieper G, Nguyen P, et al. Serum fetuin-A levels are associated with vascular calcifications and predict cardiovascular events in renal transplant recipients. Clin J Am Soc Nephrol. 2011;6(5):974–985. doi:10.2215/CJN.06150710
96. Koca N, Ersoy A, Şensoy B, et al. The association between cardiac valvular calcification and fetuin-A levels in kidney transplant recipients. Clin Exp Nephrol. 2019;23(10):1250–1256. doi:10.1007/s10157-019-01761-2
97. Price PA, Lim JE. The inhibition of calcium phosphate precipitation by fetuin is accompanied by the formation of a fetuin-mineral complex. J Biol Chem. 2003;278(24):22144–22152. doi:10.1074/jbc.M300744200
98. Holt SG, Smith ER. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrol Dial Transplant. 2016;31(10):1583–1587. doi:10.1093/ndt/gfw048
99. Ter Braake AD, Shanahan CM, de Baaij JHF. Magnesium counteracts vascular calcification: passive interference or active modulation? Arterioscler Thromb Vasc Biol. 2017;37(8):1431–1445. doi:10.1161/ATVBAHA.117.309182
100. Montezano AC, Zimmerman D, Yusuf H, et al. Vascular smooth muscle cell differentiation to an osteogenic phenotype involves TRPM7 modulation by magnesium. Hypertension. 2010;56(3):453–462. doi:10.1161/HYPERTENSIONAHA.110.152058
101. Louvet L, Metzinger L, Buchel J, Steppan S, Massy ZA. Magnesium attenuates phosphate-induced deregulation of a microRNA signature and prevents modulation of Smad1 and osterix during the course of vascular calcification. Biomed Res Int. 2016;2016:7419524. doi:10.1155/2016/7419524
102. Montes de Oca A, Guerrero F, Martinez-Moreno JM, et al. Magnesium inhibits Wnt/beta-catenin activity and reverses the osteogenic transformation of vascular smooth muscle cells. PLoS One. 2014;9(2):e89525. doi:10.1371/journal.pone.0089525
103. Altura BM, Altura BT, Carella A, Gebrewold A, Murakawa T, Nishio A. Mg2+-Ca2+ interaction in contractility of vascular smooth muscle: mg2+ versus organic calcium channel blockers on myogenic tone and agonist-induced responsiveness of blood vessels. Can J Physiol Pharmacol. 1987;65(4):729–745. doi:10.1139/y87-120
104. Ter Braake AD, Tinnemans PT, Shanahan CM, Hoenderop JGJ, de Baaij JHF. Magnesium prevents vascular calcification in vitro by inhibition of hydroxyapatite crystal formation. Sci Rep. 2018;8(1):2069. doi:10.1038/s41598-018-20241-3
105. Briet M, Burns KD. Chronic kidney disease and vascular remodelling: molecular mechanisms and clinical implications. Clin Sci (Lond). 2012;123(7):399–416. doi:10.1042/CS20120074
106. Herencia C, Rodriguez-Ortiz ME, Munoz-Castaneda JR, et al. Angiotensin II prevents calcification in vascular smooth muscle cells by enhancing magnesium influx. Eur J Clin Invest. 2015;45(11):1129–1144. doi:10.1111/eci.12517
107. de Borst MH, Vervloet MG. Ter Wee PM, Navis G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. J Am Soc Nephrol. 2011;22(9):1603–1609. doi:10.1681/ASN.2010121251
108. Goldfarb S, Martin K. Disorders of divalent ions, renal bone disease and nephrolithiasis. Nephrol Self Assess Program. 2018;17(3):205–209.
109. Lu KC, Wu CC, Yen JF, Liu WC. Vascular calcification and renal bone disorders. Sci World J. 2014;2014:637065. doi:10.1155/2014/637065
110. Kukida M, Mogi M, Kan-No H, et al. AT2 receptor stimulation inhibits phosphate-induced vascular calcification. Kidney Int. 2019;95(1):138–148. doi:10.1016/j.kint.2018.07.028
111. Wang J, Zhou JJ, Robertson GR, Lee VW. Vitamin D in vascular calcification: a double-edged sword? Nutrients. 2018;10(5):652. doi:10.3390/nu10050652
112. Nitta K, Ogawa T, Hanafusa N, Tsuchiya K. Recent advances in the management of vascular calcification in patients with end-stage renal disease. Contrib Nephrol. 2019;198:62–72.
113. Schantl AE, Verhulst A, Neven E, et al. Inhibition of vascular calcification by inositol phosphates derivatized with ethylene glycol oligomers. Nat Commun. 2020;11(1):721. doi:10.1038/s41467-019-14091-4
114. Schantl AE, Ivarsson ME, Leroux JCJAT. Investigational pharmacological treatments for vascular calcification. Adv Ther. 2019;2(1):1800094. doi:10.1002/adtp.201800094
115. Chao CT, Yeh HY, Tsai YT, et al. Natural and non-natural antioxidative compounds: potential candidates for treatment of vascular calcification. Cell Death Discov. 2019;5:145. doi:10.1038/s41420-019-0225-z
116. Henze LA, Luong TTD, Boehme B, et al. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging (Albany NY). 2019;11(15):5445–5462. doi:10.18632/aging.102130
117. Chen B, Zhao Y, Han D, et al. Wnt1 inhibits vascular smooth muscle cell calcification by promoting ANKH expression. J Mol Cell Cardiol. 2019;135:10–21. doi:10.1016/j.yjmcc.2019.07.008
118. Wei R, Enaka M, Muragaki Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci Rep. 2019;9(1):10366. doi:10.1038/s41598-019-46824-2
119. Li Z, Wu J, Zhang X, et al. CDC42 promotes vascular calcification in chronic kidney disease. J Pathol. 2019;249(4):461–471. doi:10.1002/path.5334
120. Paloian NJ, Leaf EM, Giachelli CM. Osteopontin protects against high phosphate-induced nephrocalcinosis and vascular calcification. Kidney Int. 2016;89(5):1027–1036. doi:10.1016/j.kint.2015.12.046
121. Kuo TH, Lin WH, Chao JY, et al. Serum sclerostin levels are positively related to bone mineral density in peritoneal dialysis patients: a cross-sectional study. BMC Nephrol. 2019;20(1):266. doi:10.1186/s12882-019-1452-5
122. Carracedo M, Witasp A, Qureshi AR, et al. Chemerin inhibits vascular calcification through ChemR23 and is associated with lower coronary calcium in chronic kidney disease. J Intern Med. 2019;286(4):449–457. doi:10.1111/joim.12940
123. Hawkins CL. Protein carbamylation: a key driver of vascular calcification during chronic kidney disease. Kidney Int. 2018;94(1):12–14. doi:10.1016/j.kint.2018.03.022
124. Mori D, Matsui I, Shimomura A, et al. Protein carbamylation exacerbates vascular calcification. Kidney Int. 2018;94(1):72–90. doi:10.1016/j.kint.2018.01.033
125. Kaesler N, Goettsch C, Weis D, et al. Magnesium but not nicotinamide prevents vascular calcification in experimental uraemia. Nephrol Dial Transplant. 2019. doi:10.1093/ndt/gfy410
126. Nagy A, Petho D, Gall T, et al. Zinc Inhibits HIF-Prolyl hydroxylase inhibitor-aggravated VSMC calcification induced by high phosphate. Front Physiol. 2019;10:1584. doi:10.3389/fphys.2019.01584
127. Shin MY, Kwun IS. Zinc restored the decreased vascular smooth muscle cell viability under atherosclerotic calcification conditions. Prev Nutr Food Sci. 2014;19(4):363–366. doi:10.3746/pnf.2014.19.4.363
128. Voelkl J, Tuffaha R, Luong TTD, et al. Zinc inhibits phosphate-induced vascular calcification through TNFAIP3-mediated suppression of NF-kappaB. J Am Soc Nephrol. 2018;29(6):1636–1648. doi:10.1681/ASN.2017050492
129. Liu H, Zhang X, Zhong X, et al. Puerarin inhibits vascular calcification of uremic rats. Eur J Pharmacol. 2019;855:235–243. doi:10.1016/j.ejphar.2019.05.023
130. Gueiros APS, Gueiros JEB, Nobrega KT, et al. Effect of spironolactone on the progression of coronary calcification in peritoneal dialysis patients: a pilot study. J Bras Nefrol. 2019;41(3):345–355. doi:10.1590/2175-8239-jbn-2019-0009
131. Wang P, Quan Z, Luo D, Chen W, Peng D. Spironolactone dose-dependently alleviates the calcification of aortic rings cultured in hyperphosphatemic medium with or without hyperglycemia by suppressing phenotypic transition of VSMCs through downregulation of Pit1. Mol Med Rep. 2019;19(5):3622–3632. doi:10.3892/mmr.2019.10039
132. Agharazii M, St-Louis R, Gautier-Bastien A, et al. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am J Hypertens. 2015;28(6):746–755. doi:10.1093/ajh/hpu225
133. Koike S, Yano S, Tanaka S, Sheikh AM, Nagai A, Sugimoto T. Advanced Glycation End-Products Induce Apoptosis of Vascular Smooth Muscle Cells: A Mechanism for Vascular Calcification. Int J Mol Scip. 2016;17(5):pii: E1567. doi:10.3390/ijms17091567
134. Bisson SK, Ung RV, Picard S, et al. High calcium, phosphate and calcitriol supplementation leads to an osteocyte-like phenotype in calcified vessels and bone mineralisation defect in uremic rats. J Bone Miner Metab. 2019;37(2):212–223. doi:10.1007/s00774-018-0919-y
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.