Back to Journals » Vascular Health and Risk Management » Volume 17
Variable and Severe Phenotypic Expression of the “Lebanese Allele” in Two Sisters with Familial Hypercholesterolemia
Authors Chahine J, Kreykes S, Van't Hof JR ![]() , Duprez D
, Duprez D ![]() , Nijjar P
, Nijjar P ![]()
Received 13 April 2021
Accepted for publication 15 July 2021
Published 21 July 2021 Volume 2021:17 Pages 415—419
DOI https://doi.org/10.2147/VHRM.S314704
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Pietro Scicchitano
Johnny Chahine, Sarah Kreykes, Jeremy R Van’t Hof, Daniel Duprez, Prabhjot Nijjar
Cardiovascular Division, Department of Medicine, University of Minnesota Medical School, Minneapolis, MN, USA
Correspondence: Prabhjot Nijjar
University of Minnesota Medical School, 420 Delaware Street SE, MMC 508, Minneapolis, MN, 55455, USA
Tel +1 612 626-1391
Fax +1 612 626-4411
Email [email protected]
Abstract: The “Lebanese allele” {LDLR c.2043 C>A (p.cys681X)} is a nonsense mutation in the low-density lipoprotein receptor (LDLR) gene that results in a truncated non-functioning LDLR protein. We report two sisters of Lebanese descent who presented with familial hypercholesterolemia (FH) and were both heterozygous for the Lebanese allele, but had very distinct LDL-C levels and clinical phenotypes. Whereas one of the sisters had LDL-C in the expected range of Heterozygous FH (HeFH) with the Lebanese allele (LDL-C of 292 mg/dl), the other sister had a more severe LDL-C phenotype in the Homozygous FH (HoFH) range (LDL-C of 520 mg/dl) along with manifest atherosclerosis. Surprisingly, she did not demonstrate a compound heterozygote or double heterozygote status. We discuss different mechanisms that are purported to play a role in modifying the phenotype of FH, including different variants and polygenic modifiers. HeFH patients with the Lebanese allele can have a wide spectrum of LDL-C levels that range from the typical heterozygous to homozygous phenotypes.
Keywords: familial hypercholesterolemia, familial hyperlipidemia, heterozygous, Lebanese allele
Introduction
Familial hypercholesterolemia (FH) is a monogenic disorder resulting in severely elevated LDL-cholesterol (LDL-C) and increased risk of premature coronary artery disease (CAD). In its classic iteration, FH is inherited in an autosomal dominant pattern. HeFH results from a single pathogenic gene variant. A more severe form of the disease, HoFH, results from biallelic pathogenic variants, ie, one mutation from each parent. Although FH is vastly underdiagnosed with only 10% of cases diagnosed, the prevalence of HeFH is estimated to be around 0.4% (1 in 250 people) worldwide.1,2 However, it is reported to be 10 times higher in Lebanon.3 The “Lebanese allele”, named because of its high prevalence in individuals of Lebanese descent (a founder mutation), is responsible for about 82% of FH cases in Lebanon.3 Since its early description, it has also been reported in patients from other countries, with many but not all being of Lebanese origin.4 It is a nonsense mutation in the low-density lipoprotein receptor (LDLR) gene, with substitution of cytosine by adenine at the nucleotide 2043 (c.2043 C>A), creating a premature stop codon.3 This results in a truncated non-functioning LDLR and decreased LDL-C clearance from the blood by the liver. The truncated protein does not include exon 15 or beyond, and therefore lacks the clustered O-linked sugars, membrane spanning region and a cytoplasmic tail. We present two sisters of Lebanese descent who presented with FH, and were both heterozygous for the Lebanese allele, but had very distinct LDL-C levels and clinical phenotypes. Informed consent was obtained from both patients for case publication and institutional approval was not required to publish the case details.
Case Report
Case 1: A 56-year-old female of Lebanese descent was referred to the Preventive Cardiology Clinic at the University of Minnesota for an abnormal coronary calcium scan. It showed diffuse severe coronary calcifications with an Agatston score of 441 (Figure 1). Her medical history was notable for smoking with a 20-pack year history and hyperlipidemia diagnosed at age 27. The patient was prescribed atorvastatin in the past which she stopped, and now had been off treatment for >15 years. She reported a significant family history of hyperlipidemia and premature CAD. She was born of non-consanguineous parents of Lebanese origin. One brother died of a heart attack at age 39, and another brother had an ischemic stroke. Her mother, multiple siblings, and one of her 3 children have known dyslipidemia (Figure 2 shows the family pedigree tree). Her lipid panel showed very severe hyperlipidemia with a total cholesterol (TC) of 630 mg/dl [normal < 200], LDL-C of 520 mg/dl [normal <100], high-density lipoprotein cholesterol (HDL-C) of 72 mg/dl [normal >49], and triglycerides (TG) of 188 mg/dl [normal <150]. Lipoprotein(a) (Lp(a)) was normal at 17 mg/dL [normal <29]. Her exam was unremarkable and showed no stigmata of cholesterol deposits (ie, no xanthomas, corneal arcus, or xanthelasmas). She was not on any specific diet. Carotid ultrasound showed moderate atherosclerotic plaque without significant stenosis. She was started on rosuvastatin 40 mg daily and ezetimibe 10 mg daily, and was advised to stop smoking with nicotine patch assistance. She scored 11 points (LDL-C >330 mg/dl – 8 points, premature CAD – 2 points, first degree relative with premature CAD – 1 point) according to the Dutch Lipid Clinic FH criteria classifying her as Definite FH,5 and was referred for genetic testing. Based on her intermediate phenotype with severely elevated LDL-C levels and middle-aged CAD presentation, compound or double heterozygosity was suspected. Interestingly, genetic testing showed a single allele mutation (c.2043 C>A) in the LDLR gene Exon 14 (also known as p.Cys681*). No other variants (pathogenic, likely pathogenic, or of uncertain significance) were identified in the 4 genes that were tested. After 3 months of therapy and smoking cessation, her TC was 266 mg/dL, LDL-C of 141 mg/dL, HDL-C of 91 mg/DL, and TG of 170 mg/dL. Evolocumab 140 mg twice monthly was added to her regimen, and a lipid panel after 4 months of combination therapy showed a TC of 189 mg/dL, LDL-C of 79 mg/dL, HDL-C of 91 mg/dL, and TG of 96 mg/dL.
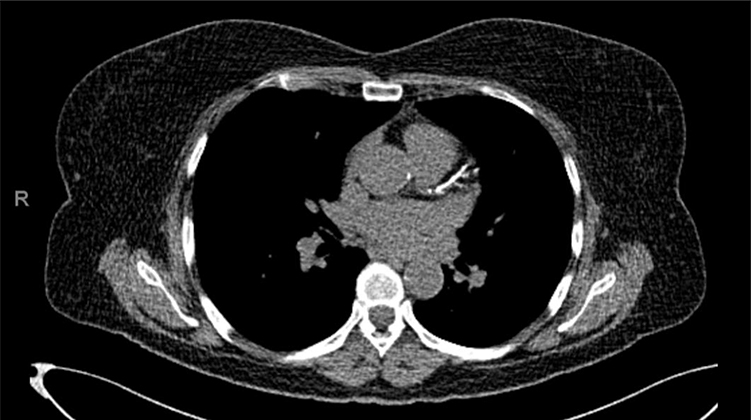 |
Figure 1 Coronary calcium scan showing diffuse severe coronary calcifications. |
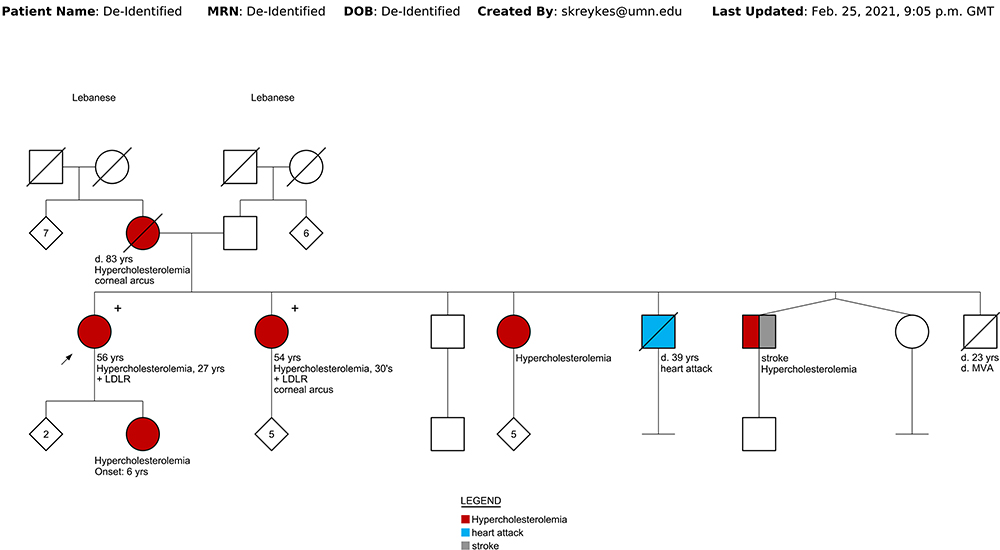 |
Figure 2 Family pedigree tree. |
Case 2: Coincidentally, her sister was referred independently to the same clinic for uncontrolled hyperlipidemia. She was a 54-year-old female with hyperlipidemia diagnosed 20 years ago. A lipid panel showed a TC of 423 mg/dL, LDL-C of 292 mg/dL, HDL-C of 59 mg/dL and TG of 360 mg/dL. Lp(a) was mildly elevated at 46 mg/dL. There was no other relevant medical history other than mild obesity with body mass index of 31, and she had no clinical atherosclerotic disease. She had been treated with atorvastatin in the past, and she reported an intolerance to ezetimibe. Her exam was remarkable for presence of corneal arcus. She was not on any specific diet. She scored 10 points according to the Dutch Lipid Clinic FH criteria (LDL-C of 250–329 mg/dl – 5 points, corneal arcus – 4 points, first degree relative with premature CAD – 1 point) classifying her as Definite FH and was referred for genetic testing. Genetic testing showed a single allele mutation (c.2043 C>A) in the LDLR gene, same as her sister, with no other variants identified. She was started on rosuvastatin 40 mg daily with rapid addition of Evolocumab 140 mg twice monthly. Lipid panel after 6 months of combination therapy showed a TC of 263 mg/dL, LDL-C of 137 mg/dL, HDL-C of 73 mg/dL, TG of 264 mg/dL, and Lp(a) of 31 mg/dL.
Genetic Testing: Next generation sequencing was performed per the Invitae FH Panel (Invitae, San Francisco, CA). The following genes commonly implicated in FH were analyzed: LDL-R, apolipoprotein B (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), in addition to low density lipoprotein receptor adaptor protein 1 (LDLRAP1) that causes autosomal recessive hypercholesterolemia.6 Genomic DNA was obtained from a blood sample for case 1 and saliva sample for case 2. Per Invitae, DNA material is enriched for targeted regions using hybridization-based protocols and sequenced using Illumina technology. Deletions and duplications are called using an in-house algorithm to look at copy number. All targeted regions are sequenced with ≥50x depth or are supplemented with additional analysis. Reads are aligned to a reference sequence (GRCh37), and sequence changes are identified and interpreted in the context of a single clinically relevant transcript (APOB transcript NM_000384.2; LDLR transcript NM_000527.4; PCSK9 transcript NM_174936.3; and LDLRAP1 transcript NM_015627.2). Enrichment and analysis focus on the coding sequence of the indicated transcripts, 10bp of flanking intronic sequence, and other specific genomic regions demonstrated to be causative of disease at the time of assay design. Promoters, untranslated regions, and other non-coding regions are not otherwise interrogated. All clinically significant observations are confirmed by orthogonal technologies, except individually validated variants and variants previously confirmed in a first-degree relative. Confirmation technologies include any of the following: Sanger sequencing, Pacific Biosciences SMRT sequencing, MLPA, MLPA-seq, Array CGH. Technical component of confirmatory sequencing is performed by Invitae Corporation (1400 16th Street, San Francisco, CA 94103, #05D2040778).
Discussion
This case highlights 2 important issues in FH:
- Different phenotype with same genotype: Despite having the same pathogenic LDLR variant, the 2 sisters had very distinct LDL-C and clinical phenotypes. As we discuss in the following section, polygenic variants and environmental factors are purported to play a role in modifying the phenotype of monogenic FH.
- Extreme hypercholesterolemia with heterozygous Lebanese allele: Though one of the sisters had LDL-C in the expected range of HeFH with the Lebanese allele, the other sister had a more severe LDL-C phenotype in the HoFH range. Surprisingly, she did not demonstrate compound heterozygote or double heterozygote status. Of note, her response to a statin is in line with heterozygous status. Because the Lebanese allele results in a null receptor function, homozygous status would show none to minimal response to a statin. In a study that included 44 families and 51 patients with FH that were heterozygous for the Lebanese allele, the mean LDL-C was 297 ± 73 mg/dL.7
Genetic testing: Genetic testing is recommended for all patients suspected to have FH based on clinical criteria like the Dutch Lipid Clinic FH criteria, as well as cascade screening to identify at-risk family members.6 The 3 primary genes implicated in FH are LDLR, APOB and PCSK9. Pathogenic variants in LDLR are by far the most common, accounting for >90% of mutations. In addition to the 3 primary genes, other gene loci have also been identified though pathogenic variants related to these are very rare (<1%), and these are not routinely analyzed in the commercially available panels. A pathogenic variant in 1 of the 3 primary genes is identified in 40–50% of all suspected patients, with a higher yield in those with a higher clinical score.6 Identifying a pathogenic variant is not necessary for the diagnosis of FH, as it may be related to a gene that was not analyzed or is not yet known, or a variant that is rare and of unknown significance at the present time. A significant proportion of patients with severe hypercholesterolemia that overlaps with the clinical FH phenotype, do not have an identifiable monogenic pathogenic variant and instead have a polygenic etiology. Polygenic hypercholesterolemia results from an accumulation of several small-effect, LDL-C raising single-nucleotide polymorphisms (SNP) that cumulatively raise LDL-C levels.8 More than 50 such genomic loci have been identified.9 Different polygenic risk scores have been developed and proposed, though currently there is no consensus about the loci and alleles to include.10 These risk scores are not readily available or used in routine clinical practice. Khera et al found that, among patients with LDL-C levels >190 mg/dL, those with identified gene mutations for FH have a >3-fold higher risk of CAD compared to those without an identifiable mutation.13 Since FH is present at birth, it results in a longer exposure time to high levels of LDL-C, compared to someone with acquired hypercholesterolemia or exposure to lower levels of LDL-C with polygenic hypercholesterolemia. However, the presence of a high polygenic risk score in patients with monogenic FH is associated with a 3-time higher risk of CAD compared to those with a low score.14
Variation in HeFH phenotype: HeFH mutation carriers show a relatively wide range of LDL-C levels,11,12 which can be due to multiple mechanisms:
- Underlying mutation: LDLR mutations cause a more severe phenotype than APOB or PCSK9 mutations. Among LDLR mutations, LDLR missense variants leading to a defective LDLR cause less severe LDL-C elevation than variants that inactivate LDLR completely, such as a nonsense or prematurely truncating variant.
- Polygenic etiology: In addition to being recognized as a common cause of severe hypercholesterolemia without an identified pathogenic variant, these polygenic SNPs can also modify the phenotype of monogenic FH. Lower LDL-C levels in the non-index family members with the pathogenic variant can be from reduced penetrance or presence of additional variants associated with lower LDL-C levels. In a study of Lebanese FH patients with pathogenic LDLR variants, those carrying an additional polymorphism of the PCSK9 gene (p.Leu21dup) had lower LDL-C levels compared to those that did not (261.6± 68.8 vs 310.9± 70.3 mg/dL, p=0.029).7
- Compound and double heterozygotes: Compound heterozygous FH results from the presence of two different mutant alleles at the same gene locus, for eg having 1 mutant allele each for 2 different LDLR mutations. Double heterozygous FH results from the presence of two different mutant alleles at two separate gene loci, for eg having 1 mutant allele each for LDLR and PCSK9. Compound heterozygotes and double heterozygotes can produce an LDL-C phenotype that overlaps with HoFH (presence of two similar mutant alleles at the same gene locus), or can be intermediate between HeFH and HoFH. Homozygous or compound heterozygous LDLR patients have a mean LDL-C >500 mg/dL.
Management: The International Atherosclerosis Society consensus document defines severe FH as LDL-C >400 mg/dL, clinical CAD, or advanced subclinical CAD (CAC score > 100).15 Guidelines recommend treatment aimed to lower LDL-C <100 mg/dL in adults, and <70 mg/dL in adults with clinical CAD.16
Conclusion
HeFH patients with the c.2043 C>A mutation in LDLR, also known as the Lebanese allele, can have a wide spectrum of LDL-C levels that range from the typical heterozygous to homozygous phenotypes. Other genetic and environmental factors may play a role in the phenotypic expression of this specific genotype.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. de Ferranti SD, Rodday AM, Mendelson MM, Wong JB, Leslie LK, Sheldrick RC. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation. 2016;133(11):1067–1072. doi:10.1161/CIRCULATIONAHA.115.018791
2. Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J. 2016;37(17):1384–1394. doi:10.1093/eurheartj/ehw028
3. Lehrman MA, Schneider WJ, Brown MS, et al. The Lebanese allele at the low density lipoprotein receptor locus. Nonsense mutation produces truncated receptor that is retained in endoplasmic reticulum. J Biol Chem. 1987;262(1):401–410. doi:10.1016/S0021-9258(19)75941-9
4. Fahed AC, Bitar FF, Khalaf RI, Moubarak EM, Azar ST, Nemer GM. The Lebanese allele at the LDLR in normocholesterolemic people merits reconsideration of genotype phenotype correlations in familial hypercholesterolemia. Endocrine. 2012;42(2):445–448. doi:10.1007/s12020-012-9669-0
5. McGowan MP, Hosseini Dehkordi SH, Moriarty PM, Duell PB. Diagnosis and treatment of heterozygous familial hypercholesterolemia. J Am Heart Assoc. 2019;8(24):e013225. doi:10.1161/JAHA.119.013225
6. Sturm AC, Knowles JW, Gidding SS, et al.; Convened by the Familial Hypercholesterolemia F. Clinical genetic testing for familial hypercholesterolemia: JACC scientific expert panel. J Am Coll Cardiol. 2018;72(6):662–680. doi:10.1016/j.jacc.2018.05.044
7. Abifadel M, Rabes JP, Jambart S, et al. The molecular basis of familial hypercholesterolemia in Lebanon: spectrum of LDLR mutations and role of PCSK9 as a modifier gene. Hum Mutat. 2009;30(7):E682–91. doi:10.1002/humu.21002
8. Wang J, Dron JS, Ban MR, et al. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically. Arterioscler Thromb Vasc Biol. 2016;36(12):2439–2445. doi:10.1161/ATVBAHA.116.308027
9. Willer CJ, Schmidt EM, Sengupta S, et al.; Global Lipids Genetics C. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283.
10. Berberich AJ, Hegele RA. The complex molecular genetics of familial hypercholesterolaemia. Nat Rev Cardiol. 2019;16(1):9–20. doi:10.1038/s41569-018-0052-6
11. Foody JM, Vishwanath R. Familial hypercholesterolemia/autosomal dominant hypercholesterolemia: molecular defects, the LDL-C continuum, and gradients of phenotypic severity. J Clin Lipidol. 2016;10(4):970–986. doi:10.1016/j.jacl.2016.04.009
12. Jansen AC, van Wissen S, Defesche JC, Kastelein JJ. Phenotypic variability in familial hypercholesterolaemia: an update. Curr Opin Lipidol. 2002;13(2):165–171. doi:10.1097/00041433-200204000-00008
13. Khera AV, Won HH, Peloso GM, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67(22):2578–2589. doi:10.1016/j.jacc.2016.03.520
14. Trinder M, Li X, DeCastro ML, et al. Risk of premature atherosclerotic disease in patients with monogenic versus polygenic familial hypercholesterolemia. J Am Coll Cardiol. 2019;74(4):512–522. doi:10.1016/j.jacc.2019.05.043
15. Santos RD, Gidding SS, Hegele RA, et al.; International Atherosclerosis Society Severe Familial Hypercholesterolemia P. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the international atherosclerosis society severe familial hypercholesterolemia panel. Lancet Diabetes Endocrinol. 2016;4(10):850–861. doi:10.1016/S2213-8587(16)30041-9
16. Catapano AL, Graham I, De Backer G, et al.; Group ESCSD. ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J. 2016;2016(37):2999–3058. doi:10.1093/eurheartj/ehw272
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.