Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 10
Update on the clinical utility of an RNA interference-based treatment: focus on Patisiran
Received 15 September 2016
Accepted for publication 28 June 2017
Published 10 November 2017 Volume 2017:10 Pages 267—278
DOI https://doi.org/10.2147/PGPM.S87945
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin Bluth
Malak Rizk, Şükrü Tüzmen
Molecular Biology and Genetics Program, Department of Biological Sciences, Faculty of Arts and Sciences, Eastern Mediterranean University (EMU), Famagusta, North Cyprus, Turkey
Abstract: RNA interference (RNAi) is a naturally existing endogenous mechanism for posttranscriptional gene regulation, nowadays commonly utilized for functional characterization of genes and development of potential treatment strategies for diseases. RNAi-based studies for therapy, after being examined for over a decade, are finally in the pipeline for developing a potential treatment for the mutated transthyretin (TTR) gene, which gives rise to a dysfunctional TTR protein. This dysfunctional protein causes TTR amyloidosis (ATTR), an inherited, progressively incapacitating, and often fatal genetic disorder. TTR is a protein produced in the liver, and functions as a carrier for retinol-binding protein and also thyroxine. This protein facilitates the transport of vitamin A around the human body. A mutation or misprint in the code of this protein results in an abnormal folding of the protein. Therefore, not only does the transportation of the vitamin A become disabled, but also there will be formation of clusters called amyloid deposits, which attack the heart and the nerves causing some patients to be unconditionally bound to bed. ATTR is a hereditary autosomal dominant disease with a 50% chance of inheritance by offspring, even with just one of the parents having a single defective allele of this gene. Alnylam Pharmaceuticals worked on the concept of RNAi therapy for years, which led to the introduction of lipid nanoparticles encircling small interfering RNAs. The drug showed extremely positive results since the first trial, and a great percentage of defective protein reduction. This drug was later named Patisiran.
Keywords: RNAi, TTR gene, TTR amyloidosis, siRNA, gene silencing, Patisiran
Introduction
RNAi notion
RNA interference (RNAi) is a posttranscriptional regulation process that has facilitated a revolution in functional genomics, which is quickly expanding into therapeutics market. RNAi is a highly conserved event in eukaryotic organisms, evidently evolved as a defense mechanism against viral infections.1–8
RNAi acts as a naturally occurring switch, which adjusts and controls the expression of specific target mRNA to knock down certain genes. RNAi mechanism is triggered by small double-stranded RNAs (dsRNAs) (<25 nucleotides long) processed from longer RNA duplexes. Upon being introduced into a cell, dsRNA is cleaved by a dicer enzyme into 21- to 23-nucleotide-long molecules called small-interfering RNAs (siRNAs).9 siRNA enables gene silencing by interacting with a protein complex referred to as the RNA-induced silencing complex (Figure 1).10 RNAi facilitated the discovery of gene function in a number of species.2–7 RNAi applications in mammalian cells include functional characterization of genes, study of protein interactions in pathways, and the development of transgenic animals and cell lines.11–13 Additionally, the RNAi mechanism has enormous potential as a therapeutic strategy, which, with the aid of bioinformatics tools, provides a better chance for choosing a drug candidate by producing siRNA and testing for its compatibility with the target gene.14–17 Furthermore, new tools such as the genome-wide reagents used for triggering RNAi and genome-compatible screening can be effectively utilized to rapidly discover gene silencing phenomenon.
 | Figure 1 The RNAi mechanism is a powerful tool for gene silencing in mammalian cells. The siRNA pathway takes place as follows: (A) long dsRNA is cleaved by a member of RNAse III family, dicer, into around 21-nucleotide-long siRNAs. The siRNAs, generated either by (A) dicer cleavage or by (B) synthetic construction, are then introduced into cells, where they integrate into the RISC. Once unwound, the antisense strand of siRNA guides RISC to the mRNA containing its complementary sequence, which triggers the destruction of the target by the endonucleolytic cleavage. Springer Methods Mol Biol. RNAi-based functional pharmacogenomics. 2011;700:271–290, Tuzmen S, Tuzmen P, Arora S, Mousses S. Copyright 2011, with permission of Springer Nature.1 Abbreviations: RNAi, RNA interference; siRNA, small interfering RNA; dsRNA, double-stranded RNA; RISC, RNA-induced silencing complex. |
In this review, our center of attention is on the updates on the clinical utility of an RNAi-based treatment, particularly concentrating on the drug Patisiran.
Potential challenges of RNAi therapeutics
Precision medicine is facilitated by the development and application of new genomic methodologies to quickly characterize genetic sequence variations and gene expression process on a genome-wide scale. Such experiments are typically accompanied by extensive association studies aimed at identifying gene targets or gene sets that predict drug response. The associations are then extensively validated and evaluated for their ability to predict response. Often, the function of these associated genes and their potential role in drug response is unknown. A better understanding of which genes are not only associated with drug response but also causally involved would add tremendous value to pharmacogenomics data, permitting a more intelligent utilization of such genes as diagnostic signatures, and as putative drug targets to improve drug response.1,18
In addition to speeding up the functional validation of gene lists from association-based pharmacogenomics data, high-throughput screening strategies for genome-scale functional analysis can potentially be a more relevant starting point for pharmacogenomics analyses. In such approaches, targets, which are involved in regulating drug response, are identified, and their putative clinical associations with drug response are subsequently validated. With this principle, comprehensive phenotype analysis enables the investigation of a wide range of genetic factors including associations with specific gene and transcript sequence variations, expression of mRNA or protein, protein modifications, and many other genetic and epigenetic variations that are often not used in primary pharmacogenomics analysis. Furthermore, this application can identify functionally relevant genes, which after undergoing in-depth functional validation may also have utility as drug targets. Hence, there is clearly an unmet need to develop and apply genome-compatible strategies and technologies to identify functional modulators of drug response.1,18
Although siRNAs are powerful silencers of gene regulation and have aided in our understanding of gene function in numerous cell lines and organisms, several challenges of the siRNA technology remain to be addressed. Specifically, the main problems related to RNAi analysis involve sequence specificity, delivery, and off-target effects. Based on the nature of these limitations, development of RNAi-based therapeutics should still be regarded to be premature.
Concise history and present therapeutic applications of RNAi in clinical trials
Song et al were the first to demonstrate the in vivo feasibility of RNAi-based therapeutic efficacy in a mouse disease model called fulminant hepatitis in the year 2003 targeting Fas gene (Tnfrsf6).19 Since then, siRNAs have been promoted as attractive therapeutic modalities. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle was performed by Davis in the year 2009.20 This nanoparticle formulation of siRNA consisted of a cyclodextrin-containing polymer, a polyethyleneglycol steric stabilization agent, and human transferrin as a targeting ligand for binding to transferrin receptor, which are typically upregulated on cancer cells.20
In 2015, Lorenzer et al illustrated the information collated in Table 121 which demonstrates the selection of ongoing clinical trials of siRNA agents. The information presented in Table 1 also illustrates the delivery systems and chemical modifications. Additionally, administration routes are also listed for demonstration of early developments, and ongoing clinical trials.21,22
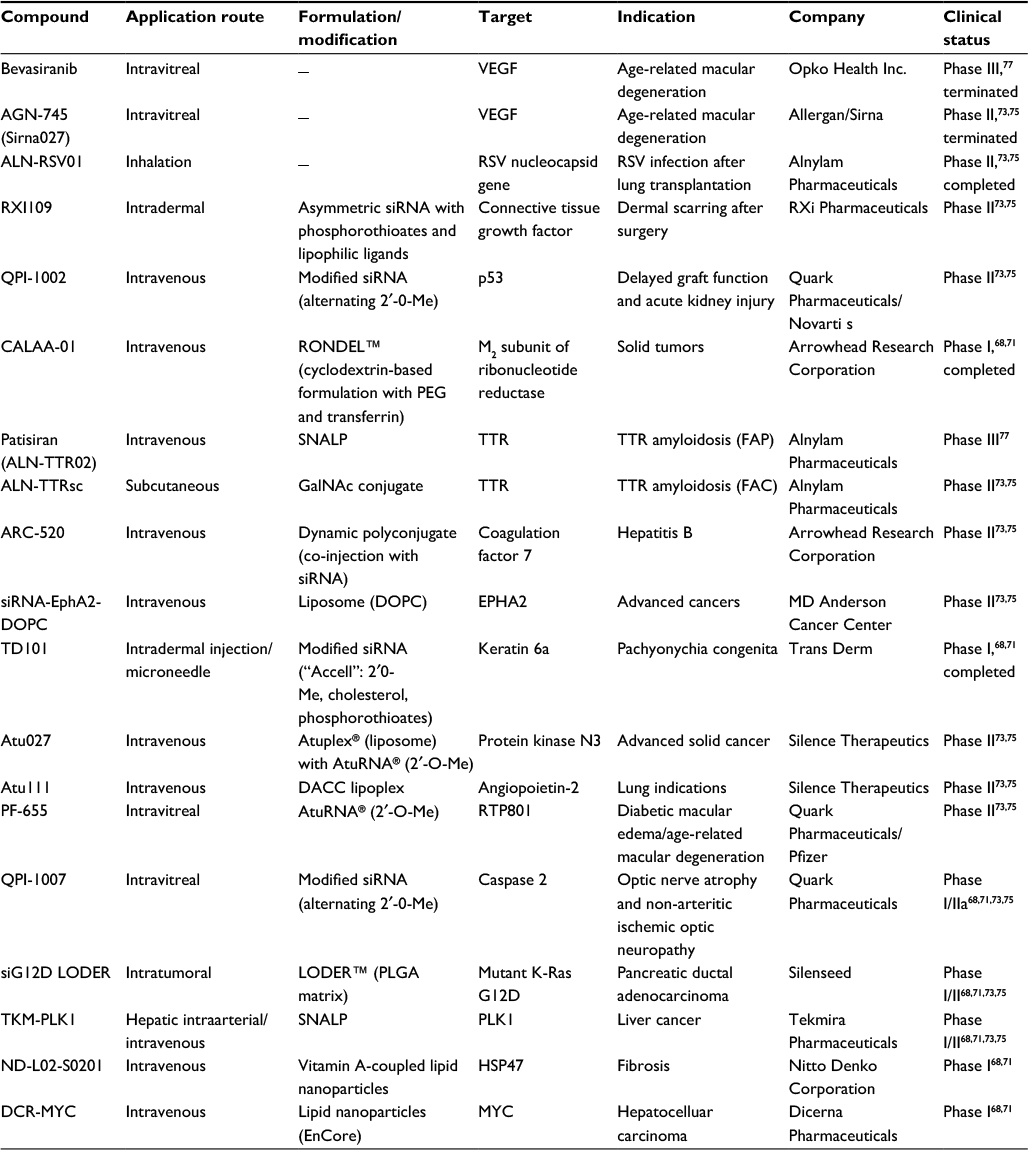 | Table 1 The selection of ongoing clinical trials of siRNA agents. Note: The information presented illustrates the delivery systems and chemical modifications. Additionally, administration routes are also listed for demonstration of early developments, and ongoing trials. Adapted from Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. Going beyond the liver: progress and challenges of targeted delivery of siRNA therapeutics. J Control Release. 2015;203:1–15.21 Abbreviations: siRNA, small interfering RNA; PEG, polyethyleneglycol; SNALP, stable nucleic acid lipid particle; TTR, transthyretin; FAP, familial amyloid polyneuropathy; FAC, familial amyloid cardiomyopathy; DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; DACC, D-alanyl-D-alanine carboxypeptidase. |
TTR gene
Transthyretin (TTR) gene acquired its name from “transports thyroxine and retinol” which is approved by the Human Genome Nomenclature Committee (HGNC).25,26 Some of the most common alternative names of the TTR gene are ATTR, TBPA, PALB, and TTHY_Human.25 TTR is a protein-coding gene and is located on the long arm of chromosome 18 (18q12.1) (Figure 2A). Human TTR gene contains four exons, and spans about 7-kb region on chromosome 18. All exons except for exon 1 contain less than 200 bp and encode a signal peptide (Figure 2B).27–30
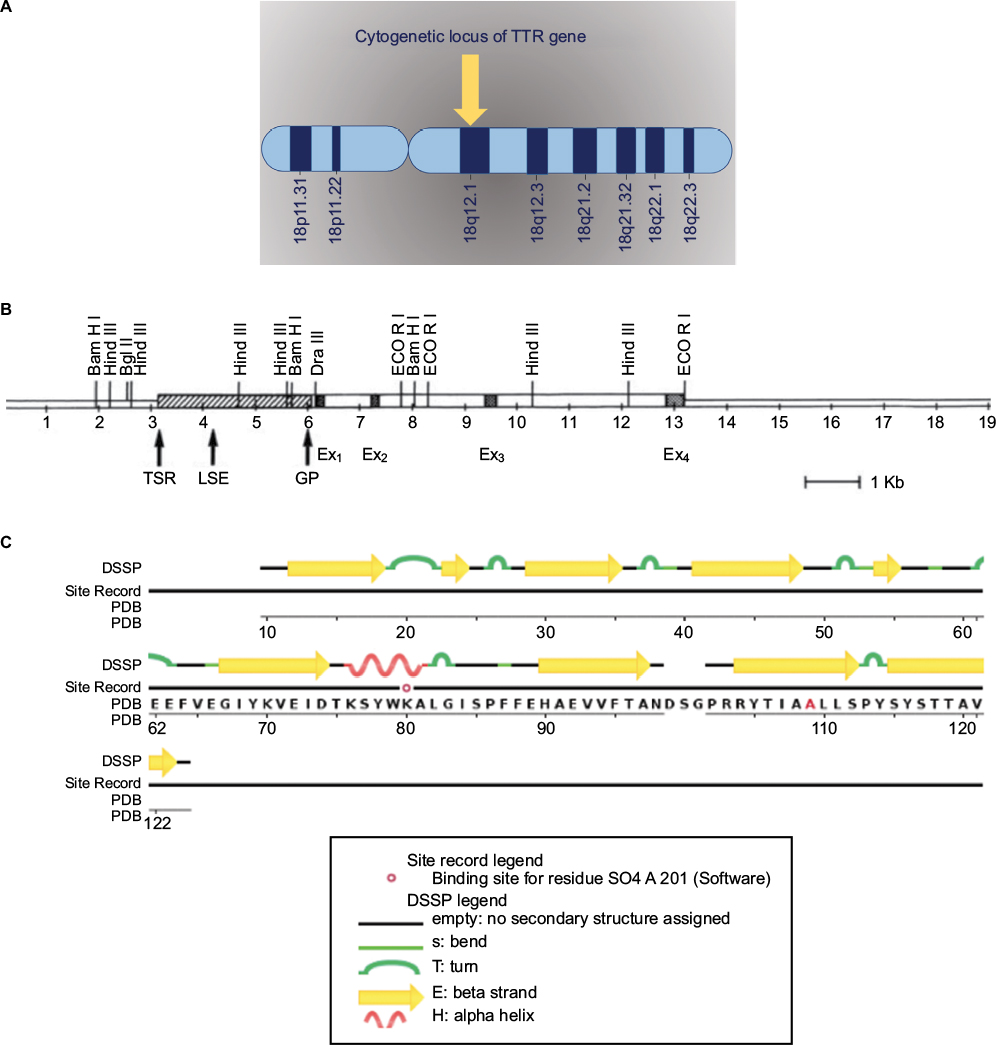 | Figure 2 (A) Cytogenetic locus of TTR gene. (B) The human TTR gene. Reprinted by permission from Macmillan Publishers Ltd: Lab Invest. Teng MH, Yin JY, Vidal R, et al. Amyloid and nonfibrillar deposits in mice transgenic for wild-type human transthyretin: a possible model for senile systemic amyloidosis. Lab Invest. 2001;81(3):385–396. Copyright 2001.88 (C) The human TTR protein structure; adapted from RCSB PDB https://www.rcsb.org/pdb/explore/remediatedSequence.do?structureId=5TZL of PDB ID 5TZL. Kabsch W, Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22(12); 2577–2637.92 Abbreviations: DSSP, database of secondary structure proteins; Ex, exon; GP, general promoter; LSE, liver specific enhancer; PDB, protein databank; TSR, tissue specific regulator; TTR, transthyretin. |
TTR gene provides instructions to produce a protein called TTR protein made of 127 identical amino acids (Figure 2C).23–26 TTR protein is an evolutionary conserved serum and cerebrospinal fluid carrier protein, and is predominantly produced in the liver; however, a small amount of TTR is produced in the part of the brain called the choroid plexus and in the retina.
The liver secretes TTR protein into the bloodstream, and the choroid plexus secretes TTR protein into the cerebrospinal fluid. The inheritance pattern of ATTR disease was first described in the Duncan family during the late 1900s.31 The first attempt in curing the disease was to utilize liver implants because doctors thought that a healthy liver would produce an unaffected version of the protein. However, this method did not have the expected positive results.
Normal allelic variants have been described in individuals of various ethnic backgrounds.27–30 Only one mutation occurs at exon 1 of TTR gene, which codes for amino acids 1–3, while the rest of the mutations are found in the other three exons.27–30 TTR protein also carries the thyroid hormone thyroxine (T4), and retinol-binding protein bound to retinol (vitamin A). In order to transport the T4 hormone, four TTR proteins need to be attached to each other to establish a tetramer, while to transport retinol (vitamin A), TTR needs to form a tetramer and also bind to retinol-binding protein.78 Retinol is necessary for growth and dull-light vision. TTR proteins assemble into a tetramer conformation in which there are four identical subunits or monomers correlated but not bonded together covalently and to have the capability to transport vitamin A around the body.33
TTR gene mutations
The most common type of mutations occurring within the TTR gene are missense mutations; however, one version of deletion has been discovered34,35,90 (Figure 3). The phenotypes related to TTR gene mutations are carpal tunnel syndrome (CTS) (MIM 115430), ATTR (MIM 105210), and dystransthyretin hyperthyroxinemia (MIM 145680).
 | Figure 3 Mutations described in TTR gene. ATTR is caused by mutations in TTR gene the protein product of which is expressed mainly in the liver. There exist more than 100 mutations in TTR protein. From Alnylam Pharmaceuticals, Inc. Alnylam Pharmaceuticals discontinues revusiran development. 2016. Available from: http://investors.alnylam.com/releasedetail.cfm?ReleaseID=992320; with permission.34 Abbreviations: TTR, transthyretin; ATTR, TTR amyloidosis. |
Carpal tunnel syndrome
CTS36–39,79 results either from a mutation within the TTR gene or from significant causes such as diabetes or pregnancy.23–26 There is also another gene associated with CTS, that is, SH3TC2 gene, which is located at the q arm of chromosome 5.40 Conditions with connections to other neuron-related diseases could manifest or imitate those symptoms of the CTS and therefore make patients more susceptible to the disease.36–39
CTS causes trembling, numbness, and very often tingling in the arms and hands.36–39,79 These conditions are due to pressure at median nerve which seems to be located in the wrist.
Amyloidosis hereditary transthyretin (or ATTR)
Amyloidosis hereditary transthyretin or ATTR occurs due to mutations in the TTR gene.23–26 ATTR has various alternative names including TTR-related hereditary amyloidosis and familial amyloid polyneuropathy (FAP).41 ATTR is inherited in an autosomal dominant fashion, and is therefore heterogeneous.41 The cause of this hereditary disease is misfolding of TTR protein.42 Amyloid fibrils are formed by proteins that are encoded by the TTR gene, and therefore, ATTR and any of the related diseases are due to the accumulation of those amyloid fibrils in the different organs of the body, especially in those tissues related to the nervous systems.41,42,80 When the misfolded proteins are exported and left to deposit in the extracellular fluid by interacting with the β-sheets, the result is a malfunction in the organs.41,42 In addition to all that, there are approximately 27 different proteins that have the tendency to produce amyloid fibrils; those different proteins are distinguished by specifying the different amyloid types produced.41,42
Expanding from the previous divisions of the ATTR, FAP is caused by the accumulation of amyloid deposits mainly due to the most common mutation of the TTR gene Val30Met. The symptoms of FAP are most common among people of age 30–40. FAP patients, in most cases, will have neuropathy.43,44 In terms of occurrence, this disease is very rarely present among the worldwide population, but it is seen commonly within Swedish, Portuguese, and Japanese populations, and among the people whose ancestors originate from those regions.43,44 Symptoms of FAP are related to the heart, kidneys, and eyes, and cause many body changes such as swelling and fatigue. FAP is closely related to familial amyloid cardiomyopathy (FAC), which occurs due to the deposition of amyloid in the heart and therefore results in shortness of breath and dizziness.45 The widely spread TTR gene mutation Val122lle is currently more prevalent among African Americans.43
In addition to all the mutations mentioned, there is one very important form of ATTR, which is the wild-type ATTR falling under the name senile systemic amyloidosis (SSA). 32,46 SSA usually affects people above age 60 and is not inherited through generations. SSA is not caused due to any mutation at the TTR gene; rather, the level of the deposition of amyloid is what causes the symptoms such as nausea, fainting, and shortness of breath to appear at the late age of the affected patients.46
As mentioned previously, the symptoms manifested by patients with ATTR depend on the variants of TTR gene. TTR gene codes for different proteins, which in turn produce precursor proteins. The peripheral nervous system (PNS), gastrointestinal system, and the heart are the most common organs within which the deposition and accumulation of the amyloid fibrils occur.47–49 Patients who suffer from the accumulation of amyloid fibrils in PNS and autonomic (orthostatic hypotension) nervous system could face symptoms such as losing perception of temperature, hyperalgesia (high sensitivity to pain), and problems in the PNS with sensorimotor impairment being the most common.47–49 Moreover, extreme weakness and impaired sensation could result due to the variance of ATTR V30M.47–49 Patients who suffer from accumulation of fibrils in the cardiac system could experience extreme postural hypotension, failure of heart function, and diastolic dysfunction.47–49 A mutation in the TTR gene affecting the central nervous system results in nystagmus and spastic paraparesis, and it very rarely causes hearing loss.49
If ATTR is suspected, there are several diagnostic methods available that can be utilized for confirmation. One of the diagnostic tests is tissue biopsy. This involves the extraction of a fat tissue sample subcutaneously or from under the skin of the patient’s body.49 The tissue is mostly extracted from the nerves and the heart, where different histological stains are used. Those stains are necessary for studying and identifying the structure of the tissue.50–53 There are different sites and types of biopsy used to show the amyloid deposition. There are different sites and types of biopsies used to show amyloid deposition such as, labial salivary gland (LSG) biopsy, nerve biopsy, abdominal fat aspiration, and finally suppression of gastrointestinal tract.50 Moreover, deciding which site is most suitable for biopsy in a certain region depends upon the population being selected in the region.50 Usually, and initially, abdominal fat is selected and is stained with Congo red, which is used specifically for identifying amyloid deposits showing green birefringence in polarized light (Westermark 2005).81 Positive Congo red staining proves the presence of the amyloid deposits, and this staining technique is followed by proteomics and immunohistochemistry in order to identify which proteins have been encoded by the different TTR gene variants so that their function could be detected.45,54,55
Different types of stains are utilized and react differently to the presence of amyloid deposits. Hematoxylin–eosin with saffron is used in various tissues and organs and reveals amyloid deposition.51 Masson Trichrome stain and Verhoff’s Van Gieson do not show amyloid deposits; however, they reveal differences between tumors affecting smooth muscles and collagen, and mark atrophies affecting the elastic tissue.50,52,53 In addition to staining, electrophoresis is used to help identify TTR gene variant when it is not inherited and is wild type; this segregation occurs according to charge.47 Besides the laboratory testing of the tissues, noninvasive testing is an alternative diagnosis for cardiac amyloidosis.91 Noninvasive testing does not require the use of medical instruments or open surgeries; such technique is done by the use of electrocardiography (ECG).91 ECG is a secure method required for all patients presumed to have amyloidosis, where electrical impulses in the heart are measured and denoted on graph paper as a tracing. The different patterns of ECG are used to distinguish between amyloid light-chain amyloidosis and TTR amyloidosis. For this technique to work properly, there should be association with many other tests and imaging procedures such as cardiac magnetic resonance, strain imaging, and serum biomarker testing.91 Genetic testing and sequencing of DNA is an alternative method utilized in order to signify and detect mutations occurring at the TTR gene; different mutations lead to different types of diseases.91
Treatment for amyloidosis hereditary TTR
The treatment focuses on three aims:
a)The first one is easing the symptoms or cardiac complications (pacemaker) related to heart failure.
b)The second one is to stop or hinder the progress of amyloid deposition.
c)The third one is genetic counseling.45
Up until recently, there is no drug or pharmacological therapy used to treat ATTR, and there is no evident effect of a diet or lifestyle that could lead to ATTR.55,56 However, there are drugs currently used to hinder and slow down the symptoms of the heart failure; when the cavities are filled with water, causing edema, diuretics are used to remove excess water in case of the absence of hypotension.55
Dysrhythmia is associated with atrial fibrillation and is a result of wild-type form of TTR, and it results in dilation of atria affecting the sinus rhythm; decreased doses of beta-adrenergic receptor antagonists are usually used as agents for controlling the rate.45,81 Besides those drugs that help alter the heart failure symptoms, there is an alternative for severe cases which is the surgical approach. Organ transplantation was most evident back in the 1990s and was mainly done on patients suffering from FAP. Patients with FAP usually undergo liver transplantation, which aids in hindering and halting the progression of neuropathy, and other neurological disabilities.56 Liver transplantation also helps stop the movement of the variant TTR mutations, and has also led to the emergence of orthotopic heart transplantation, which in turn helps stop amyloid deposition.45,82 Moreover, transplantation combinations now aid in better halting of the disease, and liver and kidney or liver and heart transplantation could be done to provide patients with better outcomes.56
TTR stabilizers
As mentioned earlier, mutant TTR and wild-type TTR amyloidogenesis results in amyloidosis. TTR stabilizer is an alternative treating technique that prohibits the dissociation of the TTR protein tetramer into a monomer. In the normal state of TTR, there are two T4 binding sites that are generated by fragile dimer–dimer interface not originally occupied. Tafamidis (2-(3,5-dichloro-phenyl)-benzoxazole-6-carboxylic acid) is a TTR-stabilizing drug, and is used orally to treat TTR-FAP. Tafamidis attaches to TTR and occupies the T4 binding sites, therefore inhibiting the dissociation of TTR tetramer by increasing the energy barrier and slowing the progression of the disease.83 Diflunisal is another nonsteroidal anti-inflammatory TTR-stabilizing drug that is orally introduced to the body. Diflunisal has shown severe side effects such as gastrointestinal bleeding, renal dysfunction, and the inhibition of COX enzyme when given with unmonitored doses. Derivatives of diflunisal, iododiflunisal and bromodiflunisal, bind to T4 binding sites in TTR, therefore stopping tetramer dissociation.84
Clinical trials
Alnylam Pharmaceuticals Inc. is a well-known biopharmaceutical company and association that was initiated in 2002. It has two main goals: the first goal is to focus on introducing RNAi therapeutics to target genes that should be halted to help treat patients with fetal diseases, and the second goal is to manufacture innovative and advanced medicines.57
ATTR is suggested to be treated through RNAi therapeutics. Patisiran drug appoints siRNAs that target and knock down the disease-causing TTR protein. In preclinical studies, one RNAi therapeutic named ALN-TTRsc02 has shown rapid and potent knockdown of serum TTR by 99%. ALN-TTsc02 was well tolerated and did not have any undesirable effects on the participants. Phase I was mainly focused on determining the safety of ALN-TTR01 and ALN-TTR02 and identifying which of them can better knock down the TTR gene causing the disease. In Phase II, volunteers were subjected to TTR stabilizers, and this phase was directed more toward knowing the tolerance of the alternate doses of Patisiran. Phase II open-label extension (OLE) helped further ensure the safety of the doses used and drug tolerance during the long run. Finally, Phase III aimed to prove the safety of Patisiran by giving a group of people Patisiran drug or placebo in a random manner. All the trials of the Patisiran drug have turned successful until the recent date, and the clinical development is still in progress.74
There are certain delivery techniques that are being used to transfect the cells with siRNA; lipid-based delivery systems are among the most commonly used methodologies.1,58,59 The utilization of lipid nanoparticles (LNP) as a transfecting technique would facilitate, encase, and enfold siRNAs, and help administer to the body intravenously; LNP transfection technique is quick and enduring and has showed great accomplishments in clinical trials and in in vivo experiments.1,58,60 More advanced and enhanced techniques such as electroporation are used along with LNP transfection; electroporation can carry and transport a vast number of siRNAs into the cells and is linked to siRNA delivery using high-throughput technologies.1 Alnylam Pharmaceuticals’ team worked with the adopted assets from Sirna Therapeutics and took delivery of siRNA into a more progressive level characterized by the formation of “GaINAc”, which is a sugar molecule administered to the body of the patient subcutaneously.58,61 This sugar molecule provides a more facilitated and rapid delivery passage, which is not very evident when using LNP.58 Some other delivery techniques could include the use of bacteria and viruses.62 Viral delivery could be used in in vivo and ex vivo treatment of certain diseases.63 Virosomes, for example, encapsulate siRNA and aid in targeting cells in vivo, while viral vectors inhibit the expression of certain receptors for ex vivo treatment.63 Besides the already mentioned advances of delivery, some other delivery technologies for treatment could include autologous cell therapy, or the injection of RNAi drugs into the targeted tumor, organ, or tissue of the body in order to break down or degrade the desired parts.62
Innovative medicines for treating TTR-related diseases
Alnylam Pharmaceuticals Inc. has been working on finding a treatment for phenotypes related to TTR gene since early 2000s. The genetic medicines initially include Patisiran (ALN-TTR02), which is responsible for targeting TTR amyloidosis within patients suffering from FAP.56 Patisiran uses RNAi to knock down TTR genes which are either wild type or mutant by the use of siRNA.65 Patisiran drug is intravenously introduced into the body of the patient through LNPs with the aid of the delivery platform MC3-LNP and is used to target the TTR genes expressed in the liver.
Tafamidis is an agent that binds to TTR and causes stabilization of the wild-type native and mutant TTR. As a result, tafamidis stops the cascade of amyloid deposition due to the dissociation of the tetramer structure of TTR protein. Moreover, this agent is used as an initial treatment resort for FAP if it is detected in the early stages.85
In addition to being approved by the US Food and Drug Administration (FDA) for treating arthritis, diflunisal stabilizes TTR tetramer as does tafamidis, and therefore enhances the quality of life and slows down the progression of neurological impairment in stage I–II ATTR-FAP patients.85
IONIS-TTRRx drug is still being clinically developed and is in Phase III trial. The aim of this trial is to ensure if IONIS-TTRRx slows down and halts the damage caused to the nerves by TTR amyloid deposits. There are two groups included in the Phase III trial; the intervention group includes patients who receive 300 mg of the drug through subcutaneous injections, and the control group includes patients receiving the placebo.86
NEOD001 is a monoclonal antibody which is designated as an orphan drug by the FDA, and identifies amyloid accumulations by reacting with the misfolded forms of the TTR protein. During Phase I and II of clinical trials, the main aim was to evaluate the safety and tolerability of NEOD001; on the other hand, Phase III clinical trial was multidimensional, double-blinded, and placebo-controlled. One of the main end points of the study includes the evaluation of biomarker NT-proBNP.87
Revusiran, alternatively called ALN-TTRsc, is an RNAi therapeutic developed for the treatment of hereditary ATTR with cardiomyopathy. ALN-TTRsc is introduced to the body subcutaneously using standard template chemistry GaINAc conjugate delivery platform, and is suitable for the treatment of FAC.64–66 The production of Revusiran has been recently stopped during Phase III because it has been shown to be unsafe in patients.
Patisiran drug was and is still used for treating FAP through RNAi therapeutics. Primarily, the only possible and trustable procedure to help treat FAP was liver transplantation, which would help slow down or stop the formation of the mutant allele; however, liver transplantation was not able to help patients with the wild-type TTR gene.67
Phase I trials
Phase I trials are usually about checking the efficacy and safety of a certain drug formula with a small number of volunteers being enrolled. In the case of Patisiran drug, Phase I was divided into two trials to check the credibility of ALN-TTR01 and ALN-TTR02; the first trial was initiated by testing the safety of ALN-TTR01. The study was randomized, and the participants were from different nationalities. The participants in the study were 18 years and above, with body mass indexes (BMI) between 18.5 kg/m2 and 33.0 kg/m2.68 A placebo was used in single-blinded method, and different doses of the drug were suggested to acknowledge the safest one to use. The study plan for ALN-TTR01 consisted of four cohorts, each subdivided into four people, where three of them received the active dosage of the drug and one of them was injected with the control drug.68,71 With the supervision of the Safety Review Committee (SRC), safe dosages of ALN-TTR01 were insured to not cause any repulsive actions in patients; thus, the decided safe dosages of the drug are 0.03, 0.1, 0.2, and 0.4 mg/kg, with the highest dosage proving nontoxic in patients suffering from cancer.71 ALN-TTR01 was proven to suppress wild-type TTR in mouse models and hTTR gene in humans.71 ALN-TTR01 silences the production of TTR gene, thus lowering the level of TTR which in turn drops the levels of thyroid hormone and vitamin A.71 Volunteers who received higher doses of ALN-TTR01 (1 mg/kg) showed better reduction of TTR levels than those administered with lower doses (0.01–0.07 mg/kg).68 Moreover, three individuals of each cohort had V30M mutation, and one had S77Y mutation; the individuals with V30M mutation showed 41% knockdown of TRR which later returned to baseline level, while S77Y showed 50% knockdown which increased to 81% and then returned back to 50%.68
The second trial was based on ALN-TTR02, where the volunteers were required to be between ages 18 and 45 with BMI of 18–31.5 kg/m2, and a normal blood pressure; they were also expected to be healthy and have not smoked for about 3 months. For this study, the intravenous ALN-TTR02 drug was encapsulated by AF-011 LNPs; moreover, eight cohorts were generated, five of which were injected with different escalating dosages and three were controls.68,71 Subsequently, each cohort was subdivided into four individuals; the first two participants were given the lowest dose of ALN-TTR02, and placebo was given to others. Once the doses were proved safe after 7 hours, the third contributor was subjected to a dose of ALN-TTR02, and the fourth contributor was given a higher dose. The three other cohorts went through the same pathway, and it was concluded that the safe doses of ALN-TTR02 are 10, 50, 150, 300, and 500 mg/kg.68 Chromosome aberration assay and Ames tests were used to test the safety of ALN-TTR02, and other regular checkups such as blood pressure, pulse rate of the heart, and body temperature were used to monitor the health of volunteers when subjected to the drug.71 Pharmacodynamics was used to evaluate the concentration of the serum TTR, vitamin A, and retinol-binding proteins, while pharmacokinetics was used to evaluate the concentration of the plasma.71 In conclusion, reduction of TTR level was more potent when ALN-TTR02 was used rather than ALN-TTR01; thus, using higher doses ch as 0.3 mg/kg resulted in 87.7% knockdown of the gene.71
Phase II trials
The second phase of the Patisiran clinical trials required from one to nine cohorts with a total number of individuals summing up to 29. Twenty of these participants were males, and the other nine were females.72 The individuals enrolled were of age ranging from 28 to 76; however, the average age of participating individuals was 62.72,73 Majority of the patients had V30M mutation, while the minority had other ATTR-causing mutations such as S77Y and S77F. Moreover, 14 out of the 29 participants received tafamidis, which was used to stabilize tetramers, six of them received diflunisal, and the other nine individuals received no tetramer stabilizers.72
The main aim of the Phase II trial was to evaluate and prove the tolerance levels of two doses of the Patisiran drug. The two doses of the Patisiran ranged from 0.01 to 0.30 mg/kg, and were administered to the patients either one time every three weeks or one time every four weeks.74 The analysis of the 29 patients revealed that there was a 96% knockdown in the TTR gene, whether mutant or wild type, with an average of 83.8% and 87% of knockdown achieved after Patisiran dose of 0.30 mg/kg was injected for the first or second time.72,74
Moreover, some additional tests, such as spectrometry, were done to calculate the levels of wild-type and mutant V30M proteins, where knockdown was evident in both cases of mutations.74,82 Patients who received tetramer stabilizers had shown an increased baseline levels in TTR genes, the stabilizing of the tetramers had not affected the knockdown of the TTR, and therefore, Patisiran still had the expected effect.74 Reduction in the levels of retinol-binding protein and vitamin A was due to the knocking down of the serum TTR.74 This study has also proven that multiple doses of Patisiran were safe and only few adverse effects were present, but very mild to cause a difference. However, there were two serious adverse events, one of which caused cellulitis, which is an inflammation in the site of infusion due to extravasation of the drug; approximately 10.3% of the patients suffered from infusion-related reactions, and the other serious symptom was long series of vomiting and nausea.74 Phase II trials have also shown a very rapid knockdown of approximately 93%.64 When 0.30 mg/kg dose of Patisiran was introduced once every four weeks, it led to a mean knockdown of about 82.6% and 84.8%, respectively, while when 0.30 mg/kg was injected once every three weeks, it led to a minimum knockdown of about 83.1% and 87.4% and a maximum knockdown of 92.8%.64
Phase II OLE
The number of patients involved in the Phase I and Phase II trials of FAP was 23. Sixteen of those 23 patients were males, and the remaining seven were females.75 The infusion of the Patisiran drug was done for about 70 minutes.75 Phase II OLE data were mentioned in the International Symposium on Amyloidosis (ISA); it was showed that multiple doses of Patisiran resulted in a knockdown of about 80% through 168 days only.64 Phase II OLE ensures tolerability of Patisiran and long-term safety of administration of different doses.64 In total, there were approximately 27 people involved in this two-year-long study, where those patients received 0.3-mg/kg dosage of Patisiran one time every three weeks.76 Clinical end points were measured every six months; modified Neuropathy Impairment Score +7 (mNIS+7) was calculated for evaluating the weakness of the muscles, and the conductance strength of the nerves.76 OLE-Phase II led to the conclusion that even after six months of using Patisiran as treatment, there was no change in impairment of the PNS in comparison to the baseline level.76 FAP patients who were not subjected to treatment revealed a very rapid increase in mNIS+7, while patients who were treated with Patisiran showed an average decrease in the level of mNIS+7.76
Phase III trials
Phase III trial was started in the year 2013, and is still ongoing until today. APOLLO Phase III trial is conducted in a random manner, where patients are given either Patisiran or placebo double-blindedly.77 Phase III trial was mainly designed to prove how effective and safe is the use of Patisiran drug in patients suffering from ATTR.70,77 Approximately 200 patients in either stage I or II of FAP were enrolled in the third phase, and were given either Patisiran drug or placebo in a ratio of 2:1; 0.3-mg/kg dose was given once every three weeks for exactly 18 months.77 The main aim of Phase III trial is to establish a comparison in mNIS+7 levels between patients who received Patisiran drug and those who received placebo, and the secondary aims included timed 10-m walk, NIS weakness, COMPASS-31 autonomic symptoms, and Norfolk quality-of-life diabetic neuropathy score.77
Conclusion
Theoretically, the therapeutic potential of RNAi has almost no limits. All annotated disease-related genes should be adaptable to RNAi-mediated posttranscriptional gene silencing. There has been rapid and synergistic progress in the fundamental and translational field of RNAi research. Pharmacokinetic studies, though currently few in number, have set an important model for further investigations. Problems including toxicity, safety, off-target effects, and delivery are currently being addressed and analyzed. Already, there are a couple of reports on clinical trials on application of siRNA for treatment of diseases that raise a lot of hope that a collection of safe and effective systemic RNAi therapies may indeed be put in use in the near future. Patisiran is one of those promising drugs, which was found to be generally safe and well tolerated by the patients. The APOLLO Phase III trial of Patisiran was designed to evaluate the efficacy and safety of Patisiran in ATTR patients. Provided that it meets all the safety guidelines for humans, Patisiran is intended to support marketing authorization for ATTR patients in the near future.
Disclosure
The authors report no conflicts of interest in this work.
References
Tuzmen S, Tuzmen P, Arora S, Mousses S. RNAi-based functional pharmacogenomics. Methods Mol Biol. 2011;700:271–290. | ||
Elbashir SM, Harborth J, Weber K, Tuschl T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods. 2002;26(2):199–213. | ||
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. | ||
Hannon GJ. RNA interference. Nature. 2002;418(6894):244–251. | ||
Huppi K, Martin SE, Caplen NJ. Defining and assaying RNAi in mammalian cells. Mol Cell. 2005;17(1):1–10. | ||
Tuschl T. RNA interference and small interfering RNAs. Chembiochem. 2001;2(4):239–245. | ||
Nakayashiki H, Nguyen QB. RNA interference: roles in fungal biology. Curr Opin Microbiol. 2008;11(6):494–502. | ||
Travella S, Keller B. Down-regulation of gene expression by RNA-induced gene silencing. Methods Mol Biol. 2009;478:185–199. | ||
Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role of a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409(6818):363–366. | ||
Filipowicz W. RNAi: the nuts and bolts of the RISC machine. Cell. 2005;122(1):17–20. | ||
Hemann MT, Fridman JS, Zilfou JT, et al. An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet. 2003;33(3):396–400. | ||
Carmell MA, Zhang L, Conklin DS, Hannon GJ, Rosenquist TA. Germline transmission of RNAi in mice. Nat Struct Biol. 2003;10(2):91–92. | ||
Tiscornia G, Singer O, Ikawa M, Verma IM. A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci U S A. 2003;100(4):1844–1848. | ||
Caplen NJ. RNAi as a gene therapy approach. Expert Opin Biol Ther. 2003;3(4):575–586. | ||
Wilda M, Fuchs U, Wossmann W, Borkhardt A. Killing of leukemic cells with a BCR/ABL fusion gene by RNA interference (RNAi). Oncogene. 2002;21(37):5716–5724. | ||
Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2(3):243–247. | ||
Scherr M, Battmer K, Winkler T, Heidenreich O, Ganser A, Eder M. Specific inhibition of bcr-abl gene expression by small interfering RNA. Blood. 2003;101(4):1566–1569. | ||
Son YA, Tüzmen Ş, Hızel C. Designing and implementing pharmacogenomics study. In: Barh D, Dhawan D, Nirmal Kumar G, editors. Omics for Personalized Medicine. New Delhi: Springer; 2013:97–122. | ||
Song E, Lee SK, Wang J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9(3):347–351. | ||
Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6(3):659–668. | ||
Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. Going beyond the liver: progress and challenges of targeted delivery of siRNA therapeutics. J Control Release. 2015;203:1–15. | ||
Vicentini FT, Borgheti-Cardoso LN, Depieri LV, et al. Delivery Systems and Local Administration Routes for Therapeutic siRNA. Pharm Res. 2013;30(4):915–931. | ||
TTR gene. Genetic Home reference. 2009. Available from: http://ghr.nlm.nih.gov/gene/TTR. Accessed June 16, 2017. | ||
NCBI. TTR transthyretin [Homo sapiens (human)]. 2015. Available from: http://www.ncbi.nlm.nih.gov/gene/7276. Accessed June 16, 2017. | ||
Online Mendelian Inheritance in Man. Transthyretin; TTR. Available from: http://www.omim.org/entry/176300. Accessed June 16, 2017. | ||
Gene Cards. TTR gene. Available from: http://www.genecards.org/cgi-bin/carddisp.pl?gene=TTR. Accessed June 16, 2017. | ||
Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10(3):160–184. | ||
Nakazato M. Genotype-phenotype relationship in familial amyloid polyneuropathy. Neurol Med (Tokyo). 1998;48:528–534. | ||
Benson MD. Amyloidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. Vol. 4. New York: McGraw-Hill; 2001:5345–5378. | ||
Saraiva MJ. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat. 2001;17(6):493–503. | ||
NOVANEXT [webpage on the Internet]. RNAi Therapies-Cure or Chimaera? Available from http://www.pbs.org/wgbh/nova/next/body/rnai/. Accessed July 11, 2017. | ||
Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–1300. | ||
Rappley I, Monteiro C, Novais M, et al. Quantification of transthyretin kinetic stability in human plasma using subunit exchange. Biochemistry. 2014;53(12):1993–2006. | ||
Alnylam Pharmaceuticals, Inc. Alnylam Pharmaceuticals discontinues revusiran development. 2016. Available from: http://investors.alnylam.com/releasedetail.cfm?ReleaseID=992320. Accessed June 16, 2017. | ||
Munar-Qués M, Saraiva MJ, Calonge JO, et al. Familial amyloid polyneuropathy in a Spanish family with a transthyretin deletion (deltaVal 122) presenting with carpal tunnel syndrome. Clin Genet. 2000;58(5):411–412. | ||
Burton C, Chesterton LS, Davenport G. Diagnosing and managing carpal tunnel syndrome in primary care. Br J Gen Pract. 2014;64(622):262–263. | ||
National Institute of Neurological Disorders and Stroke. Carpal tunnel syndrome fact sheet. Available from: http://www.ninds.nih.gov/disorders/carpal_tunnel/detail_carpal_tunnel.htm. Accessed June 16, 2017. | ||
WebMD. Carpal tunnel syndrome – topic overview. 2014. Available from: http://www.webmd.com/pain-management/carpal-tunnel/carpal-tunnel-syndrome-topic-overview#1. Accessed July 11, 2017. | ||
Shiel WC; FACP. Carpal tunnel syndrome symptoms, tests and diagnosis. Available from: http://www.medicinenet.com/carpal_tunnel_syndrome/article.htm. | ||
National Center for Biotechnology Information. SH3TC2 SH3 domain and tetratricopeptide repeats 2 [Homo sapiens (human)]. Available from: http://www.ncbi.nlm.nih.gov/gene/79628. Accessed June 16, 2017. | ||
Alnylam. Hereditary ATTR amyloidosis with polyneuropathy (hATTR-PN). Available from: http://alnylam.microartswpdev.com/product-pipeline/hereditary-attr-amyloidosis-with-polyneuropathy/. Accessed July 12, 2017. | ||
OMIM. Amyloidosis, hereditary, transthyretin-related. 2016. Available from: https://www.omim.org/entry/105210. Accessed July 12, 2017. | ||
Xu J, Yang M, Pan X, et al. Transthyretin-related hereditary amyloidosis with recurrent vomiting and renal insufficiency as the initial presentation: a case report. Medicine. 2017;96(10):e5737. | ||
Medscape. Transthyretin-related amyloidosis. 2017. Available from: http://emedicine.medscape.com/article/335301-overview. Accessed July 11, 2017. | ||
Adams D. Recent advances in the treatment of familial amyloid polyneuropathy. Ther Adv Neurol Disord. 2013;6(2):129–139. | ||
Amyloidosis Patient Information Site. ATTR amyloidosis. Available from: http://www.amyloidosis.org.uk/introduction-to-attr-amyloidosis/. Accessed June 16, 2017. | ||
Medscape. Transthyretin-related amyloidosis workup. 2017. Available from: http://emedicine.medscape.com/article/335301-workup#c6. Accessed July 12, 2017. | ||
Ando Y, Ueda M. Diagnosis and therapeutic approaches to transthyretin amyloidosis. Curr Med Chem. 2012;19(15):2312–2323. | ||
Gertz MA, Rajkumar VS, editors. Amyloidosis: Diagnosis and Treatment. Springer Science & Business Media; 2010. | ||
Adams D, Suhr OB, Hund E, et al; European Network for TTR-FAP (ATTReuNET). First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016;29 Suppl 1:S14–S26. | ||
Histalim. Hematoxylin eosin saffron. Available from: http://www.histalim.com/accueil/activities/our-services/histology/hematoxylin-eosin-saffron/. Accessed June 16, 2017. | ||
Surgical pathology – histology. Staining manual – connective tissue. Collagen – Masson’s Trichrome stain (TRI). Available from: http://library.med.utah.edu/WebPath/HISTHTML/MANUALS/MASSONS.PDF. Accessed June 16, 2017. | ||
Surgical pathology – histology. Staining manual – connective tissue. Elastic tissue fibers – Verhoeff’s Van Gieson (EVG). Available from: http://library.med.utah.edu/WebPath/HISTHTML/MANUALS/EVG.PDF. Accessed June 16, 2017. | ||
Sereikaite J, Bumelis VA. Congo red interaction with alpha-proteins. Acta Biochim Pol. 2006;53(1):87–92. | ||
Medscape. Transthyretin-related amyloidosis treatment & management. 2017. Available from: http://emedicine.medscape.com/article/335301-treatment. Accessed July 12, 2017. | ||
Adams D, Samuel D, Goulon-Goeau C, et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000;123(Pt 7):1495–1504. | ||
Alnylam. Our culture. Available from: http://www.alnylam.com/who-we-are/our-culture/. Accessed June 16, 2017. | ||
Zhou J, Shum KT, Burnett J, Rossi J. Nanoparticle-based delivery of RNAi therapeutics: progress and challenges. Pharmaceuticals (Basel). 2013;6(1):85–107. | ||
Tam YY, Chen S, Cullis PR. Advances in lipid nanoparticles for siRNA delivery. Pharmaceutics. 2013;5(3):498–507. | ||
Huang L, Liu Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu Rev Biomed Eng. 2011;13:507–530. | ||
Alnylam Pharmaceuticals, Inc. Alnylam acquires investigational RNAi therapeutic assets from Merck. 2014. Available from: http://investors.alnylam.com/releasedetail.cfm?ReleaseID=818732. Accessed June 16, 2017. | ||
Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6(9):1130–1146. | ||
Nguyen T, Menocal EM, Harborth J, Fruehauf JH. RNAi therapeutics: an update on delivery. Curr Opin Mol Ther. 2008;10(2):158–167. | ||
Alnylam Pharmaceuticals, Inc. Alnylam presents key scientific data on enhanced stabilization chemistry (ESC)-GalNAc-conjugate technology. 2014. Available from: http://investors.alnylam.com/releasedetail.cfm?ReleaseID=846985. Accessed June 16, 2017. | ||
Alnylam Pharmaceuticals. RNAi Roundtable: Patrisiran and ALN-TTRsc in Development for the Treatment of Transthyretin-Mediated Amyloidosis (ATTR). Available from http://www.alnylam.com/web/assets/RNAi_Roundtable_TTR_071514.pdf. Accessed September 25, 2017. | ||
GalNAc-siRNA with enhanced stabilization chemistry: ESC-GalNAc-siRNA. 2014. Available from:http://www.alnylam.com/web/assets/ALNY-ESC-GalNAc-siRNA-TIDES-May2014-Capella.pdf. Accessed June 16, 2017. | ||
Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. | ||
Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. | ||
Alnylam Pharmaceuticals. 2014. Available from: http://www.alnylam.com/web/assets/072214Alnylam.pdf. Accessed June 16, 2017. | ||
Mayo Clinic. APOLLO: The Study of an Investigational Drug, Patisiran (ALN-TTR02), for the Treatment of Transthyretin (TTR)-Mediated Amyloidosis. Available from: http://test.kcms.mayoclinic.org/research/clinical-trials/cls-20115984. Accessed June 16, 2017. | ||
Alnylam Presents Positive Preliminary Clinical Results for ALN-ttr01. Alnylam Pharmaceuticals, Inc. - Alnylam Presents Key Scientific Data on Enhanced Stabilization Chemistry (ESC)-GalNAc-Conjugate Technology. Available from http://investors.alnylam.com/releasedetail.cfm?ReleaseID=734340. Accessed July 12,2017. | ||
RNAi Therapeutics. International Symposium on Familial Amyloidotic Polyneuropathy Clinical Updates on ALN-TTR Programs Patisiran (ALN-TTR02) and ALN-TTRsc for the Treatment of Transthyretin Amyloidosis. 2013. Available from: http://www.alnylam.com/web/wp-content/uploads/2013/11/ALNY-ISFAP-ALN-TTRprogram-Nov2013.pdf. Accessed June 16, 2017. | ||
Alnylam Pharmaceuticals. Positive phase II data with Patisiran (ALN-TTR02). Available from: http://www.alnylam.com/capella/presentations/positive-phase-ii-data-patisiran-isfap/. Accessed June 16, 2017. | ||
Alnylam Pharmaceuticals, Inc. Alnylam reports positive phase II data for Patisiran (ALN-TTR02), an RNAi therapeutic targeting transthyretin (TTR) for the treatment of TTR-mediated amyloidosis (ATTR), and initiates phase III trial [press release]. Cambridge: Alnylam Pharmaceuticals; 2013 [November 10]. | ||
Suhr O. Clinical update on Patisiran phase 2 trials in familial amyloidotic polyneuropathy. 2014. Available from: http://www.alnylam.com/web/assets/ALNY-Clinical-Update-Patisiran-Phase2-Trials-ISA2014.pdf. Accessed July 12, 2017. | ||
Garde D. Alnylam Reports Six-Month Clinical Data from Patisiran Phase 2 Open-Label Extension (OLE) Study in Patients with Familial Amyloidotic Polyneuropathy (FAP). FierceBiotech. Available from http://www.fiercebiotech.com/biotech/alnylam-reports-six-month-clinical-data-from-patisiran-phase-2-open-label-extension-ole. Published October 13, 2014. Accessed July 11, 2017. | ||
Alnylam Pharmaceuticals. TTR amyloidosis (FAP). Available from: http://www.alnylam.com/product-pipeline/ttr-amyloidosis-fap/. Accessed July 11, 2017. | ||
Vieira M, Saraiva MJ. Transthyretin: a multifaceted protein. Biomol Concepts. 2014;5(1):45–54. | ||
Nakagawa M, Sakijima Y, Yazaki M, et al. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid. 2016;23(1):58–63. | ||
Alshehri B, D’Souza DG, Lee JY, Petratos S, Richardson SJ. The diversity of mechanisms influenced by transthyretin in neurobiology: development, disease and endocrine disruption. J Neuroendocrinol. 2015;27(5):303–323. | ||
Westermark P. Aspects on human amyloid forms and their fibril polypeptides. FEBS J. 2015;272(23):5942–5949. | ||
Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–1854. | ||
Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–9634. | ||
Almeida MR, Macedo B, Cardoso I, et al. Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem J. 2005;381(Pt 2):351–356. | ||
Cruz MW, Benson MD. A review of tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4(2):61–79. | ||
IONIS-TTRRX. IONIS-TTRRx clinical trials. Available from: http://ttrstudy.com/ionis-ttrrx-clinical-trials/. Accessed June 16, 2017. | ||
Prothena Presents New Data from Phase 1/2 Study of NEOD001 Demonstrating Improvements in Three Organ Systems in Previously-Treated Patients with AL Amyloidosis. (NASDAQ:PRTA). Available from http://ir.prothena.com/releasedetail.cfm?releaseid=978092. Accessed July 11, 2017. | ||
Teng MH, Yin JY, Vidal R, et al. Amyloid and nonfibrillar deposits in mice transgenic for wild-type human transthyretin: a possible model for senile systemic amyloidosis. Lab Invest. 2001;81(3):385–396. | ||
Yokoyama T, Hanawa Y, Obita T, Mizuguchi M. Stability and crystal structures of His88 mutant human transthyretins. FEBS Lett. Epub 2017 May 31. | ||
Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277–282. | ||
Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–2412. | ||
RCSB PDB https://www.rcsb.org/pdb/explore/remediatedSequence.do?structureId=5TZL of PDB ID 5TZL. Kabsch W, Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22(12); 2577–2637. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.