Back to Journals » International Journal of Nanomedicine » Volume 12
Two-dimensional collagen-graphene as colloidal templates for biocompatible inorganic nanomaterial synthesis
Authors Kumari D, Sheikh L, Bhattacharya S, Webster TJ ![]() , Nayar S
, Nayar S
Received 3 February 2017
Accepted for publication 3 April 2017
Published 10 May 2017 Volume 2017:12 Pages 3605—3616
DOI https://doi.org/10.2147/IJN.S133833
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Israel Rubinstein
Divya Kumari,1,* Lubna Sheikh,1,* Soumya Bhattacharya,1,* Thomas J Webster,2 Suprabha Nayar1
1Materials Science and Technology Division, CSIR-National Metallurgical Laboratory, Burmamines, Jamshedpur, India; 2Department of Chemical Engineering, Northeastern University, Boston, MA, USA
*These authors contributed equally to this work
Abstract: In this study, natural graphite was first converted to collagen-graphene composites and then used as templates for the synthesis of nanoparticles of silver, iron oxide, and hydroxyapatite. X-ray diffraction did not show any diffraction peaks of graphene in the composites after inorganic nucleation, compared to the naked composite which showed (002) and (004) peaks. Scanning electron micrographs showed lateral gluing/docking of these composites, possibly driven by an electrostatic attraction between the positive layers of one stack and negative layers of another, which became distorted after inorganic nucleation. Docking resulted in single layer-like characteristics in certain places, as seen under transmission electron microscopy, but sp2/sp3 ratios from Raman analysis inferred three-layer composite formation. Strain-induced folding of these layers into uniform clusters at the point of critical nucleation, revealed beautiful microstructures under scanning electron microscopy. Lastly, cell viability studies using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays showed the highest cell viability for the collagen-graphene-hydroxyapatite composites. In this manner, this study provided – to the field of nanomedicine – a new process for the synthesis of several nanoparticles (with low toxicity) of high interest for numerous medical applications.
Keywords: composites, graphene, collagen, lateral gluing, and inorganic nanoparticles
Introduction
The high surface area of graphene composites is being harnessed for controlled nucleation and growth of nanocrystals, with the objective of enhancing their properties or obtaining a completely new property.1 Directional growth of inorganic nanoparticles on graphene templates has already been reported. Recent work on self-assembled gold-cyanide nanowires grown on graphene templates, confirms earlier findings that assembly is facilitated due to the lattice matching and π interaction between gold atoms and graphene.2,3 Similar work has also been reported on TiO2 nanoclusters which highlight the promise of using two-dimensional (2D) graphene for the growth of 3D materials.4 Two-dimensional graphene anchors both metallic and non-metallic particles alike because of the defects introduced during its formation from the parent graphite (GR).5,6 Direct exfoliation of GR to graphene has been attempted by many, but is yet to be fool proofed.
To obtain controlled defects and specific flake sizes, here we have used collagen (CL) to exfoliate GR. CL is composed of only a few distinct amino acids and when combined with graphene, it forms a unique chemical environment. This comprises sp3 carbon domains surrounded by sp2 carbon-containing moieties that provide specific binding sites for metal ions and a constrained environment for growth.7–12 During interaction, the graphitic layers get distorted and there is a tendency for lateral docking. This CL-graphene is further used as a colloidal template for inorganic nucleation. A recent report by Cullen et al suggested that 2D materials in dispersions can extend over several nanometers in length while being atomically thin in thickness.13 This has an added advantage of increased surface area, making them attractive candidates as templates. Inorganic particles can either be synthesized and later incorporated into these templates, or grown within these templates.
The present work is a modification of our earlier report on CL-graphene synthesized by direct exfoliation of GR with CL (layer thickness ≈2–3 layers, and lateral dimensions ≈200–300 nm).14,15 Pre-treatment of CL produces better CL-graphene composites and is subsequently used as templates for the synthesis of inorganic nanoparticles of silver (Ag), iron oxide (IO) and hydroxyapatite (HA), all of which are of high interest to the medical community. CL-graphene templates give an additional advantage of enhanced mechanical properties to these nanoparticles. Various biomedical fields can benefit from this technology since, for example, CL-graphene-Ag can be used as biosensors, antimicrobial agents, catalysts, and as new generation lightweight electronic devices. CL-graphene-IO can be used as electrodes for supercapacitors and micro-fluidic platforms for drug delivery and immunoassays, and CL-graphene-HA can be used for drug delivery, tissue engineering, and bone repair and regeneration.
Materials and methods
Materials
High purity natural GR powder, 200 mesh (product number: 14734) was obtained from Alfa Aesar, Haverhill, MA, USA; CL from bovine Achilles tendon (product number: C9879) from Sigma-Aldrich Co. (St Louis, MO, USA); sodium borohydride (product number: 106371), glacial acetic acid (product number: 100056), methanol (product number: 106009), silver nitrate (product number: 101510), dextrose (product number: 617809), ferrous/ferric chloride (product numbers: 103861 and 803945), calcium nitrate tetrahydrate (product number: 102121), di-ammonium hydrogen phosphate (product number: 101207), and liquor ammonia (product number: 105423) from Merck India Limited (Mumbai, India); and graphene quantum dots (product number: 53927) from Sisco Research Laboratories Pvt. Ltd. (Maharastra, India). All chemicals were of analytical grade and were used without further purification. Double distilled water was used throughout.
Swelling and dispersing of CL in acetic acid and inorganic synthesis
A patented process for the direct exfoliation of GR to graphene was further modified by the addition of a swelling step as a pre-treatment. Exfoliation of GR is directly dependent on CL dispersion as CL is only partially dispersed in an acidic solution, the method of swelling was adopted from a US patent.16,17 The final protocol is as follows: 1% (w/v) CL was incubated in 1 M sodium borohydride at room temperature without agitation for 20–22 hrs. It was then centrifuged at 10,000 rpm for 10 min and the supernatant discarded. The swollen CL pellet was washed properly and dispersed into a 1:1 mixture of 10% acetic acid and 10% methanol in water, stirring for 2 hrs at 80°C. A 1% (w/v) stock CL dispersion was prepared using the previously mentioned protocol, after which it was diluted to 0.1% (w/v) and GR powder was added to maintain a constant GR:CL ratio of 1:1. The dispersion was stirred at 400 rpm for 24 hrs and ultra-sonicated (150 watts, 40 kHz) for 20 mins daily for 7 days, after which it was centrifuged at 10,000 rpm for 5 mins. The supernatant-containing CL-graphene composites were used as templates to synthesize HA, Ag, and IO nanoparticles. In all three cases, the supernatant was incubated overnight with the corresponding inorganic salt solution and further processed with liquor ammonia (for IO), liquor ammonia and di-ammonium hydrogen phosphate (for HA), and liquor ammonia and dextrose (for Ag).
Characterization
The hydrodynamic diameter, polydispersity index, and zeta potential were measured by dynamic light scattering (DLS), NANO 100 Zetasizer (Malvern Instruments Ltd, Malvern, UK). Surface-enhanced Raman scattering (SERS) spectra were recorded using a Nicolet, AlmegaXR dispersive spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) with laser source (λ=532 nm). X-ray photoelectron spectroscopy (XPS) spectra were recorded using a Specs spectrometer with an MgKα source (1253.6 eV) (SPECS GmbH, Berlin, Germany). A few drops of the dispersions were dried on glass substrates and then inserted into a vacuum chamber with a pressure of ~10−9 Torr. Spectra were de-convoluted using CasaXPS software (SPECS GmbH), after Shirley background subtraction. All of the peaks were calibrated with respect to the standard C1s binding energy peak of pure GR at 284.5 eV. X-ray diffraction (XRD) phase identification was completed in a Bruker D8 Discover diffractometer (Bruker Corporation, Billerica, MA, USA) operating at 40 kV with Cu-Kα radiation (λ=1.5418 Å) in the 2θ range 20°–80°, step size equal to 0.02°/step, and scan speed of 1 sec/step. The shape and morphology of the nanoparticles were recorded with a field emission gun, Nova NanoSEM 430 (FEI, Eindhoven, the Netherlands) operating at 15 kV. Transmission electron microscopy (TEM) micrographs were recorded with a JEM 2010 (JEOL, Tokyo, Japan) operating at 200 kV. UV absorbance was also measured as described in the Supplementary materials.
Cell viability studies
Cell viability, as a test of cytocompatibility of these samples, was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for assessing cell metabolic activity. NAD(P)H-dependent cellular oxidoreductase enzymes may, under defined conditions, reflect the number of viable cells present. This test was performed by seeding L929 cells (from National Centre for Cell Science, Pune, India) at densities of 5,000 cells per well in a 96-well plate, and allowing them to attach for 10 hrs in a CO2 incubator maintained at 37°C. An amount of 1 mg of each sample was weighed, sterilized with 70% ethyl alcohol (v/v), and underwent UV treatment. Powders were suspended in Dulbecco’s Modified Eagle’s Medium (DMEM) and then filter sterilized (0.2 μm; HiMedia Laboratories Pvt. Ltd., Mumbai, India). The medium was replaced with the extract in the well plate and incubated for 7 days at 37°C and 5% CO2 atmosphere to examine the growth of cells in the presence of synthesized samples. The culture medium was replaced and cells were washed with phosphate-buffered saline to remove the non-adherent cells. An amount of 100 μL per well of the MTT solution prepared in DMEM was added to the wells, and the cells were cultured for an additional 4 hrs. The medium was then removed, and 200 μL of dimethyl sulfoxide was added to each well.18 The optical density (OD) was measured at a wavelength of 595 nm using a microplate reader (Model 2030 explorer; PerkinElmer Inc., Waltham, MA, USA). The experiments were completed in triplicate with results statistically analyzed by Student’s t-tests.
Results and discussion
Direct exfoliation of GR with CL
Most reports on aqueous graphene dispersions use graphene oxide (GO) as the precursor due to its hydrophilic character.19,20 In our previous paper, we experimented with three different organic acids namely succinic, propionic, and acetic acids for dispersing CL.14 Although, acetic acid is the most widely used dispersant for CL, propionic acid was inferred to be the best amongst the three. Since the degree of GR exfoliation is dependent on how well the CL is dispersed in the acidic medium, our endeavor has been to increase the dispersibility of CL. In this work, we followed a revised protocol where instead of directly using CL in the commercially available powdered form, it was first swollen and then dispersed. Post-swelling, the dispersion stability and uniformity in acetic acid surpassed even that of the previous best, propionic acid.
After incubating for 7 days, the CL-graphene dispersions were centrifuged and the supernatant characterized using DLS, UV, Raman spectroscopy, and XPS. The exfoliated CL-graphene had a hydrodynamic diameter of 492 nm, zeta potential value of +34.1 mV, and a polydispersity index of 0.421 as compared to CL dispersions which had a hydrodynamic diameter of 705 nm, zeta potential value of +35.6 mV, and a polydispersity index of 0.842.
OD was observed at a fixed wavelength of 270 nm (Figure S1). The increase in the absorbance proves the functionalization of the G surface and gradual opening of the optical band gap. A calibration curve (Figure S2) was drawn for a known concentration of graphene quantum dots (concentration =1 mg/mL), and absorbance measured at different serial dilutions, the data were fitted with a linear profile and graph drawn; the concentration of graphene was 0.5 mg/mL. Figure 1 shows the Raman spectra of natural GR and CL-graphene composites; graphitic structures exhibit signature peaks at ≈1,350 cm−1 (D band), ≈1,580 cm−1 (G band), and ≈2,700 cm−1 (2D band) in addition to minor peaks at ≈2,450 cm−1 (G′ band), ≈2,950 cm−1 (D + G band), and ≈3,245 cm−1 (2D′ band). Natural GR (Figure 1A) did not show the D band, meaning the precursor material was almost defect free;21 however, after incubation with CL, the D peak emerged (Figure 1C). The defect content was estimated from the intensity ratio of the D band and G band (ID/IG) which increased drastically from 0.04 in GR to 1 in CL-graphene. It is reported that for 3–5 layer graphene, the mean ID/IG ratio is ≈0.45, the ratio increases to ≈0.85 for chemically reduced GO annealed at 1100°C.22,23 For graphene sheets with lateral dimensions less than 500 nm, such as ours, the defect density is much higher due to the presence of a large number of edges; the attachment of functional groups (present in CL) on graphene also increases defects.
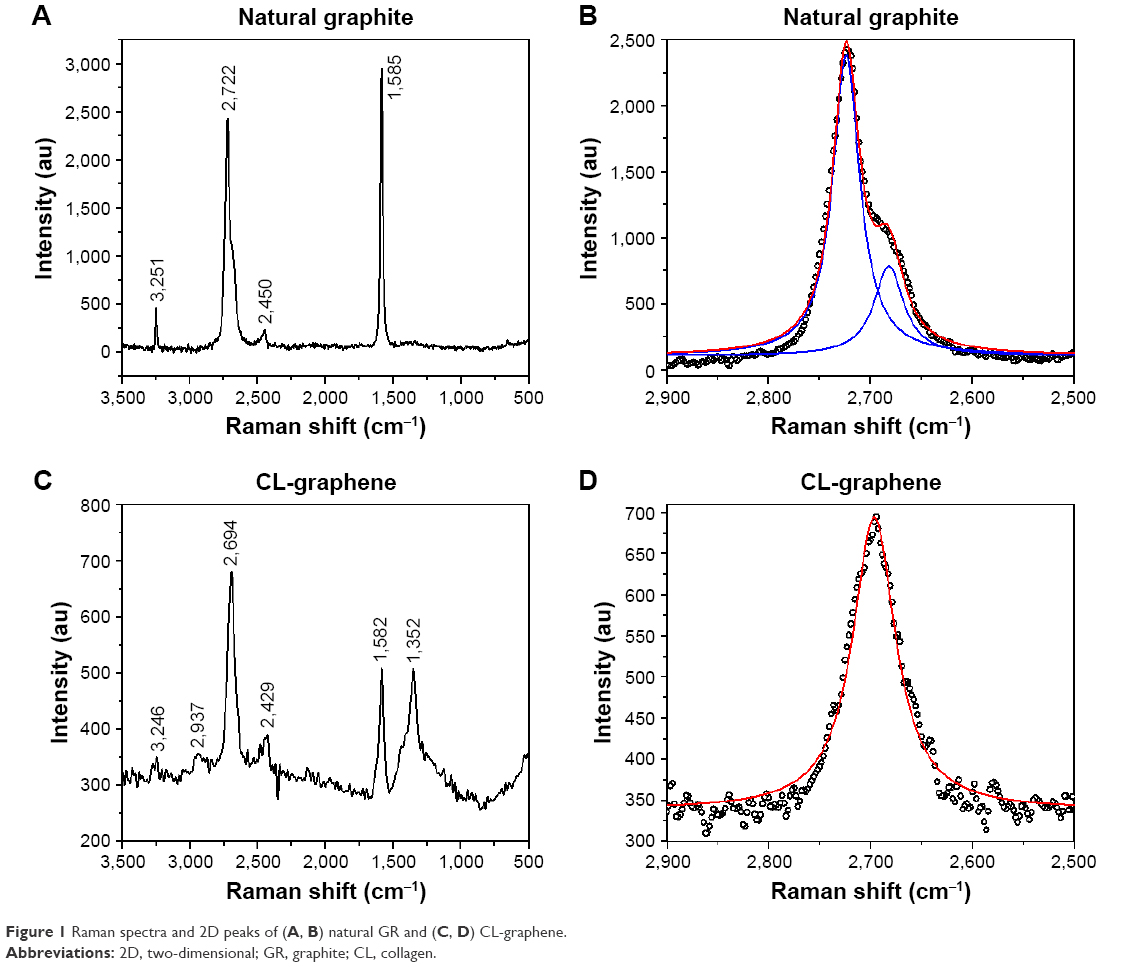 | Figure 1 Raman spectra and 2D peaks of (A, B) natural GR and (C, D) CL-graphene. |
Another interesting feature is the evolution of the 2D peak; in bulk GR, the 2D peak consists of two components, 2D1 (≈2,682 cm−1) and 2D2 (≈2,723 cm−1), roughly ¼ and ½ times the intensity of the G peak (Figure 1B). For single layer defect free graphene, it was reported that I2D/IG would be >1.5 and the 2D-band would be fitted with a highly symmetric single Lorentzian curve with full width at half maximum (FWHM) ≈30 cm−1.24 In the synthesized CL-graphene (Figure 1D), a single sharp 2D peak was obtained when fitted with a Lorentzian line-shape function, it was 1.3 times more intense than the G peak and red shifted compared to GR (Δū =28 cm−1).25 I2D/ID was equal to 1.33 and the FWHM was 51 cm−1, which suggests 2–3 layer thickness; the number of layers and in-plane crystallite size was estimated to be equal to 3 and 19.22 nm, respectively.26–29
XPS spectra reflect the chemical environment of carbon atoms in GR and CL-graphene. The shift in binding energy, change in peak width (measured as FWHM in eV), and the relative concentration are the three most important spectral parameters, shown in Table 1. The full spectra of both samples (Figure 2) show C1s (≈284.4 eV) and O1s (≈534 eV) core level peaks; CL-graphene has an additional N1s (401 eV) peak due to CL. The C1s spectra were deconvoluted using CasaXPS software and the Voigt function. In GR, the C1s peak is highly asymmetric in nature, centered at 284.5 eV (Figure 1B), and the major component which corresponds to the C-C/C=C bonds of sp2 hybridized carbon atoms. This was accompanied by a minor peak at 285.6 eV due to surface-adsorbed water molecules and a shake-up peak at higher binding energy ≈289.4 eV related to the π-π* transition typical of carbon atoms in aromatic compounds excited by impinging X-ray photoelectrons. The CL-graphene spectra (Figure 2C, D) are totally different; the most interesting being the complete disappearance of the shake-up peak brought about by weakening of the Van der Waals interactions between π-stacked graphitic layers. Another interesting feature is the broadening of the sp2 component at 284.4 eV (FWHM =0.98 eV for GR and FWHM =1.32 eV for CL-graphene) which signifies bonding with CL molecules; the peaks at 285.7 eV and 288.2 eV correspond to hydroxyl (C-OH), carbonyl (C-O), carboxyl (C=O), and amine (C-NH) functionalities; C=O bonds arising from acetic acid, C=O and C-NH are CL signatures. In addition to the broadened line profile, the relative concentration of the C-C/C=C peak also decreases sharply from 75.50 to 58.76 (Table 1), which proves the attachment of CL fibrils to the graphene substrate.
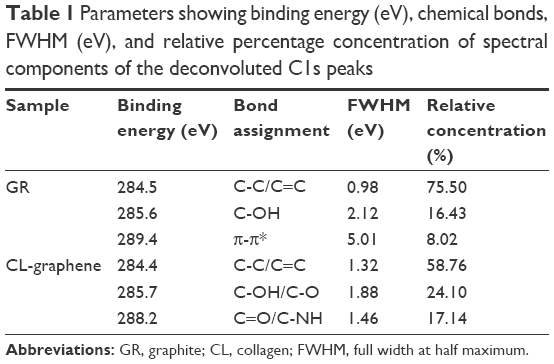 | Table 1 Parameters showing binding energy (eV), chemical bonds, FWHM (eV), and relative percentage concentration of spectral components of the deconvoluted C1s peaks |
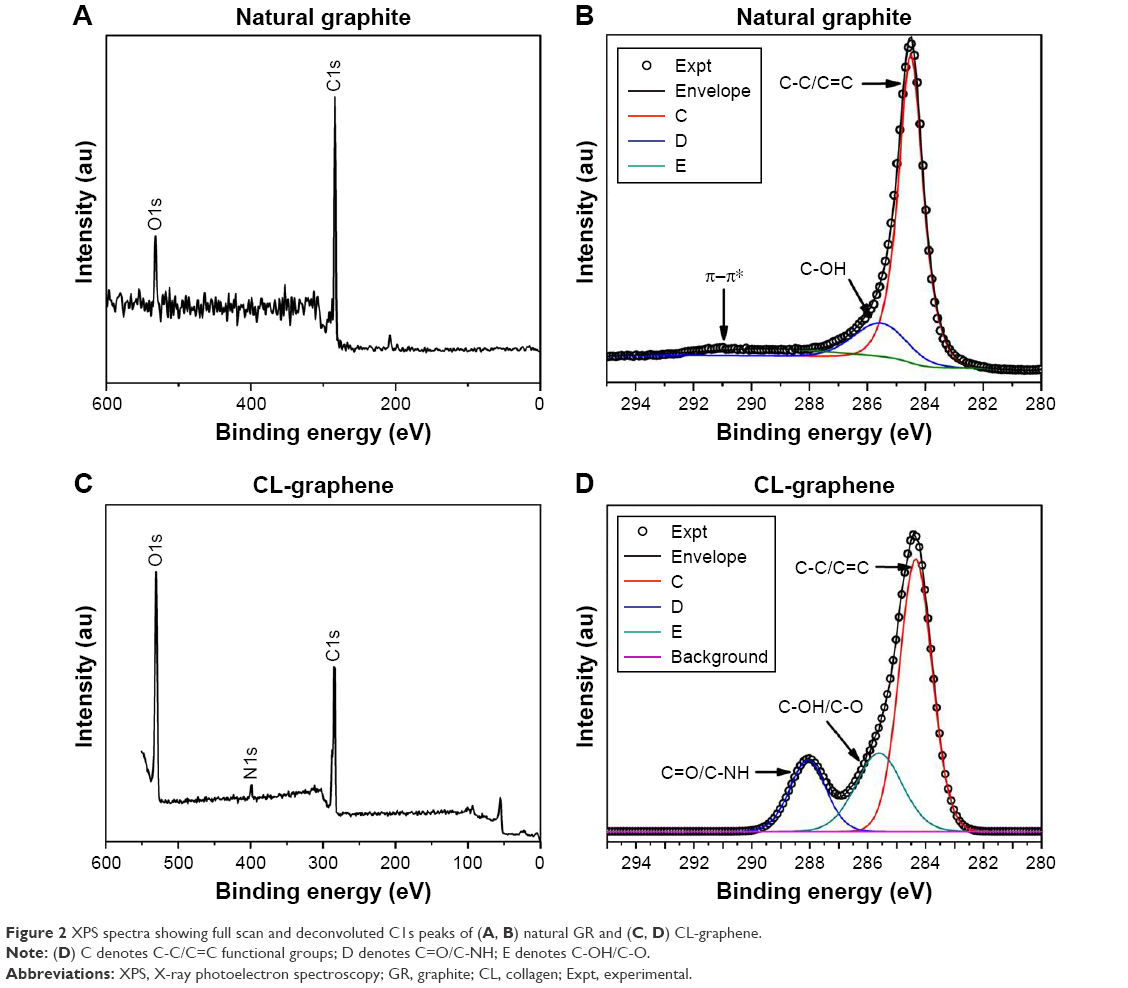 | Figure 2 XPS spectra showing full scan and deconvoluted C1s peaks of (A, B) natural GR and (C, D) CL-graphene. |
Synthesis of inorganic nanoparticles within the CL-graphene template
The XRD pattern of nanoparticles synthesized in the previously mentioned CL-graphene is shown in Figure 3A–D, indexed with reference to standard diffraction patterns (Joint Committee on Powder Diffraction Standards [JCPDS] file number 01-0646 for GR, JCPDS file number 04-0783 for Ag, JCPDS file number 09-0432 for HA, and JCPDS file number 16-0653 for ε-Fe2O3). A prominent X-ray peak was observed in the CL-graphene diffraction pattern at 2θ=22° accompanied by a minor peak at 2θ=55° corresponding to the (002) and (004) crystal planes of graphene. These peaks disappeared after inorganic nucleation; its re-appearance in CL-graphene-HA indicates the formation of HA which shares the same hexagonal unit cell as graphene. The average crystallite size was estimated from the most intense peak using Scherrer equation, where the shape factor (K) was taken as 0.89. The crystallite size was 60 nm for CL-graphene, 19 nm for CL-graphene-Ag, 54 nm for CL-graphene-IO, and 5 nm for CL-graphene-HA; previous work on inorganic nucleation without the CL-graphene template has reported much higher values, such as 65 nm for Ag,30 228 nm for IO,31 and 22 nm for HA.32 The effect could possibly be because of the strain induced in the 2D template after dense nucleation.
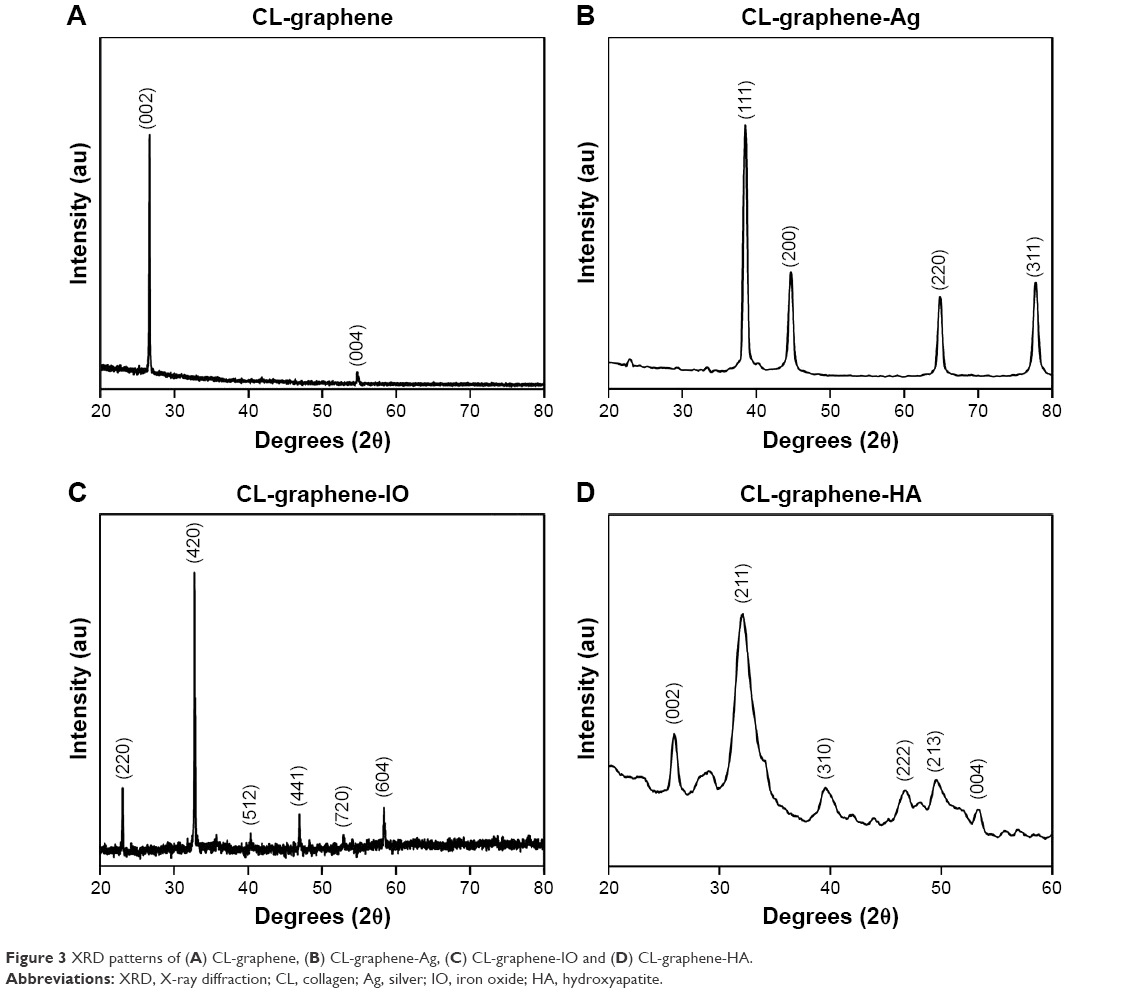 | Figure 3 XRD patterns of (A) CL-graphene, (B) CL-graphene-Ag, (C) CL-graphene-IO and (D) CL-graphene-HA. |
Post-exfoliation by CL, thin CL-graphene sheets with three layers of G, as revealed by Raman analysis, would mean less than 1 nm break-off from the parent GR stacks as their sheer thinness makes them curl to form beads. A scanning electron micrograph (Figure 4Ai) of exfoliated CL-graphene revealed bead-like structures; on increasing magnification, these beads also revealed porosity, Figure 4Aii. The mean flake and pore size was 218 nm and 28 nm, respectively; electron dispersive X-ray spectroscopy (EDS) of the beads showed very high carbon (atomic %=66.53) and nitrogen (atomic %=33.47) content; although elemental carbon can arise from both graphene and CL molecules, the presence of nitrogen proved the presence of CL. After interaction with Ag ions and subsequent reduction to metallic Ag, the bead dimensions increased (Figure 4Bi), which we believe could only result from the lateral gluing of de-stacked graphene flakes post-nucleation; the flake lengths extended to the micron range. Ag clusters were more visible at select interfaces where CL was exposed, as verified repeatedly by EDS spectra (not shown); elemental analysis of the CL-graphene-Ag composites showed a very high percentage of Ag (atomic %=80.19) with minor percentages of carbon (atomic %=14.02) and nitrogen (atomic %=5.79). The magnified image of Figure 4Bii shows a region that is densely nucleated. After IO nucleation, the CL-graphene surface was fully coated with IO nanoparticles, clearly visible in Figure 4Ci; the co-existence of many such nucleated graphene flakes was also visible in the images. The elemental composition showed a very high percentage of iron (atomic %=76.91) followed by carbon (atomic %=12.42) and nitrogen (atomic %=6.48); with increased magnification (Figure 4Cii), one can see that these flakes exist as distinct folded clusters; folding of flakes takes place after a certain threshold nucleation (a series of micrographs, not shown, suggests this). Figure 4Di, CL-graphene-HA, shows nanosized HA particles of 8 nm in size, far smaller than the usual 20–30 nm particles synthesized without graphene, possibly because of the strain induced by inorganic nucleation.32 Figure 4Dii also shows folding of nucleated sheets. The carbon and nitrogen content of CL-graphene-IO and CL-graphene-Ag is comparable, however, CL-graphene-HA has more carbon (atomic %=56.83) and nitrogen (atomic %=30.06) as compared to calcium (atomic %=5.30) and phosphorous (atomic %=7.81). This can be explained on the basis of competition between the kinetics of nucleation and adsorption; since HA is a very strong adsorbent, it is more difficult to uniformly nucleate, thus folding of sheets is also not observed. Scanning electron microscopy (SEM) was done on two different substrates to rule out any confusion with drying patterns on glass and SS304 steel.
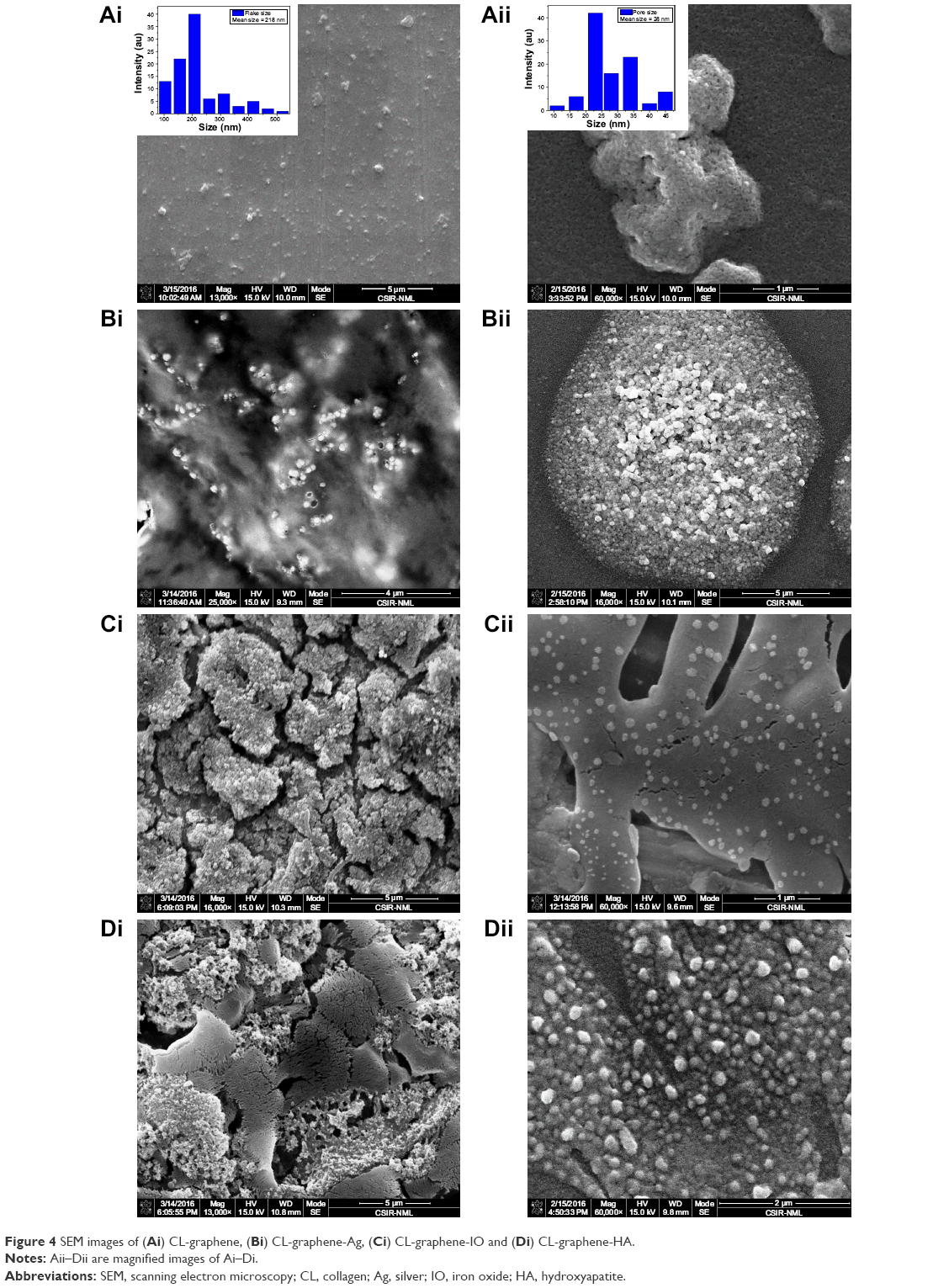 | Figure 4 SEM images of (Ai) CL-graphene, (Bi) CL-graphene-Ag, (Ci) CL-graphene-IO and (Di) CL-graphene-HA. |
The TEM image shown in Figure 5A fully corroborates the SEM findings, as perfect bead-like structures are visible in the micrograph. Figure 5B reveals details of the densely nucleated particles. The best analogy would be the beautiful inside of a cut pomegranate, membranes inside the pomegranate corresponding to the CL-graphene, with a tendency to fold because of the strain induced by nucleation. The TEM image in Figure 5C also depicts dense nucleation of IO nanoparticles which have literally coated the entire graphene surface; the uniform coating of the graphene surface makes it difficult for X-rays to differentiate individual crystallites of IO; instead, an entire graphene flake acts as one crystallite.31 The TEM image in Figure 5D shows a free-standing CL-graphene-HA film. The nucleation can be optimized to synthesize a free-standing graphene film with inorganic particles, which is expected to have many applications.
 | Figure 5 TEM images of (A) CL-graphene, (B) CL-graphene-Ag, (C) CL-graphene-IO, and (D) CL-graphene-HA. |
Raman spectra (Figure 6) of the nucleated samples qualify SEM images. After nucleation and growth of the nanoparticles, a marked difference in spectra was observed. G- and D-bands were not distinct as in the case of CL-graphene-Ag (Figure 6A); however, within the expected range 1,300–1,600 cm−1, the bands broadened considerably accompanied by splitting into three sharp peaks at 1,340 cm−1 (red shifted from 1,352 cm−1 of CL-graphene), 1,407 cm−1 (amide bonds of protein), and 1,516 cm−1 (red shifted from 1,582 cm−1 of CL-graphene) (Figure 1C). This may be attributed to the excellent SERS effect of Ag nanoparticles in the composites; such peak splitting and broadening can also be attributed to crystal nucleation and strain-induced folding of graphene sheets. The complete disappearance of the 2D band33 is due to a loss of Bernal stacking, resulting in the formation of amorphous graphene and/or doping of the graphene surface with CL.29 Another important feature is the appearance of a sharp peak at 2,930 cm−1, which is an overtone reported in covalently functionalized graphene with increasing defects; its presence also proves enhanced exfoliation.26 Apart from these, peaks at 661 and 807 cm−1 are due to the presence of AgNO3; peaks at 929, 1,034, and 1,160 cm−1 are that of Ag2O.34 Detection of Ag2O and AgNO3 suggests the possibility of intermediary products and reactants also attaching to graphene. Similar features were observed in the CL-graphene-IO spectra (Figure 6B), D- and G-bands appeared at 1,397 and 1,596 cm−1, both were blue shifted compared to CL-graphene, there was no sharp 2D peak, only a broad hump within 2,000–3,000 cm−1. The blue shift of the G-band which normally occurs at 1,580 cm−1, may be attributed to the in-plane isotropic compressive strain. Such strain-induced growth has been reported for thin film deposited graphene,35 where graphene acts as substrate and IO as film. The D+D* overtone was present in this sample too; the intensity is much less compared to CL-graphene-Ag. Peaks at 694, 564, and 426 cm−1 (not shown) represent characteristic Fe–O bond vibrations of IO.31,36 In CL-graphene-HA spectra (Figure 6C) D-band splits into 1,415 and 1,330 cm−1 and the G-band and 2D-band completely disappeared, which indicates an ability of these nucleating particles to further exfoliate CL-graphene. Peaks at 1,042 cm−1 and 1,786 cm−1 correspond to the PO43− stretching and OH bending in HA. CL-graphene had the lowest ID/IG ratio of 1.00 which increased slightly to 1.10 for both CL-graphene-Ag and CL-graphene-IO; the highest ratio of 1.60 was for CL-graphene-HA, an increase in the ratio indicates the best exfoliation as well as functionalization. However, the intensity value of the D-band reduced substantially in the composites (CL-graphene-Ag, -IO and -HA) after nucleation with respect to the template (CL-graphene); this decrease also substantiates the hypothesis of lateral gluing of flakes.
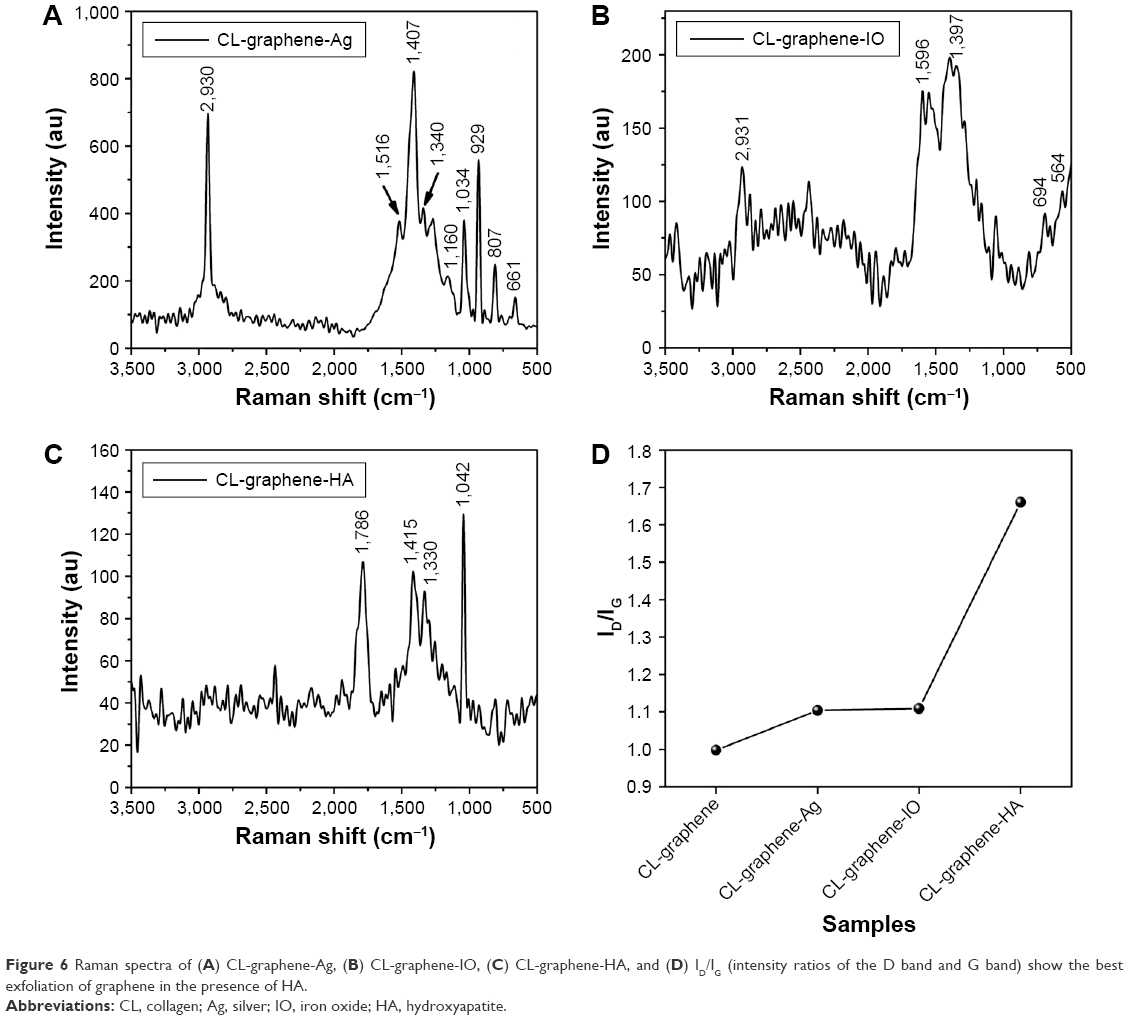 | Figure 6 Raman spectra of (A) CL-graphene-Ag, (B) CL-graphene-IO, (C) CL-graphene-HA, and (D) ID/IG (intensity ratios of the D band and G band) show the best exfoliation of graphene in the presence of HA. |
MTT assays for assessing cell growth in the presence of the three inorganic extracts for 7 days are shown in Figure 7. None of the tested extracts showed signs of cytotoxicity for the cells. A change in cell morphology, as well as cell growth and proliferation for the different extracts was captured using an inverted microscope after 7 days, as shown in Figure 7. The micrographs suggested that the cells were healthy and were unstressed without any significant change in their morphology. Furthermore, the rate of proliferation or cell viability was enhanced in the inorganic composites with regard to the control. The extensive proliferation of cells suggests that these inorganic-CL-graphene composites act as a cell growth substrate. The results are in agreement with the crystallite size calculated from XRD, which shows that as the size of the nanoparticles decreased, the cell proliferation increased, ie, CL-graphene (60 nm) < CL-graphene-IO (54 nm) < CL-graphene-Ag (19 nm) < CL-graphene-HA (5 nm).
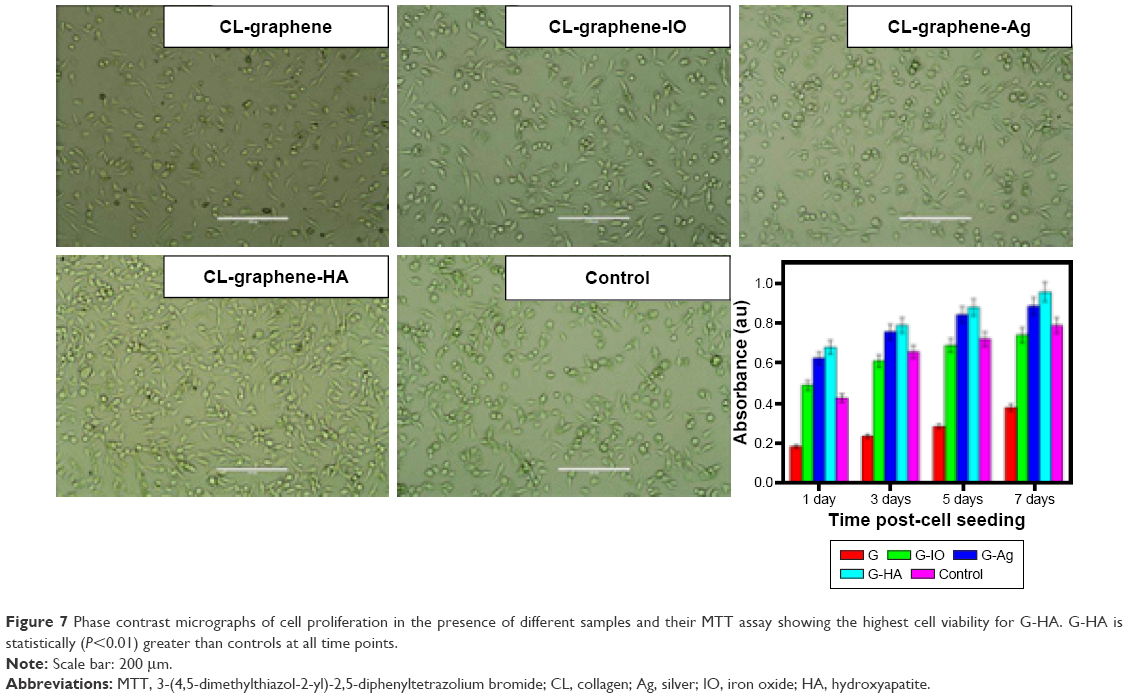 | Figure 7 Phase contrast micrographs of cell proliferation in the presence of different samples and their MTT assay showing the highest cell viability for G-HA. G-HA is statistically (P<0.01) greater than controls at all time points. |
Conclusion
This work can safely conclude that composites of graphene are fascinating materials not just for electronic applications, but also for its use as a colloidal aromatic platform for chemical reactions. CL-graphene composites as templates, define specific binding sites for the synthesis of inorganic nanoparticles which have self-assembled along hexagonal edges, as concluded from Raman analysis. The nucleation is dense and coats the 2D colloidal template to stabilize it; material characterization results described previously, support this hypothesis. This enables better anchorage and proliferation of cells with respect to the control. The immense potential of proteins like CL to form complex hierarchical structures, is yet to be explored synthetically. A better understanding of the interface between CL and growing inorganic matter is expected to open up even more medical applications.
Acknowledgments
The authors thank Yashabanta N Singhbabu for XPS measurements. The authors acknowledge the Council of Scientific & Industrial Research (CSIR), India and ESC-103 for financial support.
Disclosure
The authors declare no conflicts of interest in this work.
References
Mitra S, Banerjee S, Datta A, Chkaravorty D. A brief review on graphene/inorganic nanostructure composites: materials for the future. Indian Journal of Physics. 2016;90(9):1019–1032. | ||
Wang S, Goh BM, Manga KK, Bao Q, Yang P, Loh KP. Graphene as atomic template and structural scaffold in the synthesis of graphene-organic hybrid wire with photovoltaic properties. ACS Nano. 2010;4(10):6180–6186. | ||
Lee WC, Kim K, Park J, et al. Graphene-templated directional growth of an inorganic nanowire. Nat Nanotechnol. 2015;10(5):423–428. | ||
Jiang Z, Wang M, Cheng H, et al. facile preparation of tio 2 nanoclusters on graphene templates for photodegradation of organic compounds. J Mater Sci Technol. 2015;31(8):840–844. | ||
Kamat PV. Graphene-based nanoassemblies for energy conversion. J Phys Chem Lett. 2011;2(3):242–251. | ||
Moussa SO, Panchakarla LS, Ho MQ, El-Shall MS. Graphene-supported, iron-based nanoparticles for catalytic production of liquid hydrocarbons from synthesis gas: the role of the graphene support in comparison with carbon nanotubes. ACS Catal. 2014;4(2):535–545. | ||
Shi JL, Zhang XD, Wang XF, et al. The nanostructure preservation of 3D porous graphene: New insights into the graphitization and surface chemistry of non-stacked double-layer templated graphene after high-temperature treatment. Carbon. 2016;103:36–44. | ||
Loh KP, Bao Q, Eda G, Chhowalla M. Graphene oxide as a chemically tunable platform for optical applications. Nat Chem. 2010;2(12):1015–1024. | ||
Lerf A, He H, Forster M, Klinowski J. Structure of graphite oxide revisited. J Phys Chem B. 1998;102(23):4477–4482. | ||
Hu H, Allan CK, Li J, et al. Multifunctional organically modified graphene with super-hydrophobicity. Nano Research. 2014;7(3):418–433. | ||
Pham TA, Kim JS, Kim D, Jeong YT. Preparation of gold nanoparticles/graphene hybrid using 4-mercaptobenzoyl functionalized graphene nanosheets as templates. Synthesis and Reactivity in Inorganic, Metal-Organic, and Nano-Metal Chemistry. 2013;43(1):40–45. | ||
Preiss LC, Landfester K, Muñoz-Espí R. Biopolymer colloids for controlling and templating inorganic synthesis. Beilstein J Nanotechnol. 2014;5:2129–2138. | ||
Cullen PL, Cox KM, Bin-Subhan MK, et al. Ionic solutions of two-dimensional materials. Nat Chem. 2017;9(3):244–249. | ||
Bhattacharya S, Mishra S, Gupta P, et al. Liquid phase collagen modified graphene that induces apoptosis. RSC Adv. 2015;5(18):44447–44457. | ||
Bhattacharya S, Dhar P, Das SK, Ganguly R, Webster TJ, Nayar S. Colloidal graphite/graphene nanostructures using collagen showing enhanced thermal conductivity. Int J Nanomedicine. 2014;9:1287–1298. | ||
Nayar S, Bhattacharya S, inventor; Council of Scientific and Industrial Research, assignee. A single pot biomimetic process for the synthesis of collagen-graphite composite. Indian patent 3566DEL. 2013. | ||
Gister S, Park H, Kronenthal RL, inventors; Ethicon, Inc., assignee. Method for swelling and dispersing collagen. United States patent US patent. 1969 Jul 15. | ||
Sheikh L, Tripathy S, Nayar S. Biomimetic matrix mediated room temperature synthesis and characterization of nano-hydroxyapatite towards targeted drug delivery. RSC Adv. 2016;6:62556–62571. | ||
Hummers WS Jr, Offeman RE. Preparation of graphitic oxide. J Am Chem Soc. 1958;80(6):1339. | ||
Marcano DC, Kosynkin DV, Berlin JM, et al. Improved synthesis of graphene oxide. ACS Nano. 2010;4(8):4806–4814. | ||
Niyogi S, Bekyarova E, Itkis ME, et al. Spectroscopy of covalently functionalized graphene. Nano Lett. 2010;10(10):4061–4066. | ||
Liu WS, Zhao CY, Zhou R, Zhou D, Liu ZL, Lu XH. Lignin-assisted exfoliation of molybdenum disulfide in aqueous media and its application in lithium ion batteries. Nanoscale. 2015;7(21):9919–9926. | ||
Gao W, Alemany LB, Ci L, Ajayan PM. New insights into the structure and reduction of graphite oxide. Nat Chem. 2009;1(5):403–408. | ||
Ferrari AC, Meyer JC, Scardaci V, et al. Raman spectrum of graphene and graphene layers. Phys Rev Lett. 2006;97(18):187401. | ||
Wall M. The raman spectroscopy of graphene and the determination of layer thickness. Madison, WI: Thermo Fisher Scientific; 2011. Available from: https://tools.thermofisher.com/content/sfs/brochures/AN52252_E%201111%20LayerThkns_H_1.pdf. Accessed April 24, 2017. | ||
Pimenta MA, Dresselhaus G, Dresselhaus MS, Cançado LG, Jorio A, Saito R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys Chem Chem Phys. 2007;9(11):1276–1291. | ||
Wang G, Zhang M, Liu S, Xie X, et al. Synthesis of layer-tunable graphene: a combined kinetic implantation and thermal ejection approach. Adv Funct Mater. 2015;25(24):3666–3675. | ||
Mohiuddin TM, Lombardo A, Nair RR, et al. Uniaxial strain in graphene by Raman spectroscopy: G peak splitting, Grüneisen parameters, and sample orientation. Phys Rev B. 2009;79(20):205433. | ||
Gong L, Young RJ, Kinloch IA, et al. Reversible loss of Bernal stacking during the deformation of few-layer graphene in nanocomposites. ACS Nano. 2013;7(8):7287–7294. | ||
Mishra S, Nayar S. Protein-polymer matrix mediated synthesis of silver nanoparticles. Nanobiomedicine. 2014;1:3. | ||
Bhattacharya S, Roychowdhury A, Das D, Nayar S. Multi-functional biomimetic graphene induced transformation of Fe3O4 to ε-Fe2O3 at room temperature. RSC Adv. 2015;5:89488–89497. | ||
Sinha A, Nayar S, Agarwal A, Bhattacharyya D, Rao PR. Synthesis of nanosized and microporous precipitated hydroxyapatite in synthetic polymers and biopolymers. J Am Ceram Soc. 2003;86(2):357–359. | ||
Casiraghi C, Pisana S, Novoselov KS, Geim AK, Ferrari AC. Raman fingerprint of charged impurities in graphene. Appl Phys Lett. 2007;91(23):233108. | ||
Martina I, Wiesinger R, Jembrih-Simbürger D, Schreiner M. Micro-Raman characterisation of silver corrosion products: instrumental set up and reference database. e-PS. 2012;9:1–8. | ||
Shioya H, Craciun MF, Russo S, Yamamoto M, Tarucha S. Straining graphene using thin film shrinkage methods. Nano Lett. 2014;14(3):1158–1163. | ||
Cornell RM, Schwertmann U. The Iron Oxides: Structure, Properties, Occurences and Uses. Wiley-VCH Verlag GmbH & Co KGaA; 2003. |
Supplementary materials
UV absorbance spectroscopy
Optical density was observed at a fixed wavelength of 270 nm (Figure S1). The peaks measured at 270 nm correspond to the n → π* transition of C=O functional groups, the presence of oxygen could be from water and acylated collagen (CL). The increase in absorbance proves the functionalization of the G surface and the gradual opening of the optical band gap. The aromatic residues of proteins also show absorbance in this region (270–280 nm), but there is no doubt that the absorbance increased after inorganic nucleation in the synthesized composites, which was even more than the commercially available quantum dots (QDs). The increase followed the sequence CL-graphene-hydroxyapatite (HA) > CL-graphene-ironoxide (IO) > CL-graphene-silver (Ag) > CL-graphene > QD. There is not much of a difference between CL-graphene-Ag and CL-graphene-IO. CL-graphene-HA however, exhibited a steep increase in absorbance. Previous reports on exfoliated graphene in organic solvents like o-dichlorobenzene have shown that with increased sonication time, the optical density increases, which reflects an effective functionalization and exfoliation.1 The concentration of CL-graphene was calculated from the Beer-Lambert law
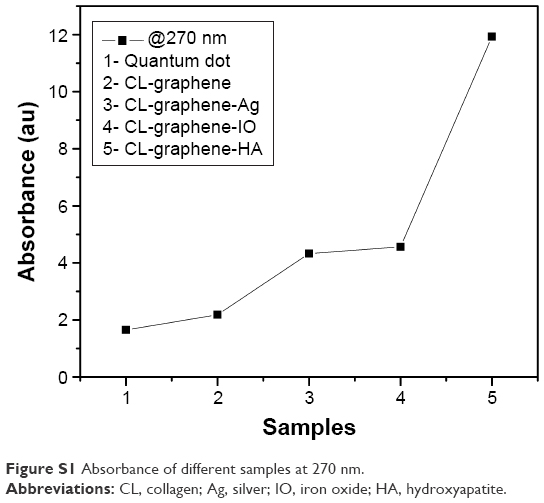 | Figure S1 Absorbance of different samples at 270 nm. |
|
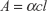
where A is the optical density, l is the length of the optical path (1 cm), and α is the absorption coefficient (mg−1mLcm−1). The calibration curve for Figure S2 was drawn for a known volume of QDs (concentration =1 mg/mL) and absorbance measured with serial dilution, the data were fitted with a linear profile and the concentration was found to be 0.2 mg/mL from the graph.
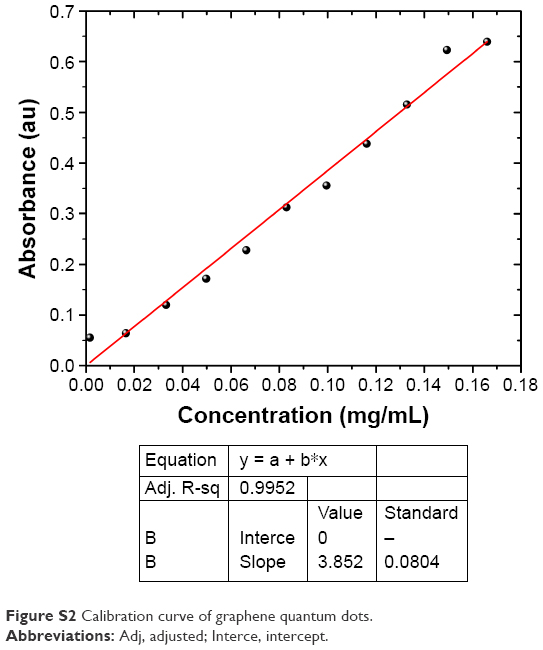 | Figure S2 Calibration curve of graphene quantum dots. |
Reference
Chul L, Leconte N, Jiho K, Cho D, Lyo I, Choi EJ. Optical spectroscopy study on the effect of hydrogen adsorption on graphene. Carbon. 2016;103:109–114. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.