Back to Journals » International Medical Case Reports Journal » Volume 18
Transthyretin-Related Familial Amyloidosis Polyneuropathy with Spinal Cord Damage: A Case Report
Authors Liu H ![]() , Huang C, Du Y, Liu J, Ren X, Wang H, Ye J, Zhou H, Duan Z
, Huang C, Du Y, Liu J, Ren X, Wang H, Ye J, Zhou H, Duan Z
Received 15 October 2024
Accepted for publication 18 December 2024
Published 4 January 2025 Volume 2025:18 Pages 1—5
DOI https://doi.org/10.2147/IMCRJ.S486387
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xudong Zhu
Hailin Liu,1,* Chao Huang,1,* Yanjiao Du,1 Jiacheng Liu,1,2 Xiangyang Ren,1 Huilin Wang,1 Jingna Ye,1 Haitao Zhou,1 Zhihui Duan1
1Department of Neurology, Luoyang Central Hospital Affiliated to Zhengzhou University, Luoyang Cerebrovascular Disease (Stroke) Clinical Medical Research Center, Regional Medical Center for Neurological Diseases of Henan Province, Luoyang, People’s Republic of China; 2Xinxiang Medical College, Xinxiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhihui Duan, Department of Neurology, Luoyang Central Hospital Affiliated to Zhengzhou University, Luoyang Cerebrovascular Disease (Stroke) Clinical Medical Research Center, Regional Medical Center for Neurological Diseases of Henan Province, Xigong District, Luoyang, Henan, 471000, People’s Republic of China, Tel/Fax +0086-0379-63892044, Email [email protected]
Introduction: Transthyretin protein-related familial amyloidosis polyneuropathy (TTR-FAP) is an autosomal dominant genetic disease caused by mutations in the TTR gene. The disease is characterized primarily by peripheral and autonomic nerve damage. Disease progression is associated with frequent involvement of the heart, lungs, kidneys, eyes, and other organs. The most common TTR mutation is c.148G>A (p.Val50Met), although the FAP resulting from the mutation rarely involves the spinal cord.
Patient Concerns: A 68-year-old man was diagnosed with the TTR c.148G>A (p.Val50Met) mutation by ultrasound, pathological, and genetic analyses. He presented with a late-onset, complicated spinal cord injury. The diagnostic process was tortuous, and despite the administration of regular treatment (conventional drugs, cardiac pacemaker, and the specific drug clofenadifen), the patient died.
Interventions: To confirm TTR-FAP, ultrasound, MRI, pathological, and genetic tests were performed.
Outcomes: The patient ultimately died of heart failure 7.5 years after the initial onset of symptoms.
Conclusion: The patient presented with unusual symptoms of spinal cord injury, and despite a long and arduous diagnostic process and administration of standard treatment for over seven years, the outcome was poor. It is thus recommended that clinicians pay attention to the identification of rare diseases with timely imaging, pathological, and genetic testing, to avoid poor outcomes.
Keywords: transthyretin familial amyloid polyneuropathy, myelopathy, case
Introduction
TTR-FAP is an autosomal dominant disease associated with mutations in the TTR gene resulting in peripheral and autonomic nerve damage. Disease progression often entails symptoms associated with the involvement of organs including the eyes, lungs, kidneys, and heart.1 The TTR gene c.148G>A (p.Val50Met) mutation is the most common mutation known to be associated with this disease, but reports of spinal cord involvement associated with this form of TTR-FAP are rare. Here, we report a case of a TTR-FAP patient exhibiting spinal cord damage.
Methods and Results
A 68-year-old male patient was admitted to the hospital on January 10, 2022 owing to a history of progressive weakness and numbness of the limbs for over 7 years. The patient had initially presented with numbness and swelling pain in his toes 7 years prior, followed by the development of glove and stocking-like changes in the limbs, limb weakness, and instability when walking. Despite the successive administration of neurotrophic and immunoregulatory treatments, his condition continued to progress. Two years ago, he presented with bradycardia and gastrointestinal dysfunction. One month ago, he developed paresthesia below T10, urinary retention, and orthostatic hypotension occurred 1 month ago. He presented to our hospital after the development of pulmonary embolism, heart failure, renal insufficiency, respiratory and urinary system infections, and other symptoms. He reported having no history of heavy metal or poison exposure, and his past and family history was unremarkable. Physical examination revealed a supine blood pressure of 130/70 mmHg and a standing blood pressure of 80/47 mmHg. The proximal muscle strength of his bilateral upper limbs was grade IV, his distal muscle strength was grade III, and the muscle strength of both lower limbs was grade III. Numbness at the distal end of both elbow joints and below chest 10 was evident together with hypoesthesia. Cardiac color Doppler ultrasonography revealed a rough myocardial ecco with some spot-like changes potentially consistent with amyloidosis (Figure 1). Biopsy samples of skin from the abdominal wall and lower right extremity did not reveal any deposition of an apple-green substance under polarized light. Genetic testing revealed a heterozygous mutation in exon 2 of TTR c.148G > A (p.Val30Met) (Figure 2). As related examinations excluded other diseases (Table 1), the patient was diagnosed with TTR-FAP with spinal cord damage. The patient’s condition gradually worsened, and he had malignant arrhythmia. After being transferred to the intracardiac intensive care unit, he was treated with drugs and a pacemaker and was discharged. Oral clofenadifen soft capsules were provided for treatment after discharge. The patient ultimately died of heart failure 7.5 years after initial symptom onset.
 |
Table 1 Abnormal Results of Pathological, Imaging, and Electrophysiological Examinations |
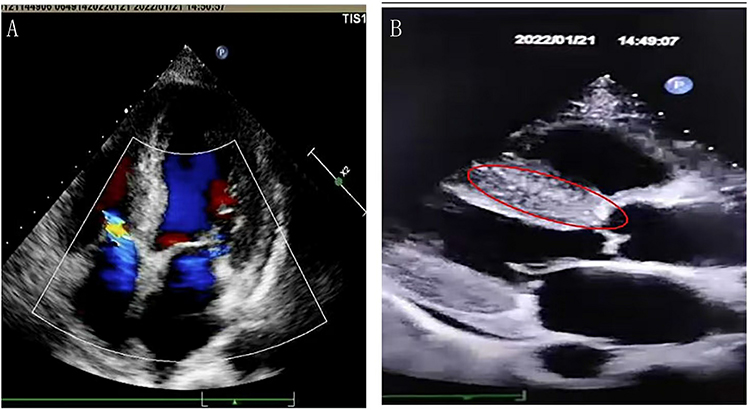 |
Figure 1 Cardiac color Doppler ultrasound results and map of the TTR gene sequence. (A) Uncoordinated movement with slight thickening of the atrial septum. (B) The myocardial echo was rough with spot-like changes; uneven enhancement of the ventricular wall is highlighted with a red circle. |
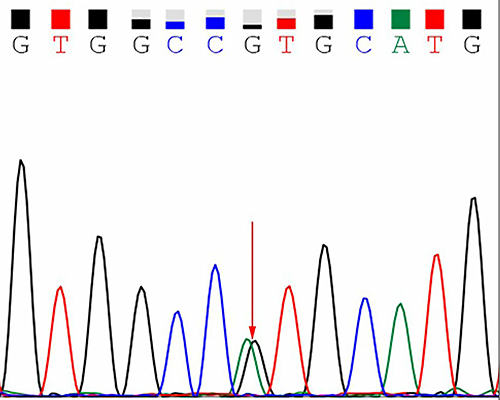 |
Figure 2 Map of the TTR gene sequence. The red arrow highlights the heterozygous mutation at exon 2 c.148G > A (p.Val30Met). |
Discussion
The patient in this report was a male who was 61 years of age at time of initial symptom onset. He did not exhibit any family history of TTR-FAP, and exhibited a chronic, progressive disease course with limb symmetry characterized by primarily distal flaccid paralysis, sensory and motor involvement, and damage to the peripheral sensory and motor peripheral nerves. Symptoms developed below the T10 spinal level and were associated with abnormal damage in the thoracic spinal cord. The patient also experienced autonomic nervous symptoms including vomiting, diarrhea, and urinary retention, as well as elevated BNP levels, blood urea nitrogen dysregulation, and lesions involving the circulatory and urinary systems. Cerebrospinal fluid, electromyography, and ganglioside autoantibody results did not support a diagnosis of chronic inflammatory demyelinating polyneuropathy. Abdominal color Doppler ultrasound and chest CT results did not reveal any space-occupying lesions, and tumor marker levels were normal, which did not support a tumor diagnosis. Serological and other marker test results did not support a diagnosis of connective tissue disease-related neurological damage. Bone marrow examination, serum protein levels, and other tests did not support a diagnosis of light chain amyloidosis peripheral neuropathy. The patient thus exhibited peripheral neuropathy with sensory, motor, and autonomic nerve damage, combined with damage to the heart, kidney, and spinal cord. The disease was also characterized as having a high possibility of being hereditary. Echocardiography revealed typical FAP cardiomyopathy manifestations, and genetic testing identified a heterozygous TTR c.148G > A (p.Val30Met) mutation. These results, together with other relevant examinations and the exclusion of other diseases, ultimately led to the diagnosis of TTR-FAP.
TTR is primarily synthesized in the liver, although 5% of the total TTR output is believed to be synthesized in the choroid plexus and retinal pigment epithelium in the brain. TTR gene mutations result in the misfolding of the TTR protein into amyloid fibrils, which are deposited in different tissues and cause disease.2 The clinical manifestations of TTR-FAP can vary widely. In areas where this disease is not endemic, it usually exhibits a late onset at 50 + years of age. Many patients experience bilateral upper limb symptoms that manifest in the form of progressive polymotor sensory neuropathy and severe movement disorders, often accompanied by heart disease, with relatively mild autonomic symptoms.3,4 In the present case, the patient was from a non-endemic area and exhibited late-onset disease that first manifested at age 61. He experienced initial onset in both upper extremities and progressive sensorimotor neuropathy in the extremities, as well as heart and kidney involvement, consistent with prior reports. This patient presented with spinal cord damage, in contrast to these previous reports. TTR-FAP damage to the central nervous system is rare and usually occurs 10 years after disease onset.5 Oshima et al6 observed a low proportion of TTR in the spinal cord of TTR-FAP patients, with this potentially being related to the independent origin of TTR in the central nervous system from the choroid plexus. This patient had clear symptoms of spinal cord damage, but no corresponding lesions were detected upon spinal cord MRI, which may be attributable to the lack of any further cervical or thoracic spinal enhanced MRI scans.Unfortunately, due to refusal by the family, no autopsy was performed for further confirmation of the myelopathy.
Typical echocardiographic findings in patients harboring TTR mutations include the thickening of the ventricular wall, granular myocardial signal, and restricted diastolic function.7,8 Tissue biopsy and pathology are also important components of the approach to FAP diagnosis, with liver and nerve tissue biopsy being performed when conditions allow for such testing.9,10 Skin biopsy offers a sensitivity of 70% and a specificity of 100% for this condition.11 TTR gene sequencing is a gold standard approach to TTR-FAP diagnosis. In the present case, the patient’s cardiac color Doppler ultrasound results were consistent with the typical presentation associated with TTR-FAP, although his skin pathology results did not yield any direct evidence of amyloid deposition, potentially as a result of the selected sampling site. Genetic testing revealed a heterozygous Val30Met mutation in the TTR gene. While this mutation is relatively common, associated spinal cord damage is rarely detected. Further testing of related family members would provide further insight, but the family members of this patient refused to undergo such genetic testing.
Liver transplantation is the most commonly suggested approach to the treatment of TTR-FAP patients, and it is most often recommended for patients with stage 1 peripheral neuropathy and an age at onset of under 50 years.12 In the present case, the patient exhibited severe stage 3 and grade 4 TTR-FAP,13 arrhythmia, and pacemaker implantation, and he was thus not a candidate for liver transplantation. While this patient briefly achieved stability, he ultimately died 7.5 years after symptom onset. As such, efforts to treat TTR-FAP patients require vigilance to facilitate early diagnosis and appropriate interventions with the potential to improve prognostic outcomes.
Conclusion
In conclusion, TTR-FAP is a rare disease that is difficult to diagnose in its early stages, particularly when combined with spinal cord symptoms. Echocardiography is a reproducible and noninvasive approach to assessing cardiac features and function in patients affected by cardiac amyloidosis, and genetic testing plays a critical role in TTR-FAP diagnosis.
Abbreviations
TTR-FAP, Transthyroxin protein-related familial amyloidosis polyneuropathy; TTR, Transthyretin.
Ethics and Consent Statements
Publication of case details does not require Institutional approval. Written informed consent was obtained from the guardian of the patient for the publication of the case details and accompanying images.
Acknowledgments
The authors would like to thank all the reviewers who participated in the review and MJEditor (www.mjeditor.com) for its linguistic assistance during the preparation of this manuscript. This paper has been uploaded to ResearchSquare as a preprint: https://www.researchsquare.com/article/rs-3458524/v1.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Supported by Luoyang Applied Technology Research and Development Project (2001024A).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Plante-Bordeneuve V, Ferreira A, Lalu T, et al. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology. 2007;69:693–698. doi:10.1212/01.wnl.0000267338.45673.f4
2. Faria TQ, Almeida ZL, Cruz PF, Jesus CSH, Castanheira P, Brito RMM. A look into amyloid formation by transthyretin: aggregation pathway and a novel kinetic model. Phys Chem Chem Phys. 2015;17:7255–7263. doi:10.1039/c4cp04549a
3. Luigetti M, Romano A, Di Paolantonio A, Bisogni G, Sabatelli M. Diagnosis and treatment of hereditary transthyretin amyloidosis (hATTR) polyneuropathy: current perspectives on improving patient care. Ther Clin Risk Manag. 2020;16:109–123. doi:10.2147/TCRM.S219979
4. Tojo K, Tsuchiya-Suzuki A, Sekijima Y, Morita H, Sumita N, Ikeda SI. Upper limb neuropathy such as carpal tunnel syndrome as an initial manifestation of ATTR Val30Met familial amyloid polyneuropathy. Amyloid. 2010;17:32–35. doi:10.3109/13506121003619369
5. Dardiotis E, Andreou S, Aloizou AM, et al. The frequency of central nervous system complications in the Cypriot cohort of ATTRV30M neuropathy transplanted patients. Neurol Sci. 2020;41:1163–1170. doi:10.1007/s10072-019-04176-9
6. Oshima T, Kawahara S, Ueda M, et al. Changes in pathological and biochemical findings of systemic tissue sites in familial amyloid polyneuropathy more than 10 years after liver transplantation. J Neurol Neurosurg Psychiatry. 2014;85:740–746. doi:10.1136/jnnp-2013-305973
7. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872–2891. doi:10.1016/j.jacc.2019.04.003
8. Bourque PR, McCurdy AR, Mielniczuk LM, Dennie C, Veinot JP, Warman Chardon J. Cardiac amyloidosis phenotype associated with a Glu89Lys transthyretin mutation. Can J Cardiol. 2017;33:
9. Yang NCC, Lee MJ, Chao CC, et al. Clinical presentations and skin denervation in amyloid neuropathy due to transthyretin Ala97Ser. Neurology. 2010;75:532–538. doi:10.1212/WNL.0b013e3181ec7fda
10. Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36:411–423. doi:10.1002/mus.20821
11. Adams D, Ando Y, Beirao JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268:2109–2122. doi:10.1007/s00415-019-09688-0
12. Vollmar J, Schmid JC, Hoppe-Lotichius M, et al. Progression of transthyretin (TTR) amyloidosis in donors and recipients after domino liver transplantation-a prospective single-center cohort study. Transpl Int. 2018;31:1207–1215. doi:10.1111/tri.13326
13. Adams D, Suhr OB, Hund E, et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016;29(Suppl 1):S14–S26. doi:10.1097/WCO.0000000000000289
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.