Back to Journals » Journal of Inflammation Research » Volume 8
Translational utility of experimental autoimmune encephalomyelitis: recent developments
Authors Guerreiro-Cacais AO, Laaksonen H, Flytzani S, N’diaye M, Olsson T, Jagodic M
Received 2 September 2015
Accepted for publication 30 September 2015
Published 13 November 2015 Volume 2015:8 Pages 211—225
DOI https://doi.org/10.2147/JIR.S76707
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Andre Ortlieb Guerreiro-Cacais, Hannes Laaksonen, Sevasti Flytzani, Marie N'diaye, Tomas Olsson, Maja Jagodic
Neuroimmunology Unit, Department of Clinical Neuroscience, Center for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden
Abstract: Multiple sclerosis (MS) is a complex autoimmune condition with firmly established genetic and environmental components. Genome-wide association studies (GWAS) have revealed a large number of genetic polymorphisms in the vicinity of, and within, genes that associate to disease. However, the significance of these single-nucleotide polymorphisms in disease and possible mechanisms of action remain, with a few exceptions, to be established. While the animal model for MS, experimental autoimmune encephalomyelitis (EAE), has been instrumental in understanding immunity in general and mechanisms of MS disease in particular, much of the translational information gathered from the model in terms of treatment development (glatiramer acetate and natalizumab) has been extensively summarized. In this review, we would thus like to cover the work done in EAE from a GWAS perspective, highlighting the research that has addressed the role of different GWAS genes and their pathways in EAE pathogenesis. Understanding the contribution of these pathways to disease might allow for the stratification of disease subphenotypes in patients and in turn open the possibility for new and individualized treatment approaches in the future.
Keywords: autoimmunity, multiple sclerosis, risk genes, EAE, knockouts, pathways
Introduction
Multiple sclerosis (MS) is a debilitating chronic inflammatory disease of the central nervous system (CNS) characterized by autoimmune destruction of myelin and subsequent loss of neurons. The cause of disease remains unknown, but epidemiological studies have clearly established genetic factors in MS etiology.1–3 The first genetic risk factor has been described in early 1970s and mapped to the human leukocyte antigen (HLA) complex,4,5 which encodes numerous genes with immune functions. More recently, this strongest genetic influence was refined to HLA-DRB1*1501 that confers threefold increased risk to develop MS5,6 and encodes molecules involved in the presentation of antigens to T-cells. With the advent of genome-wide association studies (GWAS) and large international efforts to gather sufficiently powered cohorts, more than 100 non-HLA variants have been identified to predispose for MS5,7–9 together with multiple variants and alleles within the HLA locus itself.5,10 The identified MS risk variants collectively indicate genetically regulated immune functions that control disease susceptibility and they have set the stage for molecular characterization of mechanisms causing MS. Nevertheless, apart from few examples,11–13 interpretation of the causal variants is limited and their mechanisms are still largely unknown.
Experimental autoimmune encephalomyelitis (EAE) is an animal model widely used to study mechanisms of inflammation in the CNS.14 EAE can be induced in a variety of species either by active immunization with CNS antigens in adjuvant or by passive transfer of CNS-specific T-cells. Although no single EAE model can recapitulate the complexity of MS, EAE has been successfully used to study mechanisms of relevance for MS and translate them into therapeutic interventions.15 The knowledge regarding the role of MS risk genes in vivo largely comes from EAE models, primarily owing to the possibility of gene targeting in mice. In this review, we summarize the current knowledge of the mechanisms of well-established MS risk genes5,9 (Table 1) and discuss more thoroughly those for which more abundant EAE data are available. Where possible, the genes were grouped, in the text, according to the pathways or cellular functions they fulfill. Additionally, we address in Table 1 whether data on the functional consequence of the human single-nucleotide polymorphism (SNP) is available as well as whether any clinical trials targeting these molecules are underway.
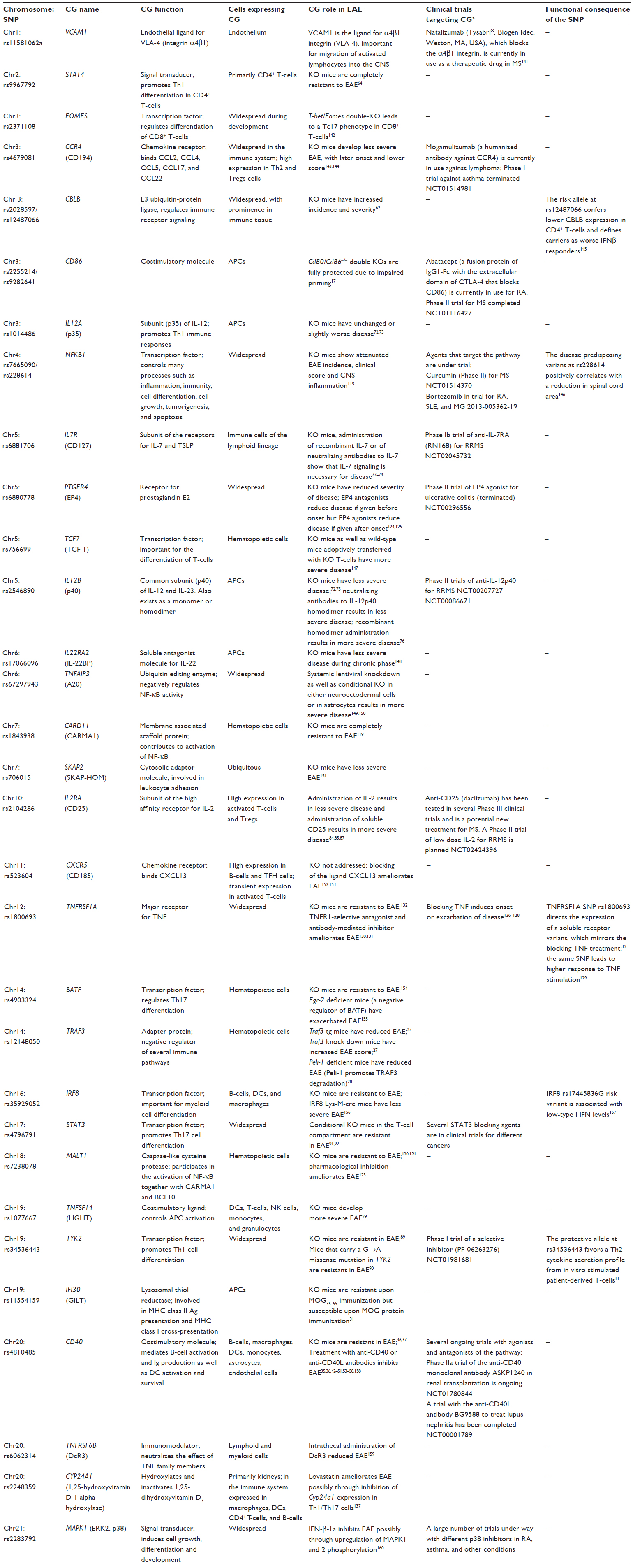 | Table 1 MS risk genes for which EAE data are available |
APC function and costimulation
CD86
CD86 (B7.2), together with the structurally homologous CD80 (B7.1), are important costimulatory molecules that regulate the crosstalk between antigen presenting cells (APCs) and T-cells, delivering “signal 2” for T-cell activation. They are upregulated upon APC activation in specific and distinct temporal patterns and bind to both CD28 and CTLA-4 on T-lymphocytes, leading either to enhancement or inhibition of T-cell function, respectively.16 In EAE, because of greatly overlapping and compensatory effects between CD86 and CD80,17 double-knock-out (KO) animals (Cd80/Cd86–/–) have been used to address the role of the receptors in disease development. Cd80/Cd86–/– animals immunized with myelin oligodendrocyte glycoprotein (MOG) show an impaired induction phase of EAE. However, transfer of MOG-specific wild-type (WT) T-cells into Cd80/Cd86–/– mice, in which any defects in priming are overridden, also leads to less severe disease with eventual complete remission, while WT recipients present with chronic progressive disease. These experiments point to a role of this costimulatory pathway in the priming of the response as well as in the effectors phase.17 Administration of antibodies against CD2818 or CTLA-4Ig fusion protein,19 which both block the pathway, lead to a reduction of disease severity during the effector phase both when given systemically as well as intrathecally, for the latter.20 Abrogation of the pathway attenuates the immune response at least partly due to death in situ of encephalitogenic T-cells.21 Because of the widespread expression of CD80/CD86 in the CNS during EAE, it is difficult to discriminate whether local APCs (microglia, dendric cells [DCs]) or infiltrating cells (monocytes) are responsible for the costimulatory events that sustain inflammation. However, even though microglia in preactive and remyelinating MS lesions do express CD86,22 the expression levels are much lower than on classical DCs or monocyte-derived DCs as judged from EAE experiments,23,24 suggesting the latter as crucial cells in the aforementioned restimulation events.
TRAF3
TRAF3 is part of the TNF receptor-associated factor family and is an adapter protein. It is a potent inhibitor of different signaling pathways including CD154 (CD40L), toll-like receptors (TLR), and IL-17R.25–27 TRAF3 can negatively regulate IL-17 signaling; Traf3 transgenic mice, which express significantly higher levels of TRAF3, have reduced EAE score and later onset. Accordingly, Traf3 knock down mice have exacerbated disease.27 Peli-1, which promotes degradation of TRAF3, is abundantly expressed in microglia. Peli-1-deficient mice, in which levels of TRAF3 remain high, have reduced EAE as well, in spite of normal peripheral T-cell activation.28 This reduction in EAE is due to an impaired response of microglia to inflammatory stimuli. In summary, TRAF3 is a negative regulator of signaling pathway in multiple cell types, affecting both peripheral as well as CNS immune activation stages.
TNFSF14
TNFSF14 encodes for LIGHT, a newly identified costimulatory ligand expressed on DCs, T-cells, natural killer (NK) cells, monocytes, and granulocytes.29 LIGHT binds to three receptors, DcR3, herpes virus entry mediator (HVEM), and lymphotoxin b receptor (LTbR), and drives increased T-cell proliferation and Th1 cytokine expression.
LIGHT has been shown, in one study, to be an important factor for the recovery phase of EAE.29 Light-deficient C57BL/6 mice develop a more severe EAE after immunization with MOG35–55 peptide compared to WT mice. While KO mice have more activated microglia/macrophages in the CNS, CD4+ T-cells from lymph nodes draining the immunization site exhibit lower IFNγ and IL-17 production. The paradoxical effect of disease exacerbation in Light-deficient mice in spite of lower Th1/Th17 effector functions is explained by adoptive transfer of encephalitogenic T-cells into KO mice, showing that LIGHT is not essential for disease induction but plays a major role in limiting disease progression and tissue damage by controlling activated macrophages/microglia in the CNS during inflammation.29
IFI30
IFI30 encodes for GILT, an enzyme that functions in MHC class II-restricted antigen processing and MHC class I-restricted cross-presentation.30 GILT may alter the character of immune responses and affect central tolerance.
Gilt KO mice are resistant to EAE induced with MOG35–55 as they fail to induce a proper antigen-specific CD4+ T-cell response.31 KO mice immunized with whole MOG protein are, however, susceptible to EAE. However, while T-cells from WT mice respond to MOG35–55, T-cells from KO animals proliferate against a different array of peptides. Furthermore, Gilt KO mice develop a disease characterized by antibody-mediated effects, indicating a switch in the pathogenic mechanism due to peptide repertoire change. The role of GILT as an endosomal reductant has also been shown by Burrows et al32 by using RTL550-CYS-MOG, a recombinant TCR (T-cell receptor) ligand (RTL) bearing cysteine-tethered antigenic peptides, to treat EAE. RTL550-CYS-MOG inhibits EAE in WT mice but not in Gilt KO mice, since RTLs must be endocytosed and presented by MHC class II and since GILT is required to liberate these cysteine-tethered peptide ligands in late endocytic compartments.
CD40
CD40 is a costimulatory molecule on APCs. The interaction of CD40 with its ligand CD40L (CD154), expressed on activated T-cells, influences a variety of immune functions including B-cell activation and Ig production, and DC survival.33
It has been shown that CD40 and CD40L expression in inflammatory cells infiltrating the CNS of mice is significantly increased during acute EAE and relapses, and decreased during remission.34 Furthermore, perivascular infiltrates of mononuclear cells have abundant expression of CD40 and CD40L in the CNS of marmoset monkeys with demyelinating EAE.35
Experiments using Cd40 KO mice have shown the importance of CD40-CD40L pathway in EAE development and Th polarization.36,37 Cd40 KO mice are resistant to EAE development, fail to drive Th17 differentiation, and exhibit reduced IL-6 production by DCs. Furthermore, mice that receive Cd40–/– DC cells prior to EAE induction exhibit an impaired ability to prime a MOG-specific IL-17 response even though their ability to induce IFN-γ production is similar to mice injected with control DCs.38 The administration of Cd40–/– DC loaded with MOG prior to standard MOG immunization also prevents the onset of EAE.39 Treatment of mice with EAE with bone marrow-derived dendritic cells (BMDC) transduced with lentiviral vectors encoding CD40 shRNA results in significant decrease of EAE compared to mice treated with BMDCs transduced with control vectors.40 EAE inhibition is even more profound when mice are injected with BMDCs cotransduced with shRNA to both CD40 and the IL-23 p19 subunit, leading to further dampening of the Th17 response. Ablation of signaling by deletion of the ligand, CD40L, using Cd40l KO mice that carry a myelin basic protein (MBP)-specific transgenic TCR also leads to EAE resistance and lack of CNS inflammation.41
Treatment with anti-CD40L monoclonal antibody (mAb) concomitantly to myelin antigen immunization completely prevents EAE development. When anti-CD40L mAb is administered after EAE onset and before peak of the disease, it significantly reduces EAE symptoms.35,36,42 When anti-CD40L mAb is administered during EAE remission, it prevents further clinical relapses.43 In addition, several reports have shown the short- and long-term inhibition of EAE in different EAE animal models using anti-CD40L treatments, suggesting that CD40-CD40L interactions may play a role in the ability of encephalitogenic T-cell to interact with APCs in the CNS and increase Th effector functions.44–51 Treatment with a combination of anti-CD40L Ab and CTLA4Ig confers additive protection against EAE and is associated with complete absence of inflammatory cell infiltrates in the CNS.52 These observations have been further corroborated by studies in marmoset monkeys and mice that show that antibodies that block CD40 inhibit EAE53–57 and suppress magnetic resonance imaging-detectable inflammation and enlargement of brain lesions.58 Finally, Ichikawa and Williams59 have shown that activation of the CD40-CD40L pathway is sufficient to overcome tolerance against self-antigens.59 In this study, myelin-reactive T-cells from tolerized donors are converted into pathogenic effector cells upon reactivation of specific lymph node cells with anti-CD40 agonists and are able to proliferate, secrete cytokines, and induce passive EAE in SJL mice.
TCR signaling
CBLB
CBLB is an E3 ubiquitin-protein ligase, which negatively regulates TCR, B-cell receptor (BCR), and FCµR1 signal transduction pathways, playing an important role in peripheral tolerance maintenance. In naïve T-cells, it inhibits VAV1 activation upon TCR engagement, but not other pathways such as Zap-70 and Lck, Ras/MAPK, PLC-γ1, or Ca2+ mobilization. In this way, CBLB imposes a requirement for CD28 costimulation for proliferation and IL-2 production, heightening the activation threshold for T-cells.60 An additional observation linking CBLB to tolerance induction is that CBLB expression in T-cells is controlled by CD28 and CTLA-4. CD28 costimulation induces CBLB ubiquitination and proteasomal degradation, while CTLA-4-B7 interaction induces Cblb expression.60 Independent of the aforementioned mechanism, CBLB has also been shown to control the generation of peripheral inducible Treg cells in response to TGFβ signaling.61 The control of tolerance at multiple levels is revealed in Cblb–/– mice immunized with MBP, which show a higher incidence and higher EAE score than their WT counterparts.62 These animals also present T-cell abnormalities in lymph node trafficking patterns, with increased expression of S1P1 on T-cells, which do however not impact their sensitivity to FTY720 (fingolimod) treatment.63
Cytokine signaling and Th phenotype
STAT4
STAT4 is a transcription factor essential for CD4+ T-cell differentiation to the Th1 phenotype. CD4+ T-cells respond to the cytokines IL-27 and IL-12 through STAT1 and STAT4 phosphorylation, respectively, leading to subsequent nuclear translocation, where they induce IFNγ production and expression of the master transcriptional regulator T-bet.
Both Th1 and Th17 T-cells can induce EAE and appear implicated in MS. However, while mice deficient in IL-12, STAT1, and IFNγ not only still develop EAE, but often present with exacerbated disease in the case of the latter; animals knocked out for Stat464 and T-bet are resistant to EAE.65 Deletion of T-bet specifically on CD4+ T-cells does not abrogate encephalitogenicity,66 implying expression in other cells as essential, and leaves STAT4 as a major player in disease establishment. Additionally, the regulation of immunity by STAT4 goes beyond mere induction of gene transcription in that STAT4 can promote active epigenetic marks.67 Recently, a study has shown that STAT4 is essential for the induction of GM-CSF secretion in both Th1 and Th17 by binding directly to the Csf2 promoter.68 Since GM-CSF is the only T-cell effector cytokine shown to date to be absolutely essential for EAE induction,69,70 the results of this study come to resolve the conundrum.
While no data on SNP influence on expression or splicing of STAT4 is available, one study has addressed the role of an alternative isoform of STAT4 (STAT4β), which lacks 44 amino acids in the C-terminus, in the development of EAE. Transgenic expression of either STAT4α or STAT4β isoforms exclusively leads to reduced EAE in STAT4α expressing animals and exacerbated disease in STAT4β expressing mice as compared to controls. STAT4β expression drives increased levels of both IFNγ and IL-17 within cellular infiltrates in the CNS of immunized animals.71
IL12B (p40)
IL12B codes for IL-12p40 that together with IL12A (IL-12p35) and IL-23p19 forms IL-12 and IL-23 heterodimers, respectively. IL-12 and IL-23 are secreted primarily by APCs and influence the differentiation of T-cells into a Th1 or a Th17 phenotype, respectively. Both Il-12p40 and Il-12p35 KO mice fail to produce IL-12 heterodimer and lymph node cells from these mice show deficiencies in primary IFNγ-responses. However, IL-12p40 deficiency renders mice completely resistant to MOG-induced EAE, whereas Il-12p35 KOs have unaltered or more severe disease compared to wild types.72,73 It was later shown that IL-12p40 is essential for EAE as a component of IL-23 rather than of IL-12.74 Bone marrow chimeras revealed that full disease is dependent on IL-12p40 being expressed by CNS resident cells.75 IL-12p40 also has the capacity to homodimerize, and administration of neutralizing antibodies to this homodimer results in less severe EAE in SJL/J mice, while treating mice with recombinant IL-12p40 homodimer gives more severe disease.76
IL7R
IL7R codes for the IL-7 receptor α chain (IL-7Rα), which together with the common γ chain, forms the receptor for IL-7. IL-7Rα is also part of the receptor for thymic stromal lymphopoietin (TSLP). IL-7 is important for the survival and differentiation of cells of the lymphoid lineage such as B-, T-, and NK cells. IL-7rα–/– mice have a marked reduction in incidence of MOG-induced EAE. Interestingly, available data point toward a sex difference with low incidence in females, while males are completely resistant and have barely any priming of T-cells toward the CNS antigen.77 Treating MOG-induced EAE in mice with recombinant IL-7 exacerbates disease and treatment with a blocking antibody to IL-7Rα ameliorates disease, both when given before onset or at peak of disease.78 The antibody treatment reduces primarily the number of peripheral T-cells, whereas B- and NK cells are relatively spared, which also correlates to a lower expression of IL-7Rα on these cells. Among the T-cells, naïve and effector T-cells are the most affected, whereas central memory T-cells are largely spared. The treatment also results in an increase in absolute numbers of MOG-specific Foxp3+ Tregs in the lymph nodes. Another study later confirmed the effect on EAE using a KO mouse model in which the IL-7Rα is still present in the thymus to avoid disturbing the development of a functional immune system.79 These mice are also protected, although to a lesser extent than full KOs. Bone marrow chimeras revealed that EAE pathology is dependent on IL-7Rα expression on both hematopoietic and nonhematopoietic cells and that IL-7Rα is expressed in the CNS by oligodendrocytes and astrocytes. The effects seen in EAE after manipulating IL-7Rα could also be due to it being part of the receptor complex for TSLP. Tslp–/– mice, however, have a seemingly normal lymphocyte distribution in the naïve state, and there is no effect on EAE onset or progression. One of the MS-associated SNPs in the locus (rs6897932) has been shown to promote expression of an alternatively spliced soluble variant, thus increasing the ratio of soluble to membrane bound forms of IL-7Rα.80 This soluble form binds to IL-7 and potentiates its activity.81
IL2RA
The IL-2 receptor alpha chain (IL2RA), also known as CD25, is a part of the high-affinity receptor complex for IL-2, which can be expressed on both hematopoietic as well as nonhematopoietic cells. High expression is found on Foxp3+ Treg and transiently on activated effector T-cells. IL-2 is important for the expansion of T-cells during an immune response, but it also influences their differentiation. As a result of the strict IL-2 dependency of Tregs, Il2ra KO mice spontaneously develop a progressive lymphoproliferative disorder82 and have therefore not been a useful tool to study the role of this gene in EAE. It has, however, been shown in a model of spontaneous EAE that transfer of Il2ra KO T-cells results in little or no protection, whereas WT or Il2 KO T-cells do. Thus, protection from disease by Tregs requires IL-2 signaling, but is not mediated by autocrine IL-2 production.83 Similarly, injection of IL-2 coupled to a nonneutralizing antibody to increase the half-life results in an increase in Treg numbers and resistance to EAE.84 In combination with rapamycin, this treatment also reduces severity of ongoing EAE. IL-2 treatment experiments point to the protective effect being associated to an expansion of NK cells in the periphery and in the CNS. Moreover, using a human variant of IL-2/anti-IL-2 antibody complex, a defective CD56+ NK cell compartment from MS patients was restored in a human/mouse chimera model.85 Soluble CD25 (sCD25) is elevated in MS patients compared to control, and there is a positive correlation with disease severity and progression.86 Treating mouse EAE with sCD25 exacerbates disease and increases Th17 responses.87 This is consistent with the aforementioned studies as sCD25 acts as a decoy receptor for IL-2. IL-2 was recently shown to be a potent inducer of GM-CSF, a cytokine crucial for the development of EAE. An MS-associated polymorphism in IL2RA (rs2104286) gene specifically increases the frequency of GM-CSF-producing Th cells from risk allele carriers as compared to Th cells from control individuals.13 Daclizumab is an antibody directed toward CD25 that has shown efficacy in several Phase III clinical trials for relapsing-remitting MS (RRMS) and is a potential new treatment.
TYK2 (Tyrosine Kinase 2) and STAT3
The Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling pathway is the predominant signal transduction cascade in innate and adaptive immunity.88 TYK2 is a member of the JAK/STAT signaling pathway and contributes mainly to the IL-12-induced Th1 cell differentiation.88 STAT3 functions mainly as a signaling molecule and transcription factor for Th17 cell differentiation.88 Dysregulation of the JAK/STAT pathway contributes to numerous autoimmune diseases, including MS/EAE.
Tyk2–/– C57BL/6 mice are resistant in MOG35–55-induced EAE with complete lack of inflammation in the CNS.89 Adoptively transferred Tyk2–/– pathogenic CD4+ T-cells fail to induce EAE in WT animals pointing to a role for TYK2 in T-cells, rather than in APCs or target tissue cells, for the phenotype. B10.D1-H2q/SgJ (Tyk2A) mice that carry a 2538 G→A missense mutation in Tyk2 gene are also resistant in MOG79–96-induced EAE compared to B10.Q/Ai (Tyk2G) mice.90 Ex vivo restimulation of splenocytes and lymph node cells from B10.D1 (Tyk2A) leads to lower IFNγ, IL-6, and RANTES production and a trend for lower IL-17 compared to B10.Q. Since Tyk2A mutation impairs the IL-12R and the IL-23R pathways, the authors speculate that EAE resistance of B10.D1 (Tyk2A) mice might be due to their inability to upregulate encephalitogenic levels of IFNγ and IL-17 on T-cells (via IL-12R and IL-23R pathways respectively). On the other hand, conditional deletion of Stat3 in the T-cell compartment renders animals resistant to EAE, highlighting the importance of STAT3 in Th17 differentiation during EAE development.91,92
Different treatments such as COX-2 inhibitors, 1,25-dihydroxyvitamin D3, COP-1, lovastatin, and AZD1480 ameliorate EAE symptoms, CNS inflammation, and demyelination.93–97 In addition, several herbal compounds such as quercetin, curcumin, berberine, embelin, cornel iridoid glycoside, and plumbagin have been shown to dampen EAE.98–104 All the aforementioned treatments dampen Th1 and Th17 differentiation through inhibition of JAK/STAT pathway.
Suppressors of cytokine signaling proteins (SOCS) inhibit JAK/STAT, inhibit JAK/STAT signaling by various mechanisms.105 SOCS3 inhibits STAT3 activation and cytokine signaling in macrophages/microglia.106 Mice with conditional KO of Socs3 in myeloid cells develop atypical EAE compared to control mice.107,108 Adoptive transfer of SOCS-3 transduced DCs significantly suppresses EAE and associates with impaired IL-23/STAT3 and IL-12/STAT4 signaling and further decreases Th17 and Th1 differentiation and increases Th2 induction.109
Additionally, Glia maturation factor (GMF),110 miR-20b,111 miR-125a,112 and the organotellurium compound AS101113 modulate EAE by directly affecting the function or transcriptional levels of STAT3.
NF-ºB signaling
NFKB1 (p50)
NF-ºB is a generic name for a protein complex of five protein subunits, NF-ºB1 (p50), NF-ºB2 (p52), RelA, RelB, and c-Rel, that act as either homo- or heterodimers, functioning primarily as transcription factors for cytokine production and cell survival. Being quite central to all immune processes, the involvement of NF-ºB proteins with EAE and MS is expected as a surrogate for immune activation in most cell types. Additionally, NF-ºB is constitutively active in neurons and expressed in all glial cell types, being crucial for nervous system plasticity, learning, and memory.114 While deletions in immune system cells generally lead to reduced inflammation during EAE, CNS-restricted expression ablation has revealed both neuroprotective or detrimental roles, depending on the type of insult. Specifically for EAE, general NF-ºB pathway inhibition did not modify disease progression when targeted on either neurons or oligodendrocytes, while targeting of astrocytes and microglia led to reduced inflammation.114
While effects on EAE have, through selective deletion of one of the five subunits or additional regulatory proteins, upstream or downstream of the activation cascade been thoroughly documented and give partially overlapping results (for an extensive review refer to Mc Guire et al114). In specific, NFKB1 (p50) is part of the canonical NF-ºB pathway that is triggered by activation of receptors such as TNFR1, TLRs, IL-1R, TCR, and BCR. Deletion of NFKB1 in mice attenuates EAE incidence, clinical score, and CNS inflammation due at least in part to a reduction in T-cell activation (both Th1 and Th2).115 Target-tissue-specific effects are also evidenced by reovirus infection experiments, in which p50–/– mice fare better with reduced CNS apoptosis.116 A similar effect can be observed in ischemia induction, in which damage is significantly reduced in p50–/– mice.117
While limited information is available for humans on NFKB1 specifically, GWAS results have implicated other players in the NF-ºB cascade, such as TNFRSF1A (TNFR1), CARMA1 (CARD11), MALT1, BCL10, PLEKHG5, and TNFAIP3 as MS-susceptibility loci.9
MALT1-BCL10-CARD
Triggering of antigen receptors on the surface of lymphocytes leads to the initiation of signaling pathways that regulate the activation, proliferation, and survival. One of the major pathways leads to NF-ºB activation and translocation to the nucleus, where it acts as a transcriptional regulator. The so-called classical pathway of activation, in response to antigen receptors, requires the signaling molecule MALT1 and its binding partners BCL10 and CARMA1 (CARD11),118 all three associated to MS, as well as NF-ºB p50 (NFKB1) (see “NFKB1 (p50)” section).9 While no studies have addressed the role of BCL10 directly on EAE, Carma1 KO animals are completely protected from EAE apparently due to a strong inhibition of Th17 differentiation.119 Similarly, Malt1–/– mice immunized with MOG35–55 do not develop EAE in spite of abundant lymphocytic infiltration into the CNS. Loss of Malt1 leads to reduced IL-17 and GM-CSF secretion from infiltrating T-cells, which fail to further recruit myeloid cells and sustain neuroinflammation, while no impact on Th17 lineage-related transcription factors or Th1 differentiation can be observed.120 This is, however, inconclusive, since another study reveals impairment in lymphocyte activation already in the periphery under a similar EAE induction protocol.121 Transgenic mice expressing a catalytically inactive form of MALT1, which conserves its scaffolding function, also present a strong defect in lymphocyte activation and protection from EAE.122 Surprisingly, ablation of catalytic activity leads to an impairment in Treg cell generation and spontaneous autoimmune gastritis, which was not seen in complete KOs in the same study. Lastly, treatment of EAE in mice with the reversible MALT1 inhibitor mepazine either prophylactically or after onset of symptoms ameliorates disease.123
Other pathways
PTGER4 (EP4)
PTGER4 codes for EP4, which is one of the four receptors for prostaglandin E2 (PGE2). PGE2 is produced by cyclooxygenase-2 and has both pro- and anti-inflammatory effects. Lipidomic analysis of the arachidonic acid cascade in the spinal cord of mice with EAE shows that the PGE2 pathway is favored over other eicosanoids and that the expression of the PGE2 receptors EP1, EP2, and EP4 correlates with clinical symptoms.124 The same study also revealed that daily administration of EP4 antagonist ONO-AE3-208 before EAE onset suppresses MOG-induced EAE, likely due to reduced T-cell proliferation as well as diminished IFNγ and IL-17 expression. Ablation of all the eight prostaglandin receptors individually revealed that only Ep4 KOs present with a significant effect, leading to decreased disease severity.125 Inhibition of EAE is also achieved by treating mice with an EP4 antagonist during the priming phase. Paradoxically, treatment with an EP4 agonist starting at onset of disease reduces disease severity. Agonists for EP1, EP2, and EP3 have no effect.
TNFRSF1A
TNFR1, encoded by the TNFRSF1A gene, is the major receptor for TNF. As a pleiotropic cytokine, the role of TNF is not clearly understood and seems to have both pathogenic and protective functions in neuroinflammation. Blocking TNF in a clinical trial for MS resulted in an exacerbation of symptoms, while concomitantly, treatment of other autoimmune diseases with anti-TNF resulted in cases of neuroinflammation.126–128 Analysis of MS GWAS data in conjunction with the 1,000 Genomes Project data implicates SNP rs1800693 as the causal variant in the TNFRSF1A region, leading to the production of a soluble TNFR1 in MS patients carrying the predisposing genotype.13,129 This soluble TNF receptor acts in the same manner as the blocking treatment and could, therefore, promote neuroinflammation. TNF also has a second receptor, TNFR2, which can be inducibly expressed in endothelium and immune cells. TNFR2 has a protective effect in EAE since Tnfr2 deficient mice have exacerbated disease while Tnfr1 KO animals or mice treated with TNFR1 antagonists are protected.130–134 Taken together, both human and mouse data would point to blocking of TNFR1 rather than TNF itself as a target for a potential therapy for MS.
CYP24A1 (1,25-hydroxyvitamin D-1 alpha hydroxylase)
CYP24A1 encodes for 1,25-hydroxyvitamin D-1 alpha hydroxylase, an enzyme that inactivates 1,25-dihydroxyvitamin D3 through hydroxylation and thus regulates its levels.135
Female B10.PL mice fed with a diet with or without vitamin D3 prior to MBP immunization have significantly less clinical and immunological signs of EAE compared to ovariectomized females or intact or castrated males.136 One hypothesis for the higher levels of 1,25-dihydroxyvitamin D3 and less Cyp24a1 transcripts in vitamin-D-fed female mice is that an ovarian hormone inhibits Cyp24a1 gene expression in the spinal cord, which in turn causes 1,25-dihydroxyvitamin D3 accumulation leading to inflammation resolution before severe EAE develops.
Lovastatin treatment provides protection in EAE mice through inhibition of Cyp24a1 gene expression in Th1/Th17 cells that may allow the accumulation of 1,25-dihydroxyvitamin D3 in the peripheral lymphoid organs and spinal cord.137
Conclusion
The complexity and the heterogeneity of human MS together with inaccessibility of the target organ and events that occur prior to disease diagnosis necessitate studies in experimental models. With the tremendous progress in MS genetics, it is likely that EAE will continue to have a central role in functional in vivo complementation of human studies, especially in combination with multiple omics from human tissues that can guide the hypothesis about the nature of the causal variants.138,139 Numerous conditional knockout mice, which enable precise gene targeting in specific cell types when crossed with appropriate Cre lines (sometimes even in an inducible manner), have already been developed by the international Knockout Mouse Project. This conventional approach can now be complemented with the latest cutting-edge technology using the CRISPR-Cas9.140 In this way, multiple genes can be targeted simultaneously, which is likely more suitable for MS pathologies that are caused by subtle changes in genes that converge to shared pathways rather than variations in single genes. Such strategies might give rise to novel models with characteristics that mimic better certain MS pathologies, making them further adapted for translational research.
Disclosure
The authors report no conflicts of interest in this work.
References
Sadovnick AD, Baird PA, Ward RH. Multiple sclerosis: updated risks for relatives. Am J Med Genet. 1988;29(3):533–541. | |
O’Gorman C, Lin R, Stankovich J, Broadley SA. Modelling genetic susceptibility to multiple sclerosis with family data. Neuroepidemiology. 2013;40(1):1–12. | |
Ebers GC, Bulman DE, Sadovnick AD, et al. A population-based study of multiple sclerosis in twins. N Engl J Med. 1986;315(26):1638–1642. | |
Jersild C, Svejgaard A, Fog T. HL-A antigens and multiple sclerosis. Lancet. 1972;1(7762):1240–1241. | |
International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476(7359):214–219. | |
Oksenberg JR, Barcellos LF, Cree BA, et al. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am J Hum Genet. 2004;74(1):160–167. | |
International Multiple Sclerosis Genetics Consortium, Hafler DA, Compston A, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357(9):851–862. | |
Australia, New Zealand Multiple Sclerosis Genetics Consortium (ANZgene). Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet. 2009;41(7):824–828. | |
International Multiple Sclerosis Genetics Consortium, Beecham AH, Patsopoulos NA, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45(11):1353–1360. | |
Patsopoulos NA, Barcellos LF, Hintzen RQ, et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet. 2013;9(11):e1003926. | |
Couturier N, Bucciarelli F, Nurtdinov RN, et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain. 2011;134 (Pt 3):693–703. | |
Gregory AP, Dendrou CA, Attfield KE, et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature. 2012;488(7412):508–511. | |
Hartmann FJ, Khademi M, Aram J, et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun. 2014;5:5056. | |
Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb Clin Neurol. 2014;122:173–189. | |
Ben-Nun A, Kaushansky N, Kawakami N, et al. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J Autoimmun. 2014;54:33–50. | |
Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2(2):116–126. | |
Chang TT, Jabs C, Sobel RA, Kuchroo VK, Sharpe AH. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med. 1999;190(5):733–740. | |
Perrin PJ, June CH, Maldonado JH, Ratts RB, Racke MK. Blockade of CD28 during in vitro activation of encephalitogenic T cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J Immunol. 1999;163(3):1704–1710. | |
Cross AH, San M, Keeling RM, Karr RW. CTLA-4-Fc treatment of ongoing EAE improves recovery, but has no effect upon relapse rate. Implications for the mechanisms involved in disease perpetuation. J Neuroimmunol. 1999;96(2):144–147. | |
Croxford JL, O’Neill JK, Ali RR, et al. Local gene therapy with CTLA4-immunoglobulin fusion protein in experimental allergic encephalomyelitis. Eur J Immunol. 1998;28(12):3904–3916. | |
Chang TT, Sobel RA, Wei T, Ransohoff RM, Kuchroo VK, Sharpe AH. Recovery from EAE is associated with decreased survival of encephalitogenic T cells in the CNS of B7-1/B7-2-deficient mice. Eur J Immunol. 2003;33(7):2022–2032. | |
Peferoen LA, Vogel DY, Ummenthum K, et al. Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J Neuropathol Exp Neurol. 2015;74(1):48–63. | |
Codarri L, Greter M, Becher B. Communication between pathogenic T cells and myeloid cells in neuroinflammatory disease. Trends Immunol. 2013;34(3):114–119. | |
Vainchtein ID, Vinet J, Brouwer N, et al. In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia. 2014;62(10):1724–1735. | |
Hildebrand JM, Yi Z, Buchta CM, Poovassery J, Stunz LL, Bishop GA. Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunol Rev. 2011;244(1):55–74. | |
Bishop GA, Xie P. Multiple roles of TRAF3 signaling in lymphocyte function. Immunol Res. 2007;39(1–3):22–32. | |
Zhu S, Pan W, Shi P, et al. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J Exp Med. 2010;207(12):2647–2662. | |
Xiao Y, Jin J, Chang M, et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat Med. 2013;19(5):595–602. | |
Mana P, Linares D, Silva DG, et al. LIGHT (TNFSF14/CD258) is a decisive factor for recovery from experimental autoimmune encephalomyelitis. J Immunol. 2013;191(1):154–163. | |
West LC, Cresswell P. Expanding roles for GILT in immunity. Curr Opin Immunol. 2013;25(1):103–108. | |
Bergman CM, Marta CB, Maric M, Pfeiffer SE, Cresswell P, Ruddle NH. A switch in pathogenic mechanism in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in IFN-γ-inducible lysosomal thiol reductase-free mice. J Immunol. 2012;188(12):6001–6009. | |
Burrows GG, Meza-Romero R, Huan J, et al. Gilt required for RTL550-CYS-MOG to treat experimental autoimmune encephalomyelitis. Metab Brain Dis. 2012;27(2):143–149. | |
Chitnis T, Khoury SJ. Role of costimulatory pathways in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. J Allergy Clin Immunol. 2003;112(5):837. | |
Issazadeh S, Navikas V, Schaub M. Kinetics of expression of costimulatory molecules and their ligands in murine relapsing experimental autoimmune encephalomyelitis in vivo. J Immunol. 1998;161(3):1104–1112. | |
Laman JD, Maassen CB, Schellekens MM, et al. Therapy with antibodies against CD40L (CD154) and CD44-variant isoforms reduces experimental autoimmune encephalomyelitis induced by a proteolipid protein peptide. Mult Scler. 1998;4(3):147–153. | |
Becher B, Durell BG, Miga AV, Hickey WF. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med. 2001;193(8):967–974. | |
Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, Kopf M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci U S A. 2009;106(3):876–881. | |
Perona-Wright G, Jenkins SJ, O’Connor RA, et al. A pivotal role for CD40-mediated IL-6 production by dendritic cells during IL-17 induction in vivo. J Immunol. 2009;182(5):2808–2815. | |
Hochweller K, Anderton SM. Systemic administration of antigen-loaded CD40-deficient dendritic cells mimics soluble antigen administration. Eur J Immunol. 2004;34(4):990–998. | |
Kalantari T, Karimi MH, Ciric B, Yan Y, Rostami A, Kamali-Sarvestani E. Tolerogenic dendritic cells produced by lentiviral-mediated CD40- and interleukin-23p19-specific shRNA can ameliorate experimental autoimmune encephalomyelitis by suppressing T helper type 17 cells. Clin Exp Immunol. 2014;176(2):180–189. | |
Grewal IS, Foellmer HG, Grewal KD, et al. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 1996;273(5283):1864–1867. | |
Gerritse K, Laman JD, Noelle RJ, et al. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A. 1996;93(6):2499–2504. | |
Howard LM, Miga AJ, Vanderlugt CL. Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J Clin Invest. 1999;103(2):281–290. | |
Howard LM, Miller SD. Autoimmune intervention by CD154 blockade prevents T cell retention and effector function in the target organ. J Immunol. 2001;166(3):1547–1553. | |
Nagelkerken L, Haspels I, van Rijs W, et al. FcR interactions do not play a major role in inhibition of experimental autoimmune encephalomyelitis by anti-CD154 monoclonal antibodies. J Immunol. 2004;173(2):993–999. | |
Howard LM, Dal Canto MC, Miller SD. Transient anti-CD154-mediated immunotherapy of ongoing relapsing experimental autoimmune encephalomyelitis induces long-term inhibition of disease relapses. J Neuroimmunol. 2002;129(1–2):58–65. | |
Howard LM, Neville KL, Haynes LM, Dal Canto MC, Miller SD. CD154 blockade results in transient reduction in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J Virol. 2003;77(3):2247–2250. | |
Drescher KM, Zoecklein LJ, Pavelko KD, Rivera-Quinones C, Hollenbaugh D, Rodriguez M. CD40L is critical for protection from demyelinating disease and development of spontaneous remyelination in a mouse model of multiple sclerosis. Brain Pathol. 2000;10(1):1–15. | |
Samoilova EB, Horton JL, Zhang H, Chen Y. CD40L blockade prevents autoimmune encephalomyelitis and hampers TH1 but not TH2 pathway of T cell differentiation. J Mol Med. 1997;75(8):603–608. | |
Du C, Bright JJ, Sriram S. Inhibition of CD40 signaling pathway by tyrphostin A1 reduces secretion of IL-12 in macrophage, Th1 cell development and experimental allergic encephalomyelitis in SJL/J mice. J Neuroimmunol. 2001;114(1–2):69–79. | |
Ding Q, Si X, Liu D, et al. Targeting and liposomal drug delivery to CD40L expressing T cells for treatment of autoimmune diseases. J Control Release. 2015;207:86–92. | |
Schaub M, Issazadeh S, Stadlbauer THW. Costimulatory signal blockade in murine relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 1999;96(2):158–166. | |
Laman JD, t Hart BA, Brok H, et al. Protection of marmoset monkeys against EAE by treatment with a murine antibody blocking CD40 (mu5D12). Eur J Immunol. 2002;32(8):2218–2228. | |
Boon L, Brok HPM, Bauer J, et al. Prevention of experimental autoimmune encephalomyelitis in the common marmoset (Callithrix jacchus) using a chimeric antagonist monoclonal antibody against human CD40 is associated with altered B cell responses. J Immunol. 2001;167(5):2942–2949. | |
Boon L, Laman JD, Ortiz-Buijsse A, et al. Preclinical assessment of anti-CD40 Mab 5D12 in cynomolgus monkeys. Toxicology. 2002;174(1):53–65. | |
Hong GU, Kim NG, Jeoung D, Ro JY. Anti-CD40 Ab- or 8-oxo-dG-enhanced Treg cells reduce development of experimental autoimmune encephalomyelitis via down-regulating migration and activation of mast cells. J Neuroimmunol. 2013;260(1–2):60–73. | |
Kim DY, Jeoung D, Ro JY. Signaling pathways in the activation of mast cells cocultured with astrocytes and colocalization of both cells in experimental allergic encephalomyelitis. J Immunol. 2010;185(1):273–283. | |
t Hart BA, Blezer EL, Brok HP, et al. Treatment with chimeric anti-human CD40 antibody suppresses MRI-detectable inflammation and enlargement of pre-existing brain lesions in common marmosets affected by MOG-induced EAE. J Neuroimmunol. 2005;163(1–2):31–39. | |
Ichikawa HT, Williams LP. Activation of APCs through CD40 or Toll-like receptor 9 overcomes tolerance and precipitates autoimmune disease. J Immunol. 2002;169(5):2781–2787. | |
Zhang J, Liu Q, Langdon WY. Cbl-b: roles in T cell tolerance, proallergic T cell development, and cancer immunity. Inflamm Cell signal. 2014;1(4):e146. | |
Harada Y, Harada Y, Elly C, et al. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med. 2010;207(7):1381–1391. | |
Chiang YJ, Kole HK, Brown K, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403(6766):216–220. | |
Fujiwara M, Anstadt EJ, Khanna KM, Clark RB. Cbl-b-deficient mice express alterations in trafficking-related molecules but retain sensitivity to the multiple sclerosis therapeutic agent, FTY720. Clin Immunol. 2015;158(1):103–113. | |
Chitnis T, Najafian N, Benou C, et al. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108(5):739–747. | |
Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochim Biophys Acta. 2011; 1812(2):246–251. | |
Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E. Cutting edge: the pathogenicity of IFN-gamma-producing Th17 cells is independent of T-bet. J Immunol. 2013;190(9):4478–4482. | |
Wei L, Vahedi G, Sun HW, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32(6):840–851. | |
McWilliams IL, Rajbhandari R, Nozell S, Benveniste E, Harrington LE. STAT4 controls GM-CSF production by both Th1 and Th17 cells during EAE. J Neuroinflamm. 2015;12:128. | |
Codarri L, Gyulveszi G, Tosevski V, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–567. | |
El-Behi M, Ciric B, Dai H, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–575. | |
Mo C, Chearwae W, O’Malley JT, et al. Stat4 isoforms differentially regulate inflammation and demyelination in experimental allergic encephalomyelitis. J Immunol. 2008;181(8):5681–5690. | |
Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest. 2002; 110(4):493–497. | |
Gran B, Zhang GX, Yu S, et al. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169(12):7104–7110. | |
Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421(6924):744–748. | |
Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J Clin Invest. 2003;112(8):1186–1191. | |
Mondal S, Roy A, Pahan K. Functional blocking monoclonal antibodies against IL-12p40 homodimer inhibit adoptive transfer of experimental allergic encephalomyelitis. J Immunol. 2009;182(8):5013–5023. | |
Walline CC, Kanakasabai S, Bright JJ. IL-7Ralpha confers susceptibility to experimental autoimmune encephalomyelitis. Genes Immun. 2011;12(1):1–14. | |
Lee LF, Axtell R, Tu GH, et al. IL-7 promotes T(H)1 development and serum IL-7 predicts clinical response to interferon-beta in multiple sclerosis. Sci Transl Med. 2011;3(93):93ra68. | |
Ashbaugh JJ, Brambilla R, Karmally SA, Cabello C, Malek TR, Bethea JR. IL7Ralpha contributes to experimental autoimmune encephalomyelitis through altered T cell responses and nonhematopoietic cell lineages. J Immunol. 2013;190(9):4525–4534. | |
Kreft KL, Verbraak E, Wierenga-Wolf AF, et al. Decreased systemic IL-7 and soluble IL-7Ralpha in multiple sclerosis patients. Genes Immun. 2012;13(7):587–592. | |
Lundstrom W, Highfill S, Walsh ST, et al. Soluble IL7Ralpha potentiates IL-7 bioactivity and promotes autoimmunity. Proc Natl Acad Sci U S A. 2013;110(19):E1761–E1770. | |
Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3(4):521–530. | |
Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J Exp Med. 2002;196(6):851–857. | |
Webster KE, Walters S, Kohler RE, et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med. 2009;206(4):751–760. | |
Hao J, Campagnolo D, Liu R, et al. Interleukin-2/interleukin-2 antibody therapy induces target organ natural killer cells that inhibit central nervous system inflammation. Ann Neurol. 2011;69(4):721–734. | |
Maier LM, Anderson DE, Severson CA, et al. Soluble IL-2RA levels in multiple sclerosis subjects and the effect of soluble IL-2RA on immune responses. J Immunol. 2009;182(3):1541–1547. | |
Russell SE, Moore AC, Fallon PG, Walsh PT. Soluble IL-2Ralpha (sCD25) exacerbates autoimmunity and enhances the development of Th17 responses in mice. PloS One. 2012;7(10):e47748. | |
O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–550. | |
Oyamada A, Ikebe H, Itsumi M, et al. Tyrosine kinase 2 plays critical roles in the pathogenic CD4 T cell responses for the development of experimental autoimmune encephalomyelitis. J Immunol. 2009; 183(11):7539–7546. | |
Spach KM, Noubade R, McElvany B, Hickey WF, Blankenhorn EP, Teuscher C. A single nucleotide polymorphism in Tyk2 controls susceptibility to experimental allergic encephalomyelitis. J Immunol. 2009;182(12):7776–7783. | |
Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol. 2008;180(9):6070–6076. | |
Maris CH, Housseau F, Yu H, Pardoll DM. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179(7):4313–4317. | |
Muthian G, Raikwar HP, Johnson C, et al. COX-2 inhibitors modulate IL-12 signaling through JAK-STAT pathway leading to Th1 response in experimental allergic encephalomyelitis. J Clin Immunol. 2006;26(1):73–85. | |
Muthian G, Raikwar HP, Rajasingh J, Bright JJ. 1,25 Dihydroxyvitamin- D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res. 2006;83(7):1299–1309. | |
Chen C, Liu X, Wan B, Zhang JZ. Regulatory properties of copolymer I in Th17 differentiation by altering STAT3 phosphorylation. J Immunol. 2009;183(1):246–253. | |
Nath N, Giri S, Prasad R, Singh AK, Singh I. Potential targets of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol. 2004;172(2):1273–1286. | |
Liu Y, Holdbrooks AT, De Sarno P, et al. Therapeutic efficacy of suppressing the Jak/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J Immunol. 2014;192(1):59–72. | |
Muthian G, Bright JJ. Quercetin, a flavonoid phytoestrogen, ameliorates experimental allergic encephalomyelitis by blocking IL-12 signaling through JAK-STAT pathway in T lymphocyte. J Clin Immunol. 2004; 24(5):542–552. | |
Natarajan C, Bright JJ. Curcumin inhibits experimental allergic encephalomyelitis by blocking IL-12 signaling through Janus kinase-STAT pathway in T lymphocytes. J Immunol. 2002;168(12):6506–6513. | |
Xie L, Li XK, Funeshima-Fuji N, et al. Amelioration of experimental autoimmune encephalomyelitis by curcumin treatment through inhibition of IL-17 production. Int Immunopharmacol. 2009;9(5):575–581. | |
Qin X, Guo BT, Wan B, Fang L, Lu L, Wu L. Regulation of Th1 and Th17 cell differentiation and amelioration of experimental autoimmune encephalomyelitis by natural product compound berberine. J Immunol. 2010;179(7):4313–4317. | |
Xue Z, Ge Z, Zhang K, et al. Embelin suppresses dendritic cell functions and limits autoimmune encephalomyelitis through the TGF-β/β-catenin and STAT3 signaling pathways. Mol Neurobiol. 2014;49(2):1087–1101. | |
Yin L, Chen Y, Qu Z, et al. Involvement of JAK/STAT signaling in the effect of cornel iridoid glycoside on experimental autoimmune encephalomyelitis amelioration in rats. J Neuroimmunol. 2014; 274(1–2):28–37. | |
Jia Y, Jing J, Bai Y, et al. Amelioration of experimental autoimmune encephalomyelitis by plumbagin through down-regulation of JAK-STAT and NF-kappaB signaling pathways. PloS One. 2011;6(10):e27006. | |
Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7(6):454–465. | |
Baker BJ, Akhtar LN, Benveniste EN. SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 2009;30(8):392–400. | |
Qin H, Yeh WI, De Sarno P, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A. 2012;109(13):5004–5009. | |
Liu Y, Holdbrooks AT, Meares GP, Buckley JA, Benveniste EN, Qin H. Preferential recruitment of neutrophils into the cerebellum and brainstem contributes to the atypical experimental autoimmune encephalomyelitis phenotype. J Immunol. 2015;195(3):841–852. | |
Li Y, Chu N, Rostami A, Zhang GX. Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J Immunol. 2006;177(3):1679–1688. | |
Zaheer S, Wu Y, Bassett J, Yang B, Zaheer A. Glia maturation factor regulation of STAT expression: a novel mechanism in experimental autoimmune encephalomyelitis. Neurochem Res. 2007;32(12):2123–2131. | |
Zhu E, Wang X, Zheng B, et al. miR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORγt and STAT3. J Immunol. 2014; 192(12):5599–5609. | |
Pan W, Zhu S, Dai D, et al. MiR-125a targets effector programs to stabilize Treg-mediated immune homeostasis. Nat Commun. 2015;6:7096. | |
Xie L, Chen J, McMickle A, et al. The immunomodulator AS101 suppresses production of inflammatory cytokines and ameliorates the pathogenesis of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2014;273(1–2):31–41. | |
Mc Guire C, Prinz M, Beyaert R, van Loo G. Nuclear factor kappa B (NF-kappaB) in multiple sclerosis pathology. Trends Mol Med. 2013;19(10):604–613. | |
Hilliard B, Samoilova EB, Liu TS, Rostami A, Chen Y. Experimental autoimmune encephalomyelitis in NF-kappa B-deficient mice:roles of NF-kappa B in the activation and differentiation of autoreactive T cells. J Immunol. 1999;163(5):2937–2943. | |
O’Donnell SM, Hansberger MW, Connolly JL, et al. Organ-specific roles for transcription factor NF-kappaB in reovirus-induced apoptosis and disease. J Clin Invest. 2005;115(9):2341–2350. | |
Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5(5):554–559. | |
Thome M, Charton JE, Pelzer C, Hailfinger S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb Perspect Biol. 2010;2(9):a003004. | |
Molinero LL, Cubre A, Mora-Solano C, Wang Y, Alegre ML. T cell receptor/CARMA1/NF-kappaB signaling controls T-helper (Th) 17 differentiation. Proc Natl Acad Sci U S A. 2012;109(45):18529–18534. | |
Brustle A, Brenner D, Knobbe CB, et al. The NF-kappaB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J Clin Invest. 2012;122(12):4698–4709. | |
Mc Guire C, Wieghofer P, Elton L, et al. Paracaspase MALT1 deficiency protects mice from autoimmune-mediated demyelination. J Immunol. 2013;190(6):2896–2903. | |
Jaworski M, Marsland BJ, Gehrig J, et al. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. 2014;33(23):2765–2781. | |
Mc Guire C, Elton L, Wieghofer P, et al. Pharmacological inhibition of MALT1 protease activity protects mice in a mouse model of multiple sclerosis. J Neuroinflammation. 2014;11:124. | |
Kihara Y, Matsushita T, Kita Y, et al. Targeted lipidomics reveals mPGES-1-PGE2 as a therapeutic target for multiple sclerosis. Proc Natl Acad Sci U S A. 2009;106(51):21807–21812. | |
Esaki Y, Li Y, Sakata D, et al. Dual roles of PGE2-EP4 signaling in mouse experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107(27):12233–12238. | |
Bosch X, Saiz A, Ramos-Casals M, Group BS. Monoclonal antibody therapy-associated neurological disorders. Nat Rev Neurol. 2011;7(3):165–172. | |
van Oosten BW, Barkhof F, Truyen L, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996;47(6):1531–1534. | |
TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology. 1999;53(3):457–465. | |
Ottoboni L, Frohlich IY, Lee M, et al. Clinical relevance and functional consequences of the TNFRSF1A multiple sclerosis locus. Neurology. 2013;81(22):1891–1899. | |
Nomura T, Abe Y, Kamada H, et al. Therapeutic effect of PEGylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J Control Release. 2011;149(1):8–14. | |
Williams SK, Maier O, Fischer R, et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PloS One. 2014;9(2):e90117. | |
Suvannavejh GC, Lee HO, Padilla J, Dal Canto MC, Barrett TA, Miller SD. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35–55)-induced experimental autoimmune encephalomyelitis. Cell Immunol. 2000;205(1):24–33. | |
Eugster HP, Frei K, Bachmann R, Bluethmann H, Lassmann H, Fontana A. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur J Immunol. 1999;29(2):626–632. | |
Kassiotis G, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med. 2001;193(4):427–434. | |
Feldman D, Krishnan AV, Swami S, Giovannucci E, Feldman BJ. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer. 2014;14(5):342–357. | |
Spach KM, Hayes CE. Vitamin D3 confers protection from autoimmune encephalomyelitis only in female mice. J Immunol. 2005;175(6):4119–4126. | |
Paintlia AS, Paintlia MK, Hollis BW, Singh AK, Singh I. Interference with RhoA-ROCK signaling mechanism in autoreactive CD4+ T cells enhances the bioavailability of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis. Am J Pathol. 2012;181(3):993–1006. | |
Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–343. | |
Claussnitzer M, Dankel SN, Kim KH, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015; 373(1):895–907. | |
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. Apr 2014;32(4):347–355. | |
Sellebjerg F, Sorensen PS. Therapeutic interference with leukocyte recirculation in multiple sclerosis. Eur J Neurol. 2015;22(3):434–442. | |
Huber M, Heink S, Grothe H, et al. A Th17-like developmental process leads to CD8(+) Tc17 cells with reduced cytotoxic activity. Eur J Immunol. 2009;39(7):1716–1725. | |
Forde EA, Dogan RN, Karpus WJ. CCR4 contributes to the pathogenesis of experimental autoimmune encephalomyelitis by regulating inflammatory macrophage function. J Neuroimmunol. 2011;236(1–2):17–26. | |
Poppensieker K, Otte DM, Schurmann B, et al. CC chemokine receptor 4 is required for experimental autoimmune encephalomyelitis by regulating GM-CSF and IL-23 production in dendritic cells. Proc Natl Acad Sci U S A. 2012;109(10):3897–3902. | |
Sturner KH, Borgmeyer U, Schulze C, Pless O, Martin R. A multiple sclerosis-associated variant of CBLB links genetic risk with type I IFN function. J Immunol. 2014;193(9):4439–4447. | |
Akkad DA, Bellenberg B, Esser S, et al. Multiple sclerosis risk loci correlate with cervical cord atrophy and may explain the course of disability. Neurogenetics. 2015;16(3):161–168. | |
Ma J, Wang R, Fang X, Ding Y, Sun Z. Critical role of TCF-1 in repression of the IL-17 gene. PloS One. 2011;6(9):e24768. | |
Laaksonen H, Guerreiro-Cacais AO, Adzemovic MZ, et al. The multiple sclerosis risk gene IL22RA2 contributes to a more severe murine autoimmune neuroinflammation. Genes Immun. 2014;15(7):457–465. | |
Liu X, He F, Pang R, et al. Interleukin-17 (IL-17)-induced microRNA 873 (miR-873) contributes to the pathogenesis of experimental autoimmune encephalomyelitis by targeting A20 ubiquitin-editing enzyme. J Biological Chem. 2014;289(42):28971–28986. | |
Wang X, Deckert M, Xuan NT, et al. Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF-kappaB- and STAT1-dependent chemokine production in astrocytes. Acta Neuropathol. Epub 2013 Sep 29. | |
Togni M, Swanson KD, Reimann S, et al. Regulation of in vitro and in vivo immune functions by the cytosolic adaptor protein SKAP-HOM. Mol Cell Biol. 2005;25(18):8052–8063. | |
Bagaeva LV, Rao P, Powers JM, Segal BM. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol. 2006;176(12):7676–7685. | |
Klimatcheva E, Pandina T, Reilly C, et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. 2015;16:6. | |
Schraml BU, Hildner K, Ise W, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460(7253):405–409. | |
Miao T, Raymond M, Bhullar P, et al. Early growth response gene-2 controls IL-17 expression and Th17 differentiation by negatively regulating Batf. J Immunol. 2013;190(1):58–65. | |
Yoshida Y, Yoshimi R, Yoshii H, et al. The transcription factor IRF8 activates integrin-mediated TGF-beta signaling and promotes neuroinflammation. Immunity. 2014;40(2):187–198. | |
Chrabot BS, Kariuki SN, Zervou MI, et al. Genetic variation near IRF8 is associated with serologic and cytokine profiles in systemic lupus erythematosus and multiple sclerosis. Genes Immun. 2013;14(8):471–478. | |
Howard LM, Ostrovidov S, Smith CE, Dal Canto MC, Miller SD. Normal Th1 development following long-term therapeutic blockade of CD154-CD40 in experimental autoimmune encephalomyelitis. J Clin Invest. 2002;109(2):233–241. | |
Chen SJ, Wang YL, Kao JH, et al. Decoy receptor 3 ameliorates experimental autoimmune encephalomyelitis by directly counteracting local inflammation and downregulating Th17 cells. Mol Immunol. 2009;47(2–3):567–574. | |
Sättler MB, Demmer I, Williams SK, et al. Effects of interferon-beta-1a on neuronal survival under autoimmune inflammatory conditions. Exp Neurol. 2006;201(1):172–181. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.