Back to Journals » International Journal of Nanomedicine » Volume 11
Transformation of eutectic emulsion to nanosuspension fabricating with solvent evaporation and ultrasonication technique
Authors Phaechamud T, Tuntarawongsa S
Received 12 March 2016
Accepted for publication 30 April 2016
Published 14 June 2016 Volume 2016:11 Pages 2855—2865
DOI https://doi.org/10.2147/IJN.S108355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Thawatchai Phaechamud,1 Sarun Tuntarawongsa2
1Department of Pharmaceutical Technology, 2Pharmaceutical Intelligence Unit Prachote Plengwittaya, Faculty of Pharmacy, Silpakorn University, Nakhon Pathom, Thailand
Abstract: Eutectic solvent can solubilize high amount of some therapeutic compounds. Volatile eutectic solvent is interesting to be used as solvent in the preparation of nanosuspension with emulsion solvent evaporation technique. The mechanism of transformation from the eutectic emulsion to nanosuspension was investigated in this study. The 30% w/w ibuprofen eutectic solution was used as the internal phase, and the external phase is composed of Tween 80 as emulsifier. Ibuprofen nanosuspension was prepared by eutectic emulsion solvent evaporating method followed with ultrasonication. During evaporation process, the ibuprofen concentration in emulsion droplets was increased leading to a drug supersaturation but did not immediately recrystallize because of low glass transition temperature (Tg) of ibuprofen. The contact angle of the internal phase on ibuprofen was apparently lower than that of the external phase at all times of evaporation, indicating that the ibuprofen crystals were preferentially wetted by the internal phase than the external phase. From calculated dewetting value ibuprofen crystallization occurred in the droplet. Crystallization of the drug was initiated with external mechanical force, and the particle size of the drug was larger due to Ostwald ripening. Cavitation force from ultrasonication minimized the ibuprofen crystals to the nanoscale. Particle size and zeta potential of formulated ibuprofen nanosuspension were 330.87±51.49 nm and -31.1±1.6 mV, respectively, and exhibited a fast dissolution. Therefore, the combination of eutectic emulsion solvent evaporation method with ultrasonication was favorable for fabricating an ibuprofen nanosuspension, and the transformation mechanism was attained successfully.
Keywords: eutectic, emulsion solvent evaporation, ibuprofen, nanosuspension, ultrasonication
Introduction
Techniques of nanosuspension preparation are classified into two categories: top-down techniques and bottom-up techniques. The top-down techniques apply mechanical force to reduce large particles to nanometer scale.1 The milling or grinding technique and high-pressure homogenization technique are widely used in many research studies and in the production of commercial products.2 The top-down techniques do not use an organic solvent; however, the processes generate heat and mechanical force that impact the surface properties.1,3,4 Typically, the bottom-up techniques are divided into precipitation3 and emulsion template.5 For the emulsion template techniques, the drug dissolves in a volatile organic solvent and transforms into nanometer particles after solvent diffusion, extraction, or evaporation. For example, celecoxib dissolved in ethyl acetate or ibuprofen dissolved in ethanol as the internal phase was transformed into nanosuspensions with emulsion diffusion method with particle sizes of 140–700 nm and 18.7–161 nm, respectively.6,7
One of the combination techniques for nanosuspension preparation is solvent evaporation coupling ultrasonic. Iradipine nanosuspension was successfully prepared by antisolvent precipitation with an ultrasonic liquid processor using polyethylene oxide and hydroxypropyl methylcellulose as the stablilizer.8 Acyclovir and eudragit® RS 100 dissolved in ethanol as the internal phase was transformed into nanosuspension by emulsion solvent diffusion and an ultrasonicator.9 The particle size of the nanosuspension decreased as the power input and ultrasonication time were increased.10
During solvent removal from the emulsion, drug crystallization occurs in the internal phase droplets, but it is also possible for nucleation to occur within the aqueous phase. However, the crystallization tends to favor a formation of nuclei in the oil phase. Crystals had grown within oil droplets when wetted with the oil higher than the external phase. On the other hand, crystals that demonstrate a higher wetting with the external phase than the oil exhibit dewetting with the oil and wetting with the external phase and growth of the oil droplet. Dewetting is favorable when γOWcosθO <0 (γOW is the interfacial tension of the internal phase and external phase and θO the contact angle of the internal phase on the crystal surface).11 The mechanism of transformation for the emulsion solvent evaporation is a progressive evaporation of the solvent that is used to dissolve the drug from the emulsion droplet, leading to drug supersaturation, nucleation, and crystal growth. The parameters that affect the evaporation rate of emulsions and particle size of crystals are the diffusion coefficient of the internal phase molecules in the external phase, the surface area of evaporated droplet, and conditions of evaporation such as temperature and pressure.12,13
A eutectic system is a mixture of two or more compounds that suppress the melting point of each compound because the total entropy of the system increases.14,15 Eutectic systems have been applied in many areas in the pharmaceutical field such as the increase of drug solubility, permeation, and absorption.16–19 A eutectic mixture can be used as the oil phase in emulsions20,21 and as a deep eutectic solvent.22–27 Menthol has generally been used as a eutectic component,28,29 with a permeation enhancer property.30 Menthol forms a liquid eutectic mixture at room temperature with many sublimate compounds such as camphor, borneol, thymol, and cineole.31–33 Recently, a therapeutic deep eutectic solvent was used to prepare the drug particles from menthol and ibuprofen using supercritical fluid technology.22
Nonsteroidal anti-inflammatory drugs such as ibuprofen, ketoprofen, indomethacin, diclofenac, and piroxicam have been reported as solutes in eutectic systems.17,19,31,32,34 Ibuprofen nanosuspensions have been prepared using bottom-up techniques such as the emulsion solvent diffusion method,7,35 melt emulsification method,35 sonoprecipitation method,1 and cavi-precipitation method.31,34,36,37 However, a eutectic mixture has not been reported as a solvent for the production of nanosuspension using these processes. This solvent consists of a compound that is synthesized to mimic the natural compound with low toxicity and that is edible thus it is introduced as the green solvent.38 The eutectic mixture is a volatile liquid; therefore, it should be employed as the oil phase to prepare the emulsions and applied as a green solvent for the emulsion solvent evaporation method. The slow evaporation rate of this solvent facilitates the investigation of transformation from emulsion to nanosuspension. Therefore, the aim of this research is to characterize the ibuprofen nanosuspension prepared using the eutectic emulsion solvent evaporation technique, while the mechanistic study of transformation is also investigated.
Materials and methods
Materials
Ibuprofen, menthol (M), and camphor (C) were purchased from P.C. Drug Center Co. Ltd. (Bangkok, Thailand); Tween 80 from VWR International Ltd. (Bournemouth, UK); phosphoric acid 85% from Merck KGaA (Darmstadt, Germany); methanol, acetonitrile, and chloroform from SK chemicals Co. Ltd. (Ulsan, Korea); sodium hydroxide and potassium dihydrogen phosphate from QReC (Auckland, New Zealand); glucose and sodium benzoate from Ajax Finechem Pty. Ltd. (Auckland, New Zealand); xanthan gum (Xantural® 75) from CP Kelco Shandong Biological Company Ltd. (Shanghai, People’s Republic of China); and ibuprofen suspension (Nurofen® 100 mg/5 mL) from Reckitt Benckiser (Thailand) Ltd. (Bangkok, Thailand).
Preparation of ibuprofen eutectic solution and emulsion
The 1:1 menthol:camphor powder was mixed in an airtight glass container at room temperature overnight to completely form a eutectic mixture. Then 30% w/w ibuprofen was dissolved in this solvent to obtain the eutectic solution. For the preparation of ibuprofen eutectic emulsion, 1.667 g of ibuprofen eutectic solution (30% w/w ibuprofen in eutectic solvent) was used as the internal phase. Approximately 0.5 g of Tween 80 was used as an emulsifier, and water was used as the external phase by adjusting the volume to 25 mL. The eutectic emulsion was prepared using a homogenizer (Ultra-Turrax T10 basic; IKA, Staufen, Germany) at 20×103 rpm, 75°C in a circulator water bath (CWB-13L; Hon Yong Scientific Equipment Co., Ltd, Seoul, Korea) for 2 minutes, and then sonicated using a probe ultrasonicator (Sonics Vibra cell VCX130; Sonics & Materials Inc., USA) with probe model CV18. Each formula was sonicated at an amplitude level of 50% with a 30-second on and 2-second off pulse for 2 minutes.
Preparation of ibuprofen nanosuspension
Ibuprofen nanosuspension was prepared using a combination of a eutectic emulsion evaporation method and ultrasonication. The 25-mL ibuprofen eutectic emulsion was diluted to 125 mL with a dilution medium (12.5 g of glucose in water) to prevent water loss before the transformation. The diluted eutectic emulsion was continuously evaporated under a laminar air flow hood on a hot plate stirrer at 45°C, 200 rpm for 2 days. Finally, the volume of the suspension was adjusted to 25 mL with the addition of distilled water and then sonicated with a probe ultrasonicator (Sonics Vibra cell VCX130; Sonics & Materials Inc.) with probe model CV18. Each formula was sonicated at an amplitude level of 50% with a 30-second on and 2-second off pulse for 30 minutes in an ice bath. This product was assigned to be a nonformulated ibuprofen nanosuspension (nNS). The control nanosuspensions prepared by the eutectic emulsion solvent evaporation technique and ultrasonication techniques were assigned the names C1 and C2, respectively.
Characterization of particle size and zeta potential
The particle size and zeta potential of the ibuprofen nanosuspension (nNS), control nanosuspensions (C1 and C2), and ibuprofen powder (100 mg/5 mL dispersed in 2% Tween 80 solution) were measured using a Zeta-sizer (Zeta-sizer nano series ZS; Malvern Instruments, Malvern, UK). The morphology of the particles was captured using an attachable digital C-mount camera (Moticam2; Motic®, Hong Kong, People’s Republic of China) under an inverted microscope (LHS-H100P-1; Nikon Corporation, Tokyo, Japan).
Characterization of ibuprofen nanocrystal
The raw ibuprofen powder and dried ibuprofen nanosuspension (nNS) were observed under a scanning electron microscope with an accelerating voltage of 15 keV. The sample was strewed onto carbon double-adhesive tape that adhered to a metal stub and then sputter-coated with gold. Images of the samples at 50× and 500× magnifications were taken using secondary electron image mode. The chemical structure was analyzed with an fourier transform infrared (FTIR) spectrophotometer (Nicolet 4700; Thermo Electron Corporation, Madison, WI, USA) using the KBr disk method. The crystallinity was analyzed with an X-ray powder diffractometer (Miniflex II; Rigaku Corp., Tokyo, Japan). The X-ray source was Cu-Kα, which employed an X-radiation wavelength of λ=1.541841 Å between a 2θ angle ranging from 4° to 60° with a voltage of 30 kV and current of 15 mA. The thermal properties of ibuprofen powder and ibuprofen nanocrystals were analyzed using a differential scanning calorimeter (DSC) (Pyris Sapphire DSC, Standard 115V; Perkin Elmer Instruments, Japan) and a thermo-gravimetric analyzer (TGA) (Pyris/TGA; Perkin Elmer Instruments) in air atmosphere. DSC analyzed within the ranges of −10°C to 100°C with a heating rate of 10°C/min. TGA analyzed within the ranges of 30°C–500°C at the same rate of DSC.
Transformation of droplets into particles
During the evaporation process, an emulsion was sampled at different time intervals of 8, 16, 24, 32, 40, and 48 hours. The particle size and zeta potential of each sample were analyzed using a Zeta-sizer (Zeta-sizer nano series ZS; Malvern Instruments). Morphology changes of the particles were captured using an attachable digital C-mount camera (Moticam2; Motic®) under an inverted microscope (LHS-H100P-1; Nikon) at ×200 magnification.
Amount of menthol and camphor during evaporation process
During the evaporation process, the emulsion/suspension was sampled at 8, 16, 24, 32, 40, and 48 hours. The menthol and camphor in each sample were extracted using chloroform. Briefly, 5 mL emulsion/suspension and 10 mL chloroform were added into the airtight glass containers and continuously stirred at 50 rpm using a shaking incubator until the water part was cleared. The amounts of menthol and camphor in the chloroform were analyzed using a gas chromatography (GC) method with validation using the 6890N GC network system (Agilent Technologies, Santa Clara, CA, USA) (n=3) as method described previously.39
Thermal behavior of drug during evaporation process
The amounts of ibuprofen, menthol, and camphor in the internal phase at each time point were calculated with an assumption that ibuprofen did not evaporate during the process. The internal phase at each time point was prepared from the calculated data obtained from GC determination. The thermal properties of ibuprofen powder and ibuprofen in the internal phase at each time point were investigated using DSC (Pyris Sapphire DSC, Standard 115V; Perkin Elmer Instruments). The measurement temperature was set within the ranges of −10°C to 100°C with heating then cooling phases at a rate of 10°C/min for investigating melting and crystallizing behaviors. The determination of the glass transition temperature (Tg) of each sample was performed under air atmosphere. The measurement temperature was set within the ranges of −100°C to 0°C with a heating rate of 10°C/min.
Dewetting on crystallization process
Dewetting of ibuprofen crystals at each time point was calculated from the contact angle of the internal phase on the ibuprofen plate (θO) and the interfacial tension of the internal phase and external phase (γOW) (n=3). Dewetting is favorable when γOWcosθO <0.11 The contact angles of the internal phase (θO) and external phase (θW) at each time point during an evaporation process on an ibuprofen plate were investigated by sessile drop technique using a goniometer (FTA 1000; First Ten Angstroms, Portsmouth, NH, USA) at the 5th second of contact time. The ibuprofen plate was prepared by the crystallization of melted ibuprofen on a glass slide with a flat surface. The densities of the internal phase and the external phase at each time during evaporation process were measured using a pycnometer (n=3) used for interfacial tension measurement. The interfacial tensions of the internal phase and the external phase (γOW) at each time point during the evaporation process were investigated by drop shape analysis using a goniometer (FTA 1000; First Ten Angstroms). The heavy phase (higher density) was dropped into the light phase (lower density), and the interfacial value was measured (n=3).
Formulation and characterization of ibuprofen nanosuspension
The ibuprofen nanosuspension was prepared using a combination of a eutectic emulsion evaporation method and ultrasonication. Approximately 60 g of ibuprofen eutectic emulsion was diluted with 240 mL of the dilution medium (30 g of glucose and water qs to 240 mL) and continuously evaporated under a fume hood on a hot plate stirrer at 45°C for 2 days. Finally, the volume of suspension was adjusted to 55 mL and then sonicated with a probe ultrasonicator with probe model CV18. Each formula was sonicated at an amplitude level of 50% with a 30-second on and 2-second off pulse for 30 minutes in an ice bath. Approximately 300 mg of xanthan gum was dispersed in the nanosuspension as a viscosity inducer and stabilizer, and 60 mg of sodium benzoate, a preservative, was added with continuous stirring using a magnetic stirrer overnight. The volume of nanosuspension was adjusted to 60 mL with the addition of water and mixed using a homogenizer. This product was assigned to be a formulated ibuprofen nanosuspension (fNS). The final formulation of fNS is shown in Table 1. The fNS and commercial ibuprofen suspension (Nurofen) were investigated for viscosity and rheology using a Brookfield viscometer. The particle size and the zeta potential were measured using the Zeta-sizer as described earlier.
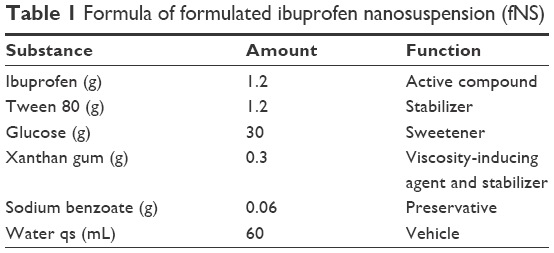 | Table 1 Formula of formulated ibuprofen nanosuspension (fNS) |
Dissolution studies
The dissolutions of ibuprofen powder, ibuprofen nanosuspension, and commercial ibuprofen suspension (Nurofen), comprising ibuprofen equivalent to 100 mg, were individually investigated using a United Stated Pharmacopeia dissolution apparatus II (paddle) (RC-6; Minhua Pharmaceutical Machinery Co. Ltd., Shanghai, People’s Republic of China) at 50 rpm, 37°C±0.5°C. Approximately 900 mL of phosphate buffer of pH 7.2 was used as the medium. The sampling time intervals were 1, 5, 10, 15, 30, and 60 minutes. Approximately 5 mL of samples was removed through a syringe with a 0.22-μm filter and an equal volume of fresh medium was replaced. The amount of dissolved ibuprofen was measured using high-performance liquid chromatography (HPLC) using an HPLC agilent 1100 series (Agilent Technologies) with ultraviolet detection at 220 nm. A C18 column (Ace, 4.6×250 mm, 5 μm particle size; Phenomenex, Torrance, CA, USA) was used. The mobile phase was 1:1 v/v of acetonitrile and 0.01 M phosphoric acid. The flow rate was 1 mL/min, and the sample volume was 20 μL. All the operations were performed at room temperature. The similarity factor (f2) and difference factor (f1) of fNS with NS and with Nurofen suspension were calculated.
Results and discussion
Physicochemical properties of ibuprofen particles
The particle size and zeta potential of nNS, C1, C2, and ibuprofen powder are shown in Table 2. The nNS prepared using the combination of a eutectic emulsion solvent evaporation method together with an ultrasonication technique exhibited the best particle properties as in the smaller size and higher zeta potential than those of the others significantly (P<0.05). Ibuprofen nanosuspension was not successfully prepared by emulsion solvent evaporation or sonication methods when employed individually, since the particles produced were in micrometer range.
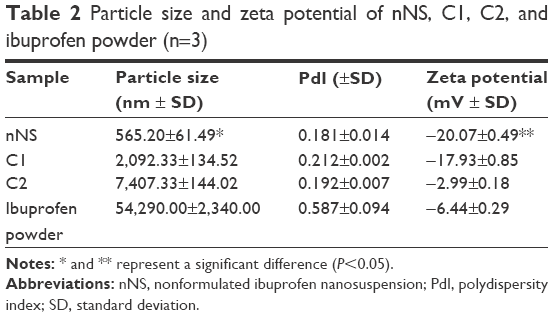 | Table 2 Particle size and zeta potential of nNS, C1, C2, and ibuprofen powder (n=3) |
The scanning electron microscope images of ibuprofen powder and ibuprofen nanocrystals are shown in Figure 1. The ibuprofen powder comprised needle-like crystal with a smooth surface as previously reported.40 The ibuprofen nanocrystals after ultrasonication comprised small fragments with a smooth surface because the cavitation force of ultrasonication reduced the crystallized ibuprofen into nanometer size. The ibuprofen nanocrystals agglomerated during filtration; however, in the dosage form of nanosuspension, the ibuprofen nanocrystals dispersed without agglomeration or sedimentation.
 | Figure 1 SEM images of ibuprofen powder and ibuprofen nanocrystal. |
There was no new peak or peak shift from the infrared spectrum of ibuprofen nanocrystals, which signified that this preparation did not affect the structure of ibuprofen (data not shown). The diffractograms of ibuprofen powder and ibuprofen nanocrystals were slightly different, and both showed the crystalline form of ibuprofen. The patterns of them nearly superposed, in which the difference was due to preferred orientation effect or varying unit cell dimension and as such shifting occurred in peak position as mentioned in BP 201541 (data not shown).
The DSC thermogram of ibuprofen powder was similar to that of ibuprofen nanocrystals (data not shown). The melting point for both powder and nanocrystals was 77°C as reported previously.24,42 TGA thermograms of ibuprofen powder and ibuprofen nanocrystals showed only one step of weight loss. Ibuprofen was melted, followed by its evaporation. The weight loss of ibuprofen powder started at 120°C and ended at 275.54°C with the dTGA peak at 249.91°C. For ibuprofen nanocrystal, the weight loss started and ended at 140°C and 236.59°C, respectively, and the dTGA peak was at 232.98°C. Similar thermal behavior of ibuprofen had been reported in previous works, which demonstrated that evaporation started at approximately 158°C–190°C and ended at approximately 245.7°C–277°C, and the dTGA peak was at 243.3°C–255°C.43,44 Therefore, this preparation technique did not alter the chemical properties and thermal behavior of ibuprofen, and the eutectic solvent was completely removed using this evaporation process. The rate of weight loss of ibuprofen nanocrystals (2.134%/°C) was higher than that of ibuprofen powder (1.924%/°C), which could be explained by the greater surface area of nanocrystal.
Transformation of emulsion droplet into particle
The ibuprofen eutectic emulsion was a white turbid emulsion with a unique flavor of menthol and camphor. Upon evaporation, the eutectic solvent in the internal phase droplet was continuously evaporated. The change of morphology under the inverted microscope could not be observed within 48 hours and the transformation of droplets into particles was observed after volume adjustment. The particle size and the zeta potential of droplets and particles at each sampling time are shown in Figure 2. It had been reported that the melted ibuprofen could retain a liquid state for a long time due to its apparently low glass transition temperature (Tg). This melted drug obtained from the melt solidification technique was induced and transformed into crystals by agitation.42,45 The investigation of a correlation of Tg and crystallization temperature (Tcr) of BaNaB9O15 glass indicated that a low Tg promoted a low Tcr.46 This correlation indicated that a lower Tg system required higher activation energy for crystallization than a higher Tg system. Ibuprofen had low Tg; therefore, the melted ibuprofen required high activation energy for crystallization, thus the adjustment to the final volume with the addition of distilled water, which disturbed the system with the higher mechanical force from this addition, triggered crystallization of ibuprofen. The droplet increased in size with time and significantly enlarged at 24 hours (P<0.05) and then transformed into particles after volume adjustment. The particle size of the obtained crystals was significantly decreased to 565±61.5 nm (P<0.05) after sonication during which the cavitation force fragmented the large particles into nanoparticles. Figure 2 showed a relatively low polydispersity index near to 0.20. The combination of the emulsion solvent evaporation (diffusion) method with sonication has been reported for fabricating the polymeric ocular nanosuspension with particle sizes of 100–300 nm.9
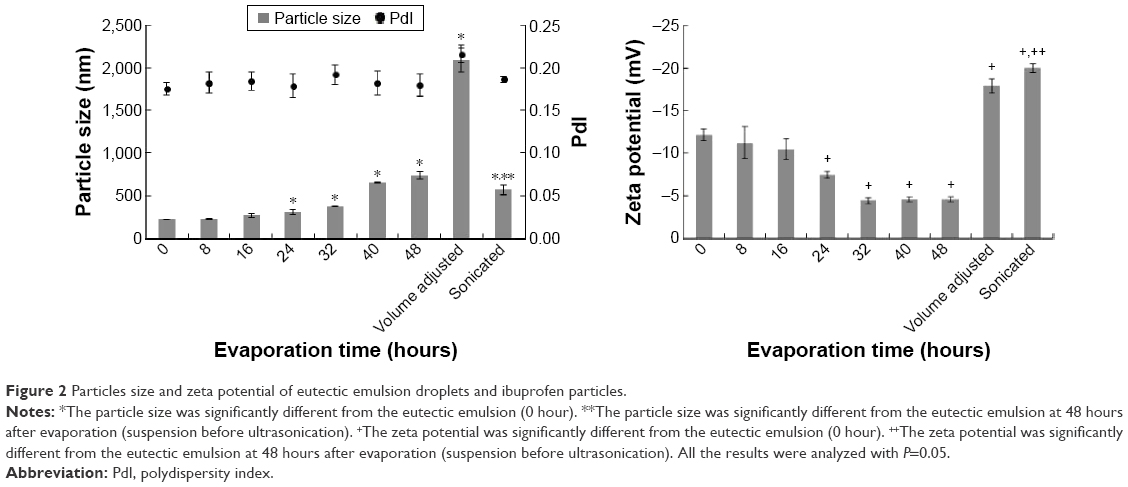 | Figure 2 Particles size and zeta potential of eutectic emulsion droplets and ibuprofen particles. |
The zeta potential of ibuprofen was negatively charged because of the carboxyl group in its molecule.47 During the evaporation process, the zeta potential of the emulsion gradually decreased prior to transformation (Figure 2). The total volume of emulsion gradually decreased due to the evaporation of water and eutectic solvent; therefore, the concentration of Tween 80 was increased and the zeta potential was decreased because of the greater adsorption of this nonionic surfactant on the droplet surface.48 When the droplet was transformed into the particle, the zeta potential was increased. During this transformation process, ibuprofen molecules were condensed and formed into solid particles,49 which exhibited a higher zeta potential than the emulsion.
Amount of menthol and camphor during evaporation
The residues of menthol and camphor in the eutectic emulsion progressively decreased. The evaporation rate of menthol was less than that of camphor from the eutectic emulsion. The molecular weight of menthol (156.27 g mol−1) was higher than that of camphor (152.23 g mol−1), and the vapor pressure of menthol (0.064 mmHg at 25°C) was notably lower than that of camphor (0.65 mmHg at 25°C). The amounts of menthol and camphor at 16 hours were 216.58±4.91 and 18.49±1.03 mg, respectively. The amounts of menthol and camphor in the eutectic emulsion were undetectable after 48 and 24 hours, respectively.
Thermal behavior of ibuprofen during evaporation
The concentrations of menthol, camphor, and ibuprofen in the internal phase at each time point of evaporation were shown in Table 3. Ibuprofen was at the supersaturation level after 8 hours of evaporation with simultaneous crystallization; however, the crystallization of ibuprofen during evaporation was not observed under the inverted microscope, and this phenomenon was confirmed by DSC data.
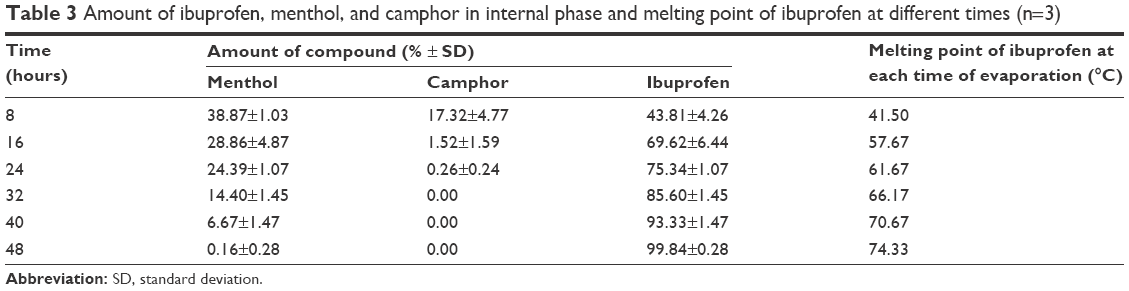 | Table 3 Amount of ibuprofen, menthol, and camphor in internal phase and melting point of ibuprofen at different times (n=3) |
The melting point of ibuprofen powder was 77.33°C (Figure 3), and during the heating step, the melting point of ibuprofen in the internal phase (prepared from the data in Table 3) increased as the evaporation time was longer because the amount of ibuprofen in the sample was increased and the melting suppression of the eutectic effect was decreased. Before 24 hours of evaporation, the melt endothermic peaks of ibuprofen crystals in the eutectic system were evident in the DSC thermograms (Figure 3). The higher ibuprofen concentration at 16 hours gave rise to a shaper peak than that at 8 hours. At 24–40 hours of evaporation, the internal phase was a mixture of menthol and ibuprofen; therefore, the DSC thermograms exhibited two melting peaks. The first peak at 10°C represented the melting of the eutectic system of menthol and ibuprofen and the second peak was the melting of excess ibuprofen as reported previously.17,19 The DSC thermograms from the heating and cooling steps of the affined internal phase at varying times of evaporation are shown in Figure 3. During the cooling step, the endothermic peak from crystallization did not occur in either of the ibuprofen powder or the affined internal phase and was also not evident under the inverted microscope. Therefore, the Tg of ibuprofen powder was not evident although this value had been previously reported at −45.15°C22 and −41°C,24 respectively. The DSC thermogram after evaporation for 24 hours indicated that the Tg of the demonstrated internal phase was at −58.95°C (data not shown) because the Tg suppression resulted from the eutectic formation between menthol and ibuprofen, indicating its amorphous state. The Tg of 3:1 menthol:ibuprofen has been reported recently at −50.1°C.22 The reducing Tg of lidocaine in the eutectic formation has also been reported.16 From these experimental data, we found that ibuprofen should be crystallized at 16 hours because the concentration of ibuprofen in the internal phase was higher than the saturation level. In addition, the melting point of ibuprofen was higher than the evaporation temperature, but the image under the inverted microscope indicated that the internal phase did not crystallize until the adjustment of the final volume was conducted as previously discussed. Thus, the crystallization of ibuprofen did not occur through the evaporation process.
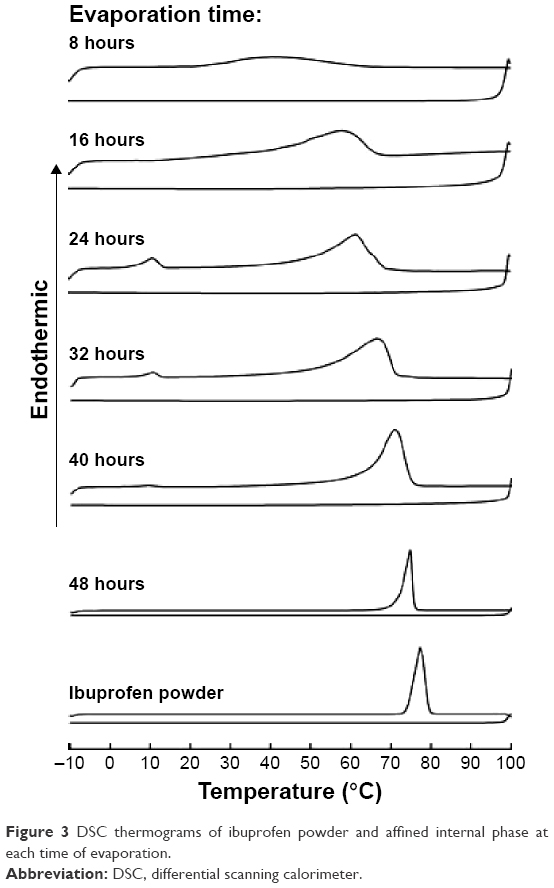 | Figure 3 DSC thermograms of ibuprofen powder and affined internal phase at each time of evaporation. |
Dewetting of ibuprofen crystals during crystallization and transformation mechanism
The contact angle of the internal phase and external phase increased with evaporation time as shown in Figure 4. During evaporation, the viscosity of the internal phase and external phase increased owing to the increased solute concentration from solvent evaporation. The contact angle of the internal phase was apparently lower than that of the external phase at all times of evaporation, indicating that the ibuprofen crystals were preferentially wetted by the internal phase than the external phase. When the crystallization occurred, the ibuprofen crystals should have formed inside the oil droplets. The interfacial tension between the internal phase and external phase reduced with evaporation time because of the increase of Tween 80 concentration as shown in Table 4. The dewetting value of ibuprofen crystals during evaporation was 4.80–7.88 (Table 4), which was not favorable in this system, thus the crystallization occurred within droplets.11 In this study, the ibuprofen crystals were larger than the emulsion droplets, which signified that the crystal growth was owing to Ostwald ripening after crystallization.49,50 The schematic of the transformation mechanism of the ibuprofen eutectic emulsion into ibuprofen nanosuspension is demonstrated in Figure 5. Ibuprofen crystallization was not found during the evaporation process, but it occurred when water was added to adjust the final volume. Ibuprofen crystallization occurred inside the droplets and then crystal growth and Ostwald ripening occurred, thus the particle size of ibuprofen was larger than the emulsion droplets. Finally, the ultrasonication was applied to obtain the ibuprofen nanosuspension.
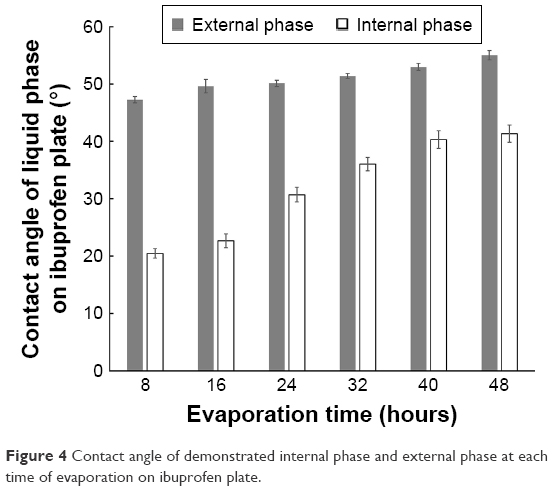 | Figure 4 Contact angle of demonstrated internal phase and external phase at each time of evaporation on ibuprofen plate. |
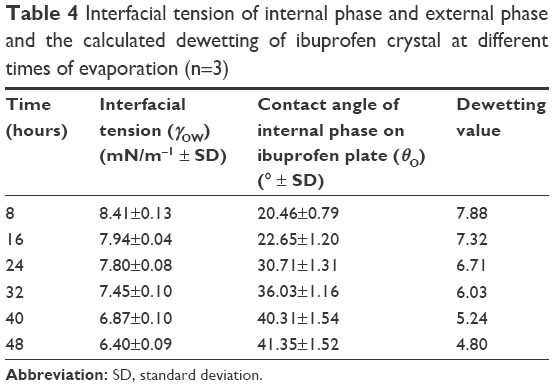 | Table 4 Interfacial tension of internal phase and external phase and the calculated dewetting of ibuprofen crystal at different times of evaporation (n=3) |
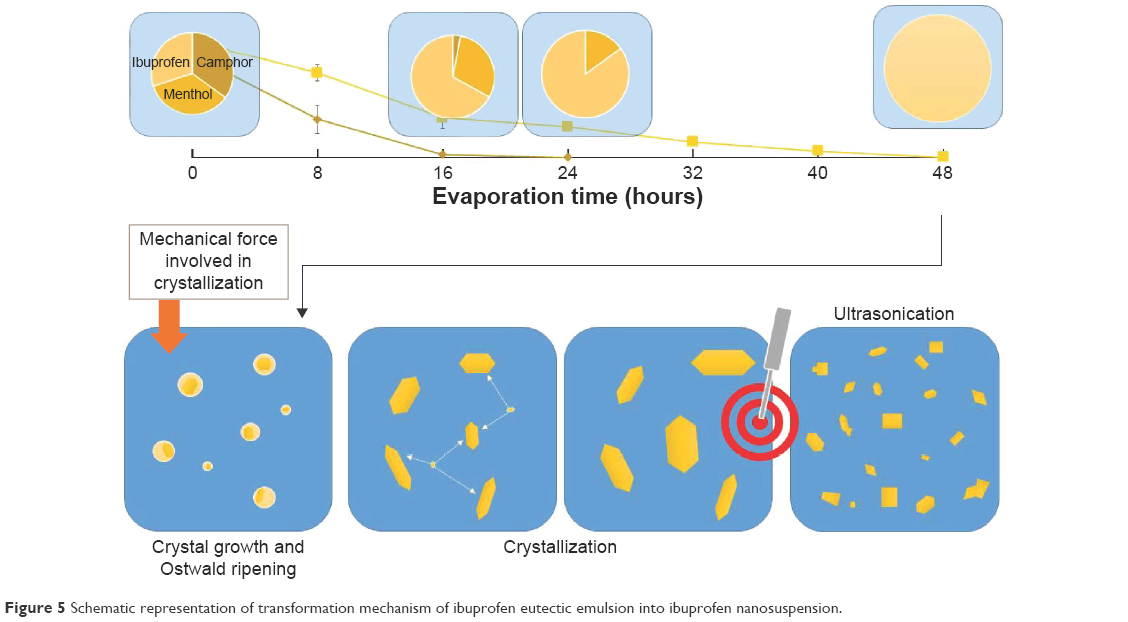 | Figure 5 Schematic representation of transformation mechanism of ibuprofen eutectic emulsion into ibuprofen nanosuspension. |
Characterization of formulated ibuprofen nanosuspension
A preparation technique utilizing a eutectic emulsion solvent evaporation method followed by sonication was used for ibuprofen nanosuspension preparation as previously described. The particle sizes of fNS (330.86±51.49 nm) and nNS (565.20±61.50 nm) were significantly lower than that of ibuprofen powder (54,290±2,340 nm) (P<0.05). However, the addition of xanthan gum to the formulation increased the viscosity, which reduced the crystal growth and Ostwald ripening after crystallization, thus the particle size of an fNS was lower than that of an nNS.51 The viscosity of fNS was 58.27±0.98 cps, which exhibited pseudoplasic flow. Moreover, the addition of xanthan gum influenced the zeta potential. The zeta potential of an fNS (−31.10±1.56 mV) was significantly higher than that of the nNS (−20.07±0.49 mV) and ibuprofen powder (6.44±0.29 mV) (P<0.05) due to the negatively charged nature of xanthan gum. The recommended zeta potential for physically stable nanosuspensions with electrostatic repulsion is at least ±30 mV, and for system combined with steric stabilization, it is recommended at ±20 mV.48,52 Therefore, the fNS became a physically stable nanosuspension by the addition of xanthan gum because there was no phase separation such as creaming or cracking after preparation for 4 months.
Dissolution studies
The dissolution rates of ibuprofen from commercial ibuprofen suspension (Nurofen), ibuprofen powder, nNS, and fNS were determined. The standard equation for ibuprofen in the range of 5–120 μg/mL using HPLC analysis was y =48.675x −7.4886 with r2=0.9997. The drug release profiles are shown in Figure 6. The releases of the fNS and nNS were 87.82% and 94.36% at 1 minute of dissolution test, respectively. Both the nNS and fNS were completely dissolved within 10 minutes with f2 =60.56 and f1 =5.08, indicating that the dissolution profiles were similar. The addition of xanthan gum increased the zeta potential and viscosity of the nanosuspension to obtain the stable nanosuspension without retarding the dissolution rate. The commercial ibuprofen suspension (Nurofen) and ibuprofen powder completely dissolved within 15 and 30 minutes, respectively. The f2 value of fNS with Nurofen suspension was 29.93, indicating the difference in dissolution profile.53 This result indicated that ibuprofen nanosuspension was a rapidly dissolved formulation with a dissolution rate faster than that of the commercial ibuprofen suspension (Nurofen) and ibuprofen powder.
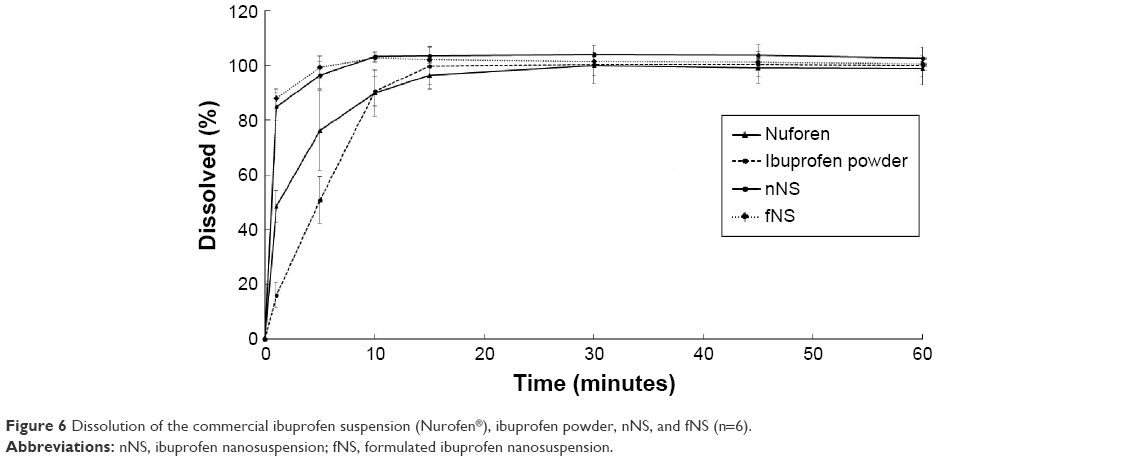 | Figure 6 Dissolution of the commercial ibuprofen suspension (Nurofen®), ibuprofen powder, nNS, and fNS (n=6). |
Conclusion
Menthol and camphor could be used as the volatile eutectic solvent for ibuprofen. This eutectic emulsion was transformed into a nanosuspension by the emulsion evaporation method followed by ultrasonication. During evaporation, menthol and camphor, as the eutectic solvent, were progressively evaporated. Camphor exhibited a higher evaporation rate than menthol; therefore, the content of the internal phase progressively changed. The concentration of ibuprofen in the internal phase reached supersaturation during the evaporation, which favored crystallization, but the crystallization of ibuprofen did not occur because ibuprofen in the internal phase required high activation energy. When the final volume was adjusted together with agitation from mixing, the crystallization of ibuprofen was triggered. The dewetting of ibuprofen was not favorable; therefore, the crystallization occurred in the droplet. After the crystallization, Ostwald ripening facilitated crystal growth, thus the drug particles were larger than the parent emulsion droplets. This Ostwald ripening should be reduced by the addition of some stabilizer. The cavitation force from the ultrasonicator reduced the size of crystals into a nanosuspension. Therefore, the mechanistic transformation from eutectic emulsion to nanosuspension was attained. The eutectic emulsion solvent evaporation coupled with ultrasonication technique could be used for the preparation of ibuprofen nanosuspension with a rapid release profile comparable to commercial ibuprofen suspension products.
Acknowledgments
This research work was kindly supported by the Faculty of Pharmacy, Silpakorn University and was also supported by a grant from the National Nanotechnology Center, National Science and Technology Development Agency (Grant P-11-00226) and the Research and Development Institute, Silpakorn University. We also thank Henry James Erskine-Hill, Dr Kamonpan Boonkit, Eugene D Kilayco, and Dr Purin Charoensuksai for their valuable comments and help.
Disclosure
The authors report no conflicts of interest in this work.
References
Verma S, Gokhale R, Burgess DJ. A comparative study of top-down and bottom-up approaches for the preparation of micro/nanosuspensions. Int J Pharm. 2009;380:216–222. | ||
Möschwitzer JP. Drug nanocrystals in the commercial pharmaceutical development process. Int J Pharm. 2012;453:142–156. | ||
Chan HK, Kwok PC. Production methods for nanodrug particles using the bottom-up approach. Adv Drug Deliv Rev. 2011;63:406–416. | ||
Chingunpituk J. Nanosuspension technology for drug delivery. Walailak J Sci Tech. 2007;4:139–153. | ||
Lakshmi P, Kumar G. Nano-suspension technology: a review. Int J Pharm Pharm Sci. 2010;2:35–40. | ||
Dolenc A, Kristl J, Baumgartner S, Planinsek O. Advantages of celecoxib nanosuspension formulation and transformation into tablets. Int J Pharm. 2009;376:204–212. | ||
Pignatello R, Bucolo C, Ferrara P, Maltese A, Puleo A, Puglisi G. Eudragit RS100 nanosuspensions for the ophthalmic controlled delivery of ibuprofen. Eur J Pharm Sci. 2002;16:53–61. | ||
Thao TT, Phuong HT, Minh NUN, et al. Amorphous isradipine nanosuspension by the sonoprecipitation method. Int J Pharm. 2014;474:146–150. | ||
Dandagi P, Kerur S, Mastiholimath V, Gadad A, Kulkarni A. Polymeric ocular nanosuspension for controlled release of acyclovir: in vitro release and ocular distribution. Iran J Pharm Res. 2009;8:79–86. | ||
Liu D, Xu H, Tia B, et al. Fabrication of carvedilol nanosuspensions through the anti-solvent precipitation–ultrasonication method for the improvement of dissolution rate and oral bioavailability. AAPS PharmSciTech. 2012;13:295–304. | ||
Spicer P, Hartel R. Crystal comets: dewetting during emulsion droplet crystallization. Aust J Chem. 2005;58:655–659. | ||
Aranberri I, Beverley K, Binks B, Clint J, Fletcher P. How do emulsion evaporate? Langmuir. 2002;18:3471–3475. | ||
Aranberri I, Binks B, Clint J, Fletcher P. Retardation of oil drop evaporation from oil-in-water emulsions. Chem Commun. 2003;21:2538–2539. | ||
Bi M, Hwang S, Morris K. Mechanism of eutectic formation upon compaction and its effects on tablet properties. Thermochim Acta. 2003;404:213–226. | ||
Woolfson A, Malcolm R, Campbell K, Jones D, Russell J. Rheological, mechanical and membrane penetration properties of novel dual drug systems for percutaneous delivery. J Control Rel. 2000;67:395–408. | ||
Lazerges M, Rietveld I, Corvis Y, Céolin R, Espeau P. Thermodynamic studies of mixtures for topical anesthesia: lidocaine-salol binary phase diagram. Thermochim Acta. 2010;497:124–128. | ||
Stott P, Williams A, Barry B. Transdermal delivery from eutectic systems: enhanced permeation of a model drug ibuprofen. J Control Rel. 1998;50:297–308. | ||
Stott P, Williams A, Barry B. Mechanistic study into the enhanced transdermal permeation of a model β-blocker; propranolol; by fatty acids: a melting point depression effect. Int J Pharm. 2001;219:161–176. | ||
Yong C, Oh Y, Jung S, et al. Preparation of ibuprofen-loaded liquid suppository using eutectic mixture system with menthol. Eur J Pharm. 2004;23:347–353. | ||
Nazzal S, Smalyukh I, Lavrentovich O, Khan M. Preparation and in vitro characterization of a eutectic based semisolid self-nanoemulsified drug delivery system (SNEDDS) of ubiquinone: mechanism and progress of emulsion formation. Int J Pharm. 2002;235:247–265. | ||
Wahlgren C, Quiding H. Depth of cutaneous analgesia after application of a eutectic mixture of the local anesthetics lidocaine and prilocaine (EMLA cream). J Am Acad Dermatol. 2000;42:584–588. | ||
Aroso IM, Craveiro R, Rocha Â, et al. Design of controlled release systems for THEDES-therapeutic deep eutectic solvents, using supercritical fluid technology. Int J Pharm. 2015;492:73–79. | ||
Dominguez de Maria P, Maugeri Z. Ionic liquids in biotransformations: from proof-of-concept to emerging deep-eutectic-solvents. Curr Opin Chem Biol. 2011;15:220–225. | ||
Dudognon E, Danède F, Descamps M, Correia NT. Evidence for a new crystalline phase of racemic Ibuprofen. Pharmaceut Res. 2008;25:2853–2858. | ||
Lindberg D, de la Fuente Revenga M, Widersten M. Deep eutectic solvents (DESs) are viable cosolvents for enzyme-catalyzed epoxide hydrolysis. J Biotechnol. 2010;147:169–171. | ||
Miller R. Deep eutectic solvents and applications. United States patent US 20090247432. 2009. | ||
Morrison HG, Sun CC, Neervannan S. Characterization of thermal behavior of deep eutectic solvents and their potential as drug solubilization vehicles. Int J Pharm. 2009;378:136–139. | ||
Patel T, Ishiuji Y, Yosipovitch G. Menthol: A refreshing look at this ancient compound. J Am Acad Dermatol. 2007;57:873–878. | ||
Al-Bayati FA. Isolation and identification of antimicrobial compound from Mentha longifolia L. leaves grown wild in Iraq. Ann Clin Microbiol Antimicrob. 2009;8:20. | ||
Kunta JR, Goskonda VR, Brotherton HO, Khan MA, Reddy IK. Effect of menthol and related terpenes on the percutaneous absorption of propranolol across excised hairless mouse skin. J Pharm Sci. 1997;86: 1369–1373. | ||
Schwarz J, Weisspapir M. Eutectic mixture of camphor, menthol, thymol and similar compounds is a powerful solvent for non-steroidal anti-inflammatory drugs; transdermal drug delivery of e.g. indomethacin, diclofenac, or ketoprofen. United States patent US20050158348 A1, 2005. | ||
Schwarz J, Weisspapir M. Vehicle for topical delivery of anti-inflammatory compounds. United States patent US20060241175 A1, 2006. | ||
Tuntarawongsa S, Phaechamud T. Menthol, borneol, camphor and WS-3 eutectic mixture. Adv Mater Res. 2012;560:355–358. | ||
Tuntarawongsa S, Phaechamud T. Polymeric eutectic drug delivery system. J Met Mat Min. 2012;22:27–32. | ||
Kocbek PB, Baumgartner S, Kristl J. Preparation and evaluation of nanosuspensions for enhancing the dissolution of poorly soluble drugs. Int J Pharm. 2006;312:179–186. | ||
Sinha B, Muller RH, Moschwitzer JP. Systematic investigation of the cavi-precipitation process for the production of ibuprofen nanocrystals. Int J Pharm. 2013;458:315–323. | ||
Tuntarawongsa S, Phaechamud T. Application of eutectic solvent to preparation of ibuprofen suspension by emulsion evaporation method. Adv Mater Res. 2015;1060:188–191. | ||
Parjikolaei B, El-Houri R, Fretté X, Christensen K. Influence of green solvent extraction on carotenoid yield from shrimp (Pandalus borealis) processing waste. J Food Eng. 2015;155:22–28. | ||
Phaechamud T, Tuntarawongsa S, Charoensuksai P. Evaporation behavior and characterization of eutectic solvent and ibuprofen eutectic solution. AAPS PharmSciTech. Epub 2015 Dec 15. | ||
Nokhodchi A, Amire O, Jelvehgari M. Physico-mechanical and dissolution behaviours of ibuprofen crystals crystallized in the presence of various additives. DARU J Pharm Sci. 2010;18:74–83. | ||
British Pharmacopoeia Commission, The stationery office, UK. British pharmacopoeia (BP); 2015, p. V-530. | ||
Paradkar AR, Maheshwari M, Ketkar AR, Chauhan B. Preparation and evaluation of ibuprofen beads by melt solidification technique. Int J Pharm. 2003;255:33–42. | ||
Tita B, Fulias A, Bandur G, Rusu G, Tita D. Thermal stability of ibuprofen. Kinetic study under non-isothermal conditions. Rev Roum Chim. 2010;55:553–558. | ||
Xu F, Sun L, Tan Z, Liang J, Li R. Thermodynamic study of ibuprofen by adiabatic calorimetry and thermal analysis. Thermochim Acta. 2004;412:33–37. | ||
Maheshwari M, Ketkar AR, Chauhan B, Patil VB, Paradkar AR. Preparation and characterization of ibuprofen–cetyl alcohol beads by melt solidification technique: effect of variables. Int J Pharm. 2003;261:57–67. | ||
Vaish R, Varma K. The glass transition and crystallization kinetic studies on BaNaB9O15 glasses. J Phys D: Appl Phys. 2009;42(1):015409. | ||
Momoh M, Adedokun M, Lawal S, Ubochi G. Formulation and in vitro evaluation of ibuprofen-loaded poly (D,L-lactide-co-glycolide) microparticles. Trop J Pharm Res. 2014;13:1571–1576. | ||
Singh SK, Srinivasan KK, Gowthamarajan K, Singare DS, Prakash D, Gaikwad NB. Investigation of preparation parameters of nanosuspension by top-down media milling to improve the dissolution of poorly water-soluble glyburide. Eur J Pharm Biopharm. 2011;78:441–446. | ||
McClements DJ. Crystals and crystallization in oil-in-water emulsions: implications for emulsion-based delivery systems. Adv Colloid Interface Sci. 2012;174:1–30. | ||
Coupland JN. Crystallization in emulsions. Curr Opin Colloid Interface Sci. 2002;7:445–450. | ||
Verma S, Kumar S, Gokhale R, Burgess DJ. Physical stability of nanosuspensions: investigation of the role of stabilizers on Ostwald ripening. Int J Pharm. 2011;406:145–152. | ||
Papdiwal A, Pande V, Sagar K. Design and characterization of zaltoprofen nanosuspension by precipitation method. Der Pharma Chemica. 2014;6:161–168. | ||
Abbirami V, Sainithya P, Shobana A, Devi D, Hari BN. A review on in-vitro bioequivalence studies and its methodologies. Int J Chem Tech Res. 2013;5:2295–2302. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.