Back to Journals » International Journal of Nanomedicine » Volume 13
Transferrin receptor-targeted HMSN for sorafenib delivery in refractory differentiated thyroid cancer therapy
Received 12 September 2018
Accepted for publication 6 November 2018
Published 6 December 2018 Volume 2018:13 Pages 8339—8354
DOI https://doi.org/10.2147/IJN.S187240
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
You Ke,1 Cheng Xiang2
1Department of Nephrology, The Second Affiliated Hospital of Zhejiang University, Hangzhou, China; 2Department of Surgery, The Second Affiliated Hospital of Zhejiang University, Hangzhou, China
Background: Thyroid cancer becomes the most common endocrine cancer with the greatest growing incidence in this decade. Sorafenib is a multikinase inhibitor for the treatment of progressive radioactive iodine-refractory differentiated thyroid cancer (DTC), while the off-target toxicity effect is usually inconvenient for patients taking.
Methods: In this study, hollow mesoporous silica nanoparticles (HMSNs) with transferrin modification (Tf-HMSNs) were loaded with sorafenib (sora@Tf-HMSNs) to help targeted delivery of sorafenib. Due to the biocompatible Tf shell, Tf-HMSNs exhibited excellent biocompatibility and increased intracellular accumulation, which improved the targeting capability to cancer cells in vitro and in vivo.
Results: Sora@Tf-HMSNs treatment exhibited the strongest inhibition effect of res-TPC-1 cells and res-BCPAP cells compared with sora@HMSNs and sorafenib groups and induced more cancer cell apoptosis. Finally, Western blot analysis was conducted to check the expression of RAF/MEK/ERK signaling pathway after sorafenib encapsulated Tf-HMSNs treatment.
Conclusion: Overall, sora@Tf-HMSNs can significantly increase the effective drug concentration in cancer cells and thus enhance the anticancer effect, which are expected to be promising nanocarriers to deliver anticancer drugs for effective and safe therapy for RAI-refractory DTC.
Keywords: sorafenib, RAI-refractory DTC, hollow mesoporous silica nanoparticles, transferrin, RAF/MEK/ERK
Introduction
Thyroid cancer is the most common endocrine malignant neoplasm and the cancer with the greatest increasing incidence rate. In 2015, it was estimated that there were 62,450 new cases of thyroid cancers in the United States and an estimate of 1,950 deaths from this disease.1 The increased prevalence of thyroid cancer may be heightened by autoimmune phenomena, genetic mutation, iodine intake alternation, and potential environmental carcinogens such as radiation exposure.2 After surgery, radioactive therapy and postoperation l-thyroxine treatment are usually in need3 after which still 7%–23% of patients develop distant metastases, and two-thirds of patients with distant metastases become radioactive iodine (RAI) refractory4,5 accompanied with tumor dedifferentiation. Dedifferentiated tumors are more aggressive and had a worse outcome.6,7 Currently, novel molecular targeting agents are gradually changing the natural history of traditional systemic chemotherapies of RAI-refractory thyroid cancer, which have showed improved progression-free survival in patients while the focus ablation rate still remains unsatisfactory.8
Sorafenib, an oral multikinase inhibitor, has been approved to treat progressive RAI-refractory DTC in 2013 by the U.S. Food and Drug Administration. It has been verified to target BRAF, RET, VEGFR, PDGF-β, chimeric RET/papillary thyroid cancer (PTC), and c-kit.9,10 The MAPK (RAS/RAF/MEK/ERK) pathway is essential to tumor angiogenesis and has been demonstrated to mediate tumor cell proliferation, differentiation, survival, and motility.11–12 More than 70% DTC patients are MAPK pathway overactivated because of BRAF and RAS mutations.13 Therefore, the RAF/MEK/ERK serine/threonine kinase cascade plays a key role in the development of cancers and is considered as a potential target for sorafenib treatment.14 However, clinical application of sorafenib in RAI-refractory DTC patients still remains limited because of its water insolubility, low oral bioavailability (~8.43%), light sensitivity, and especially high off-target toxicity such as hand–foot skin reaction, diarrhea, and alopecia.15–17 In a double-blind, Phase III clinical trial, 66.2%, 64.3%, and 18.8% of patients experienced dose interruptions, reductions, or withdrawals due to adverse effect, respectively.9 Therefore, there is an urgent need to enhance the bioavailability of sorafenib to improve the therapeutic result and overcome the side effects for the treatment of RAI-refractory thyroid cancer.
Nowadays, nanoparticle technology is being explored extensively for its ability to deliver proper drugs into tumor cells.18,19 Nanoparticles (NPs) can change the pharmacokinetic and pharmacodynamic profiles of traditional therapeutics and thus may improve the efficacy of existing anticancer drugs.20 Among them, hollow mesoporous silica nanoparticles (HMSNs) show great promise as a viable platform for effective delivery of anticancer drugs. HMSNs, with the hollow core for storage reservoir and the mesoporous shell for encapsulated compounds or substantial surface area for reactions, have a lot of advantages. In fact, they have large surface area and pore volume,21 adjustable pore size,22 versatile surface functionalization,23,24 and tunable biodistribution.25,26 More importantly, the surface of nanoparticles can be tailored with moieties, which are tumor specific such as peptides, antibodies, or ligands, and thus deliver anticancer drugs to target focus via ligand–receptor recognition without changing drug functionalities.27–29 Active targeting drug delivery enhances the drug concentration at the targeted tumor focus, achieving a better tumor suppression effect and reducing the unwanted cytotoxicity to normal tissues.30
Herein, in current study, we set out to fabricate a transferrin (Tf)-conjugated HMSN (Tf-HMSN)-based nanocarrier with tumor cell membrane Tf receptor (TfR) targeting. We expect the Tf-HMSNs loaded with sorafenib (sora@Tf-HMSNs) system would increase the delivery efficiency and reduce the off-target effect, which represents a promising strategy against RAI-refractory thyroid cancer.
Materials and methods
Chemicals
Tetraethyl orthosilicate (TEOS), cetyltrimethyl ammonium bromide (CTAB), ammonia hydroxide (NH3·H2O, 25 wt%), anhydrous ethanol, hydrochloric acid, sodium carbonate (Na2CO3), 3-triethoxysilylpropylsuccinic acid anhydride (TESPSA), (3-aminopropyl) triethoxysilane (APTES), dimethyl sulfoxide (DMSO), Tf, dimethylformamide (DMF), succinic anhydride (10 wt%) 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), fluorescein isothiocyanate (FITC), and DAPI were purchased from Sigma-Aldrich. Sorafenib was obtained from Selleckchem. FBS and DMEM were purchased from Gibco (Grand Island, NY, USA). Purified, deionized water was prepared with Heal Force Millipore Apparatus (Millipore Co., Billerica, MA, USA).
Preparation of HMSNs
HMSNs were synthesized via a template method in our lab. Briefly, 100 mL anhydrous ethanol, 8 mL deionized water, and 4 mL NH3·H2O (~37%–38%) were mixed together and stirred for a while at 30°C. After adding 3 mL of TEOS, the whole reaction was stirred for 5 hours. Then, a mixture of 1.2 g CTAB, 200 mL deionized water, and 10 mL anhydrous ethanol were added and continually stirred for 30 minutes. Subsequently, 1 mL TEOS was added and the reaction solution was maintained for reaction for another 12 hours at 30°C. The products were separated by centrifugation and redispersed in 0.4 M Na2CO3 aqueous solution. After etching at 50°C for 2 hours to obtain the hollow inner part, the resulting particles were collected. Finally, HMSNs were obtained after removal of the surfactant templates by washing three times using HCl/ethanol (v/v=1:10) with the help of sonication and washing thoroughly with water.
The FITC-labeled HMSNs (FITC-HMSNs) were synthesized by reacting FITC with HMSNs-NH2. For modification of amine groups, 10 mg HMSNs were stirred with 1.5 mL APTES in ethanol overnight and then centrifuged. After redispersed in water, 0.5 mg FITC was added into the solution and the solution was kept stirring for another 5 hours. The unlabeled FITC was removed by washing thoroughly with water.
For modification of carboxyl groups, HMSNs-NH2 nanoparticles (NPs) were reacted with 10% succinic anhydride in DMF according to a method showed by Liu et al.31 The reaction was maintained for 6 hours under N2 gas, and finally carboxyl groups were formed onto the surface of nanoparticles (denoted as HMSNs-COOH).
To obtain Tf-HMSNs, we applied a two-step EDC/NHS activation and surface grafting method. Typically, 20 mg of HMSNs-COOH were mixed in 10 mL of PBS with EDC (500 μL, 1 mg/mL) and NHS (500 μL, 1 mg/mL), and the solution was stirred for 4 hours at room temperature. Tf (200 μL, 1 mg/mL) was added dropwise after the NPs were dispersed in 4 mL of PBS. Then the solution was stirred for 2 hours and incubated overnight at 4°C before finally isolated and purified.
HMSNs characterization
The morphology and structure of HMSNs were determined by using scanning electron microscope (SEM; Sirion 200) and transmission electron microscope (TEM; Tecnai, G220). For SEM sample preparation, a dilute aqueous solution of the nanoparticles (2.5 mg/mL) was suspended adequately and then dropped on a clean silicon wafer and dried thoroughly. Then, a layer of gold was sprayed on the surface of nanoparticles for observation. Before TEM observation, the samples were dispersed in very dilute water (0.5 mg/mL) by sonication and then deposited on copper grids. Then, the as-prepared grids were dried completely at room temperature.
The diameter and zeta potential of the HMSNs and Tf-HMSNs were measured using a dynamic light scattering (DLS) apparatus. All the samples were prepared by ultrasonic dispersing.
Surface area and pore size of HMSNs were measured using surface area and porosity analyzer (NOVA 3200e). The nitrogen adsorption–desorption isotherm was detected at −196°C. The surface areas were calculated by Brunauer–Emmett–Teller (BET) method at the pressure range of 0.05–0.2. Adsorption data were collected for pore size distribution calculate by Barrett–Joyner–Halenda (BJH) method.
Tf identification
The Tf was detected using a ultraviolet–visible (UV–vis) spectrophotometer according to a modified Bradford assay. The assay was based on the fact that the Coomassie Blue G250 dye could bind to protein. Briefly, HMSNs (3 mg) and Tf-HMSNs (3 or 8 mg) samples were subjected to protein reagent (5 mL), with the final concentration of Coomassie Blue G250 dye was 0.01% (w/v). The sample volume was adjusted to 0.1 mL with appropriate buffer beforehand. Then the absorption spectra of HMSNs and Tf-HMSNs were detected using a UV–vis spectrophotometer.
Drug loading and release
Typically, by the adsorption equilibrium method, drugs (3 mg sorafenib) were dissolved in 1 mL DMSO. Subsequently, the fabricated carriers (10 mg) were added into the mixture and stirred gently for 24 hours at room temperature to achieve the largest loading of sorafenib. After centrifugation, the suspension was washed with PBS for three times to remove any remaining external surface adsorption of sorafenib. Then the supernatant was collected completely and the drug-loaded nanoparticles (sora@Tf-HMSNs) were obtained. HPLC (1100S; Agilent Technologies Inc., Santa Clara, CA, USA) with a UV–vis detector was used to measure the encapsulation efficiency (EE%) and drug loading (DL%) at 264 nm. A reversed phase-C18 column (Hypersil ODS2 C18 5 μm, 4.6×250 mm) was used for chromatographic separations. The mobile phase was methanol/water (30:70, v/v) with 0.1% trifluoroacetic acid at 1.0 mL/min flow rate. EE% and DL% were obtained by the following equations: (The drug encapsulated was calculated by the amount of total drug used minus the amount of drug in the supernatant and in the collected washing buffer.)
|

|
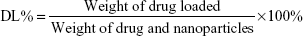
In vitro drug release was carried out by adding an amount of 10 mg sorafenib-loaded nanoparticles in phosphate buffered saline in a dialysis tube (PBS, pH 7.4). The samples were incubated and kept shaking at 200 rpm for 48 hours at 25°C and 37°C. At different intervals (0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, 72 hours), every 1 mL of sample was removed and 1 mL fresh PBS was added. The amount of released sorafenib was examined by HPLC. The experiments were carried out in triplicate and repeated three times.
Cell culture
The thyroid cancer cells TPC-1 and BCPAP were purchased from American Type Culture Collection. Cells were cultured in a humidified incubator at 37°C with 5% CO2 and cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco) supplemented with 10% FBS (Gibco). Resistant TPC-1 and BCPAP (res-TPC-1 and res-BCPAP) cells were prepared for several generations sublethal 131I concentration culture continuously, increased radioactivity concentration of 131I was in need to reach the same suppression effect for RAI-refractory cells in comparison to wild-type PTC cell lines. The median sublethal intensity of 131I increased more than two times in TPC-1 and BCPAP cell line (denoted as res-TPC-1 and res-BCPAP).
Immunofluorescence imaging
To demonstrate the TfR expression, res-TPC-1 and res-BCPAP cells were seeded on a coverslip and cultured at 37°C in RPMI-1640 medium overnight. Then cell fixation was performed with 4% paraformaldehyde for 10 minutes followed by PBS wash for three times. After fixation, the cells were permeabilized with 0.5% Triton X-100 for 20 minutes after blocked with 5% BSA for 30 minutes. Subsequently, the cells were incubated with mouse anti-TfR antibody (1:500; Thermo Fisher Scientific, Waltham, MA, USA) for 2 hours. After rinsing with PBS, the cells were reacted with secondary goat antimouse Alexa Fluor 594-conjugated antibody (1:1,000; Thermo Fisher Scientifc, Waltham, MA, USA). Nucleus was stained with DAPI for 5 minutes followed by PBS washing. The coverslips were examined by fluorescence microscope (IX71 inverted optical microscope in fluorescence mode; Olympus, Japan, Tokyo).
In vitro cytotoxicity
The cytotoxicity was determined by cell counting kit-8 (CCK-8) assay in res-TCP-1 and res-BCPAP cell lines; cells were treated with sorafenib, Tf-HMSNs, and sora@Tf-HMSNs for 48 hours. Briefly, cells were seeded in 96-well plates at a density of 5,000 cells each well. After incubation overnight, media were removed and cells were incubated in media containing sorafenib, Tf-HMSNs, and sora@Tf-HMSNs at a series of concentrations. After incubation for 24 hours and 48 hours, cells were rinsed three times with PBS, and then 10 μL of CCK-8 reagent was added into each well. After incubation for 2 hours, the absorbance was measured with a microplate reader (Infinite F50; Tecan Austria, Mannedorf, Austria) at 450 nm wavelength. All the experiments were performed three times with five replicates.
In vitro cellular uptake
For cellular uptake experiments, the glass slides were freshly made and sterilized followed by washing with PBS three times. Res-TPC-1 cells were seeded on the as-prepared slides in 96-well plates and grown overnight. Then the culture media were replaced with fresh media containing 100 μg/mL FITC-HMSNs or FITC-Tf-HMSNs. After incubation for 4 hours and 24 hours, cells were rinsed with PBS and 4% paraformaldehyde fix. Then the nuclei were stained with DAPI for 10 minutes. The cover slips were sealed and observed under confocal laser scanning microscope (CLSM, A1 Si; Nikon, Tokyo, Japan).
Cell apoptosis analysis
Res-TPC-1 cells were seeded in a 6-well plate. After 80% confluence, the medium was treated with desired compounds for 24 hours. Then cells were rinsed with PBS, trypsinized and resuspended in Annexin V binding buffer. Then 1 μL of Annexin V/FITC was added to each sample, and the mixture was incubated in the dark for 5 minutes at room temperature. Samples were then examined by flow cytometry.
Western blot
Typically, proteins from res-TCP-1 or res-BCPAP cells were extracted with RIPA lysis buffer (Beyotime, Jiangsu, China). Cell lysates were subjected to SDS-PAGE, and then transferred to polyvinylidene difluoride (PVDF) membranes. After blocking, the membranes were reacted with primary antibodies (anti-TfR, anti-MEK, anti-RAF, anti-Perk, anti-pMEK) at 4°C overnight and then secondary antibodies at room temperature for 2 hours. Band signals were detected by ChemiDocXRS (Bio-Rad, Hercules, CA, USA) and analyzed by ImageLab software.
Human cancer xenograft establishment and HMSN distribution
Human thyroid cancer cells TPC-1, collected in 0.2 mL RPMI-1640 medium, were injected into the severe combined immune deficiency (SCID) mice at the right lateral back (Shanghai SLAC Laboratory Animal Co., Shanghai, China). Mice were randomly divided into sora@HMSNs or sora@Tf-HMSNs treatment group and NC group after the tumors reach approximately a diameter of 0.5 cm (six mice each group); SCID mice bearing subcutaneous thyroid cancer tumors were then injected with DiR-labeled sora@HMSNs or 1,1‘-dioctadecyl-3,3,3’,3‘-tetramethylindotricarbocyanine (DiR)-labeled sora@Tf-HMSNs (9 mg/kg) suspended in 0.1 mL saline solution through the tail vein. For DiR-labeled nanoparticles, 10 mg nanoparticles were mixed in 1 mL DMSO. Then 3.5 mg DiR was added and the solution was stirred for 24 hours at room temperature before finally isolated and purified. After 1, 6, and 24 hours, the mice were anesthetized and placed in vivo imaging system (PerkinElmer, Waltham, MA, USA) chamber. Red fluorescent images (835 nm) were obtained with 745 nm excitation light. After euthanasia with CO2 and sacrificed, the tumors and organs were collected and embedded on slides for further imaging with fluorescent microscope. Finally, the tumor volumes were measured every 3 days, the growth rate was recorded as reproduction curve (tumor volume = length × width2/2). All animal experiments were comply with the animal research: reporting of in vivo experiments guidelines and carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines. These in vivo experiments were approved by Ethics Committee in Second Affiliated Hospital of School of Medicine, Zhejiang University.
Statistical analysis
All the values were presented as mean ± SD. Significant differences among groups were evaluated by one-way ANOVA using SPSS 13.0. P<0.05 was considered to be significant.
Results and discussion
TfR expression
TfR was a transmembrane glycoprotein commonly overexpressed on the surface of cancer cells for increased iron requirement because of cell proliferating and the fact that the key enzyme in DNA synthesis is iron required.32–35 In previous studies, to data, more researches focus on ligand-modified nanomedicines clinical management especially for cancer treatment.36–38 TfR recognition could facilitate endocytosis of the delivered particles through binding of Tf to TfR. As a result, TfR was made as excellent candidates for targeted therapeutics.39 TfR is also demonstrated aberrantly expressed in thyroid cancers.40,41 Here, we verified the overexpression of TfR in the thyroid cancer cell lines by Western blot in res-TPC-1 cells and res-BCPAP cellular experiments (Figure S1A). Furthermore, immunofluorescence confirmed that TfR (red color) was overexpressed especially on cell membranes (Figure S1B). In this regard, we aim to synthesize a Tf-conjugated nanocarrier, which can combine to the TfR on PTC cells and targeted delivery of anticancer drugs into cancer cells.
Synthesis and characterization of HMSNs and Tf-HMSNs
The chemotherapeutic effect in treating refractory thyroid cancer is relatively poor as low drug concentration in the tumor focus. In current study, we developed a HMSN-based system, which was modified with Tf protein and loaded with the anticancer drug sorafenib (Scheme 1). In our study, we aim to improve the sorafenib chemotherapeutic availability in thyroid cancer treatment by utilizing Tf-HMSNs to target TfR ligand for drug delivery. First, silica nanoparticles were synthesized according to a modified Stöber method.42 The mesoporous silica shell was coated via surfactant templating sol–gel method on the particle core. Na2CO3 aqueous solution was used to remove the solid core to form the hollow core. Finally, with ultrasonication support, the surfactant CTAB was eliminated by HCl/ethanol mixture. The structure of the resulting HMSNs was characterized using SEM accompanied with TEM. SEM indicated that the well-formed spherical nanoparticles were 160–200 nm in diameter and uniform in size (Figure 1A). After Tf modification, the TEM micrograph showed that the nanoparticle had a uniform core–shell structure and the Tf modification nanoparticles showed annular dark spots, which meant the porous-like structure formation (Figure 1B). The surface area and porosity of particle were demonstrated with nitrogen adsorption–desorption isotherms. The BET surface area was 1,125.6 m2/g and with 1.13 cm3/g pore volume (Figure S2A). The particle pore size kept uniform, which was around 2.2 nm calculated by BJH method (Figure S2B). The results above strongly indicated the potential application and loading ability for drug delivery.
 | Scheme 1 Schematic representation of the construction of hollow mesoporous silica nanoparticle (HMSN) based cancer cell-targeted nanocarriers. |
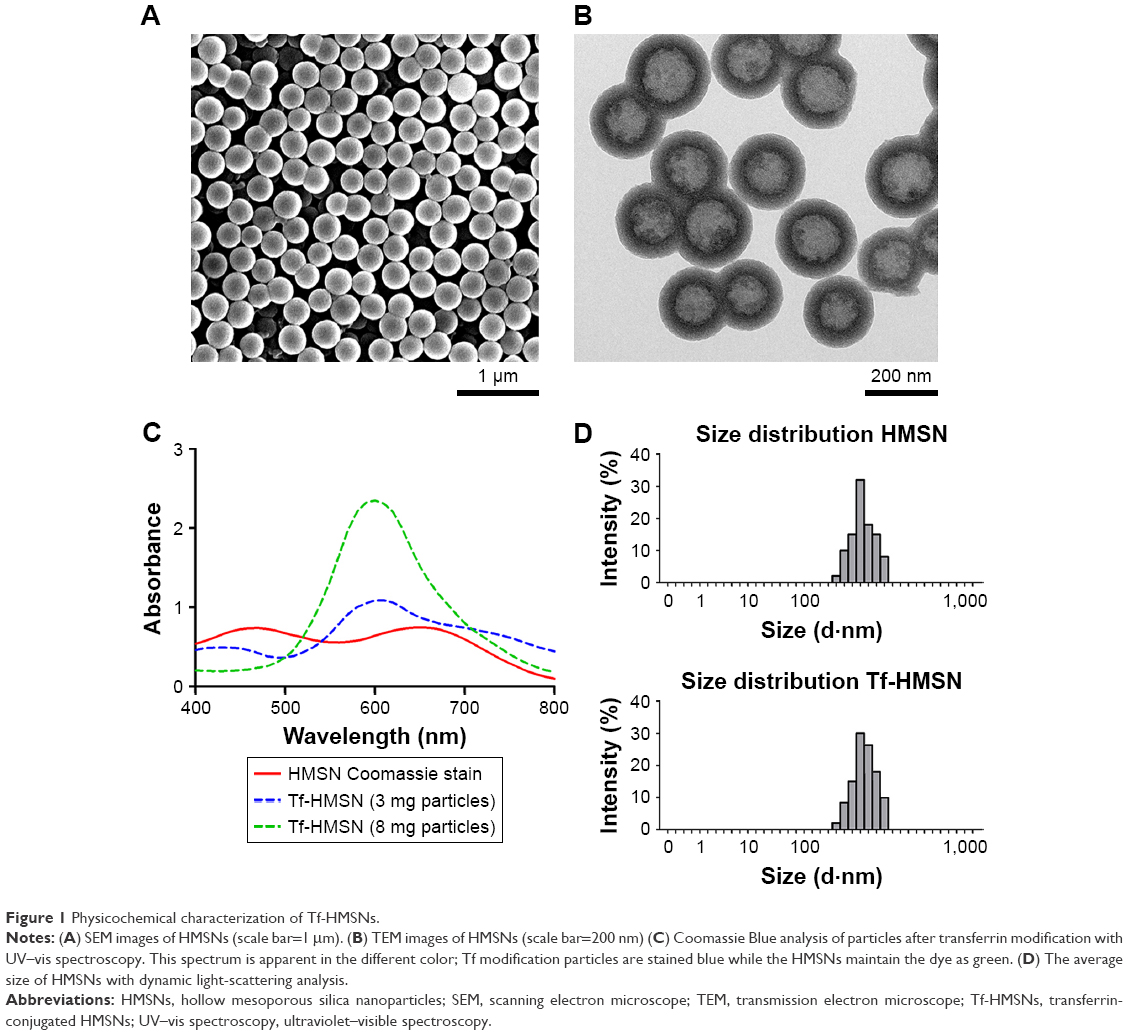 | Figure 1 Physicochemical characterization of Tf-HMSNs. |
The successful surface functionalization was further confirmed by Bradford assay. Nanoparticles were treated with the dye Coomassie Brilliant Blue and the samples relative absorbance were consecutively analyzed under UV–vis spectroscopy. Nanoparticles without protein modification show the maximum absorbance at 650 nm. After Tf-HMSNs suspension, the solution absorption spectrum exhibited a wide peak at 595 nm. The presence of the Tf on the nanoparticles was confirmed with Coomassie Blue.43 The presence of Tf surface was identified by the peak shifted from 650 to 595 nm for Tf-HMSNs, while HMSNs group had a maximum 650 nm absorption peak. The 595 nm absorbance increased proportionally when the amount of Tf-HMSNs nanoparticles increased from 3 to 8 mg (Figure 1C). This shift of UV absorbance from 650 to 595 nm was apparent in the solution color; the particles with protein modification were stained blue while the pure particles remain in the original green dye color. After Tf modification to produce the Tf-HMSNs, DLS showed that the size slightly increased to an average of 220 nm in diameter, which further proved the presence of Tf layer on the outermost surface of HMSNs. The relative HMSNs diameter interval proportion was demonstrated (Figure 1D). After Tf conjugation, we examined the average size and zeta potential of both HMSNs and Tf-HMSNs via DLS. Zeta potential of HMSNs showed −28 mV whereas Tf-HMSNs showed −36 mV at pH 7.4 (Figure S3). Since the isoelectric point of Tf is 5.2 mV, Tf decoration leads to a more negative charge for HMSNs. The Tf modification made this nanocarrier biocompatible and favorable for prolonged circulation in blood.44
In vitro sorafenib loading and release study
Ideal carrier means high drug capacity and enough EE%. For further application of Tf-HMSNs as nanocarrier for anticancer therapy, sorafenib acted as the drug of interest to treat refractory thyroid cancer in this study. Naturally, sorafenib is poor soluble in water while can be dissolved in DMSO. By the adsorption equilibrium method, sorafenib was easily absorbed and concentrated into HMSNs hollow core. In our experiment, sorafenib EE% was calculated 85%±4.6% and achieved a DL% density of 32±3.4 mg/g. Also, in anticancer treatment, the controlled and sustained drugs release typical is an important property for its clinical application. Based on pharmacology theory, fast release would increase the drug diffusion in plasma while not the concentration at the target focus, which increased the invalid drug release and the off-target toxicity. The release of sorafenib@HMSNs and sorafenib@Tf-HMSNs was investigated by the dialysis bag diffusion technique and then detected by HPLC. Normally, the majority of nanoparticles ingested by cancer cells take several hours to reach equilibrium in our previous experience. The release rate from HMSNs or Tf-HMSNs was measured until 72 hours at 12 continuous time intervals. The cumulative release curve was displayed (Figure 2). A persistent release profile of sorafenib was observed. During the first 24 hours, 28%±0.9% and 17%±0.7% of sorafenib released were recorded in HMSNs and Tf-HMSNs group, respectively. Simultaneously, sorafenib was released slower after 24 hours; the release rate of sorafenib was slower after external Tf coating (20.3%±0.8%) at 72 hours compared with HMSNs group (31.2%±1.0%) that effectively delayed its release from the mesopores of silica nanoparticle, which was similar to the previous study.45 The Tf moiety was close to the surface pore diameter could efficiently blocking aperture and slow down the premature drug release.46 The sustained release of sorafenib from Tf-HMSNs is highly beneficial to enhance anticancer effect, which increases the drug concentration at the tumor site.
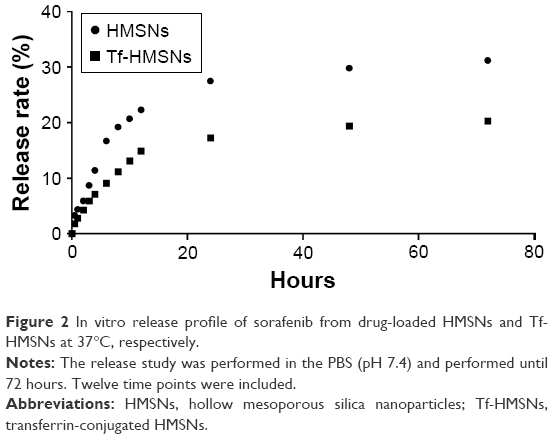 | Figure 2 In vitro release profile of sorafenib from drug-loaded HMSNs and Tf-HMSNs at 37°C, respectively. |
Construction of 131I-resistant thyroid cancer cells and in vitro cellular uptake
RAI (131I) treatment strategies on thyroid cancer have already been included in clinical guidelines; successful 131I remnant focus ablation rate is relative unsatisfactory. We mainly focused on molecular biology features in 131I resistance and tried to search more therapeutic targets in RAI-refractory PTC patients. Fortunately, we were the first to establish the 131I-resistant thyroid cancer cell lines in our latest literature.47 After successful 131I refractory thyroid cancer cell line construction and Tf-HMSNs manipulation, efficient uptake and internalization of the drug-loaded carriers are indispensable. TfR overexpression in thyroid cancer cells makes them excellent candidates for target therapeutics with Tf and TfR recognition.48 Given this, in vitro studies were carried to test the drug-targeting efficacy in res-TPC-1 and res-BCPAP cell line. The HMSNs were fluorescently labeled with FITC (a widely used bioprobe with green fluorescence) via a cocondensation method. Then the uptake of sora@HMSNs and sora@Tf-HMSNs in thyroid cancer cells was identified by CLSM. After incubated with HMSNs or Tf-HMSNs for 24 hours, as evidenced from Figure 3A and B, the HMSNs (green fluorescence, fluorescein) were internalized in significant quantities. Furthermore, the cells treated with FITC-labeled Tf-HMSNs showed a higher fluorescence absorbance rate in the cytoplasm than HMSNs treated group and control (FITC only) group, thereby clearly illustrating the higher capacity of FITC-tagged Tf-HMSNs uptake into tumor cells. Without HMSNs mediating, the green-labeled particles dispersed irregularly in the surrounding area; only little green signal was detected intracellularly. As a result, TfR on thyroid cancer cells help the Tf-modified HMSNs recognition and binding, as a result increasing the intracellular uptake efficiency. Also, someone else probed whether the increased uptake of Tf-HMSNs was indeed a result of the overexpression of TfR on the cancer cell plasma membrane or not. After transfecting the Tf receptor gene with a TfR plasmid, previous study found that more Tf-HMSNs were accumulated in the transfected cells compared to that untransfected cells.46 These results indicated that the enhanced Tf-HMSNs uptake was related to cell TfR overexpression.
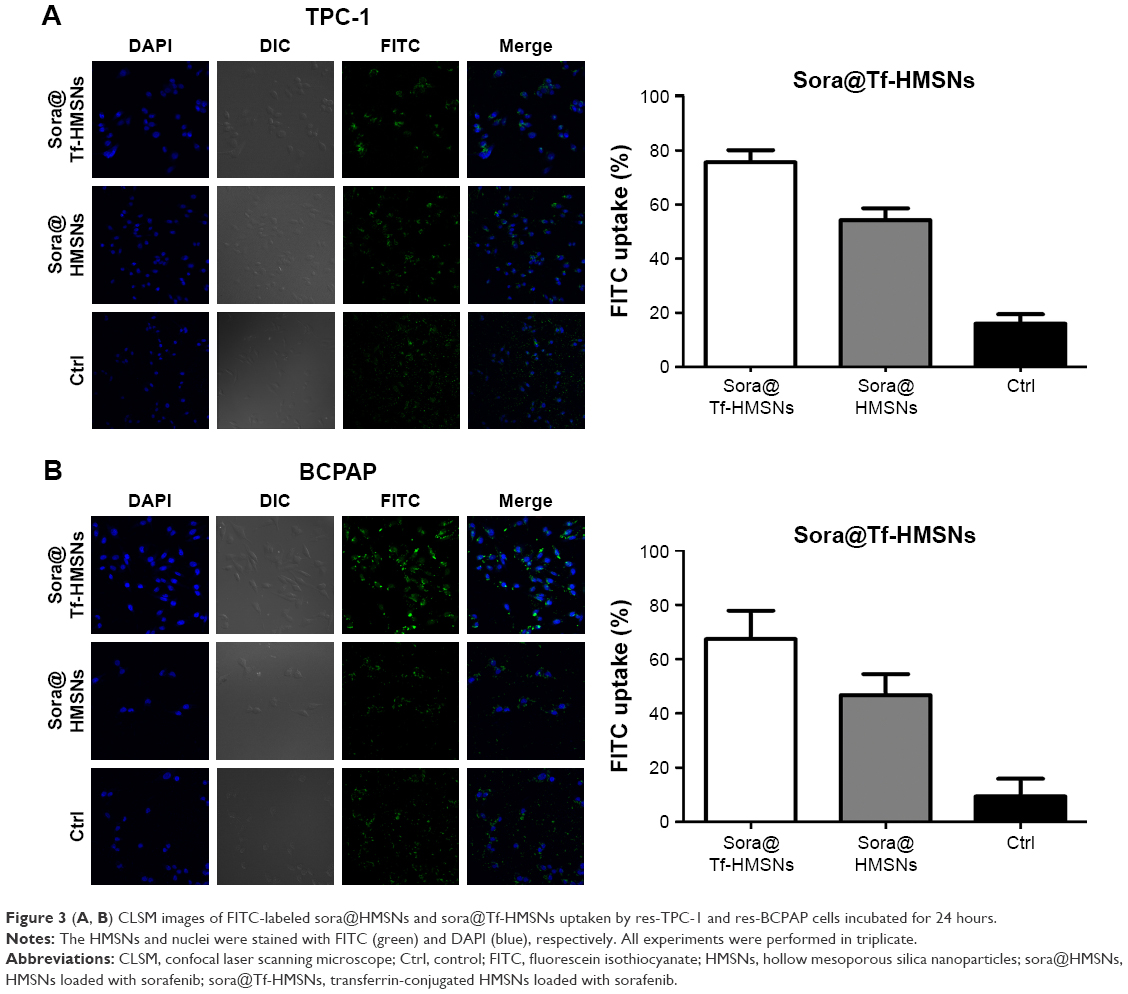 | Figure 3 (A, B) CLSM images of FITC-labeled sora@HMSNs and sora@Tf-HMSNs uptaken by res-TPC-1 and res-BCPAP cells incubated for 24 hours. |
The cytotoxicity assay
For drug delivery application, the prerequisite for a drug carrier system is minimal cytotoxicity. A previous report showed that HMSN was an ideal low-toxicity carrier even when the concentration achieved was as high as 400 μg/mL, which was more than ten times the therapeutic concentration used in our study.49 Here, we evaluated the cellular toxicity of HMSNs and Tf-HMSNs in res-TPC-1 cells and res-BCPAP cells treated with interval concentrations of HMSNs for 48 hours. As evidenced in Figure S4, the cell viability was >98% at a maximum particle concentration of 400 μg/mL after 24- and 48-hour treatment, which confirmed the good safety and biocompatibility of this carrier system.
Next, cytotoxicity of sora@HMSNs, sora@Tf-HMSNs, and free sorafenib on res-TPC-1 cells was further analyzed by CCK-8 assay. Cell viability of all groups showed a concentration-dependent pattern. There was a drastical increase in the cytotoxicity after sora@Tf-HMSNs treatment, which was in correlation to the enhanced nanoparticle intracellular uptake. At dose of 50 μg/mL sorafenib, sora@Tf-HMSNs leaded to 72.4%±3.8% of cell death compared with sora@HMSNs group (61.3%±3.7%) and (53.3%±3.2%) of free sorafenib group after 48-hour treatment. Similar results were exhibited on res-BCPAP cells after 48-hour treatment; cell cytotoxicity rate was 69.7%±2.4%, 58.4%±3.2%, and 40.5%±5.8% in sora@Tf-HMSNs, sora@HMSNs, and sorafenib groups treated with 50 μg/mL of sorafenib, respectively (Figure 4A and B). These results were in line with our hypothesis that Tf-HMSNs could enhance cellular uptake and delivered more drug to the thyroid cancer cells and thereby increased anticancer efficacy.
 | Figure 4 Sorafenib-loaded Tf-HMSNs (sora@Tf-HMSNs) increase cell cytotoxicity (A) res-TPC-1 cells and (B) res-BCPAP cells viability after incubation with different concentrations of sorafenib (sora), sorafenib-loaded HMSNs (sora@HMSNs), and Tf-HMSNs for 24 hours, respectively. |
Sora@Tf-HMSNs induce apoptosis through RAF/MEK/ERK pathway
The kinase inhibitors, which target the RAF/MEK/ERK pathway, have led to increasing studies in PCT cancer. As a multikinase inhibitor, sorafenib can alter signaling through the MAPK pathway50 and influence the tumor proliferation, tumorigenicity, and dedifferentiation processes, which lead to cell apoptosis eventually.51 It is practicable to investigate if the growth inhibition and apoptosis of thyroid cancer cells is indeed caused by the sorafenib-loaded Tf-HMSNs, and whether the modification of TfR on HMSNs can induce more cell death due to targeted delivery effect. We performed Annexin V-based flow cytometric analysis to verify the induction of apoptosis by loaded sorafenib. The sorafenib-loaded HMSNs or Tf-HMSNs were incubated with res-TPC-1 and res-BCPAP cells for 24 hours. In consistency with previous researches, sora@Tf-HMSNs treated group showed more increased apoptotic population than sora@HMSNs and control group (Figures 5 and S5). RAF-mitogen-activated protein kinase (MEK) regulated kinase (ERK) extracellular-signal usually occurs in thyroid cancer.52 Approximately 50% of PTC patients harbor activating BRAF gene mutations. As a result, MEK activates the ERK1 and ERK2 kinases.53 Abnormally increased activation of the MEK/ERK kinase pathway leads to cell proliferation inhibition, dedifferentiation, and improved cell survival ability. This pathway is important to tumor proliferation, and its frequent mutational activation in PTC has led to the research of target molecular inhibitors. In original, sorafenib was designed to inhibit the ARAF, BRAF, and RAF1 kinases, which could inhibit ERK activation in cancer cells and inhibit cancer proliferation.54 Radioiodine-refractory usually accompanied with iodine sensitive gene silencing, which is associated with MAPK over activating usually caused by the BRAF mutation.55 MAPK pathway inhibitor has already been made use on levels of clinical trials by kinds of BRAF and MEK inhibitors treatment.56 In clinical trials, MEK inhibitor, selumetinib, and BRAF inhibitor, dabrafenib, have been applied to induce redifferentiation and radioiodine sensitization in radio-resistant thyroid cancer as a result of MAPK pathway blockade.57 Next, we detected the RAF/MEK/ERK signaling pathway after sora@Tf-HMSNs treatment. Western blot analyses were performed to check the expression of these targets after drug-encapsulated HMSNs treatment. In our study, our sorafenib-encapsulated Tf-HMSNs treatment led to a remarkable reduction in the expression of RAF, pMEK, and pERK compared with HMSNs encapsulated and sorafenib-only treated group (Figure 6).
 | Figure 5 Annexin V/FITC staining indicating enhanced apoptosis and cell death by sora@Tf-HMSNs compared to sora@HMSNs and sorafenib group using flow cytometry in TPC-1 cell line. |
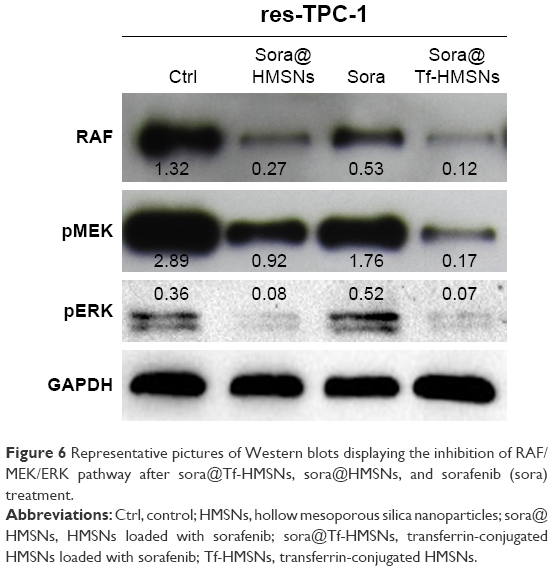 | Figure 6 Representative pictures of Western blots displaying the inhibition of RAF/MEK/ERK pathway after sora@Tf-HMSNs, sora@HMSNs, and sorafenib (sora) treatment. |
In vivo anticancer effect of sorafenib-loaded HMSNs
The HMSN system was applied in an in vivo model to study the therapeutic effect of the nanocarrier and its targeting effect. A total of 5×106 TPC-1 cells were injected into the right lateral abdominal wall subcutaneously of the SCID mice to establish human thyroid cancer xenografts. Mice bearing subcutaneous tumors were injected via the tail vein with DiR-labeled sora@Tf-HMSNs or sora@HMSNs. After 1, 6, and 24 hours, the mice were anesthetized. Then they were placed in the chamber of in vivo imaging system. As shown in Figure 7A, red fluorescence signal was captured in mice tumors treated with DiR-labeled sora@HMSNs or sora@Tf-HMSNs, indicating the nanoparticles accumulation in tumors. Compared with sora@HMSNs group, DiR-excited fluorescence signal was much more accumulated in xenografts tumor tissue in sora@Tf-HMSNs group, suggesting the effective delivery of the cargo inside the thyroid cancer cell. After euthanasia, the tumors and major organs were collected for analysis by the imaging system. The red fluorescence signal was observed from the tumors after sorafenib-loaded HMSNs treatment (Figure 7A). Also, the fluorescence intensity of major organs, including livers, spleens, kidneys, lungs, hearts, and cerebrums was analyzed. More accumulation of sorafenib-loaded HMSNs was identified in xenografts tumor tissue after Tf conjugation. The growth rate and weight of tumors in sorafenib-loaded Tf-HMSNs treated model were significantly reduced compared with sora@HMSNs treated group (Figure 7B). Consequently, the increased accumulation of drug in tumor tissue exhibited better tumor-killing effect and delayed thyroid cancer progression, which may prolong disease-free survival rate of refractory thyroid cancer patients.
 | Figure 7 Sora@Tf-HMSNs could enhance drug accumulation and inhibit tumor growth of xenograft in nude mice. |
Conclusion
We have successfully prepared a HMSN nanocarrier for the delivery of sorafenib for RAI-refractory thyroid cancer therapy. We have showed that Tf-HMSNs exhibit sustained drug release and can promote cellular uptake in vitro. The targeted HMSNs effectively suppressed the PTC cell proliferation and improved the cytotoxic efficacy than free drug or nontargeted group. The delivery sora@Tf-HMSNs enhanced by Tf modification as the targeting ligand, leading to the highest cytotoxicity. In vivo imaging system showed more accumulation of drug at the tumor focus, and it had a better antitumor effect after Tf modification.
Overall, these Tf-modified nanoparticles would be most ideal system to improve the therapeutic effect in RAI-refractory thyroid cancer. This delivery system holds great potential in DTC therapy with future nanomedicine technique. Further work is still needed to evaluate the anticancer efficacy of sora@Tf-HMSNs in vivo. Consequently, a more versatile cell-specific targeted drug delivery strategy can be anticipated in the near future.
Acknowledgment
This work was supported by National Natural Science Foundation of China (grant number: 81701727).
Disclosure
The authors report no conflicts of interest in this work.
References
Lorusso L, Pieruzzi L, Biagini A, et al. Lenvatinib and other tyrosine kinase inhibitors for the treatment of radioiodine refractory, advanced, and progressive thyroid cancer. Onco Targets Ther. 2016;9:6467–6477. | ||
Wartofsky L. Increasing world incidence of thyroid cancer: increased detection or higher radiation exposure? Hormones. 2010;9(2):103–108. | ||
American Thyroid Association Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer, Cooper DS, Doherty GM, Haugen BR, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167–1214. | ||
Shoup M, Stojadinovic A, Nissan A, et al. Prognostic indicators of outcomes in patients with distant metastases from differentiated thyroid carcinoma. J Am Coll Surg. 2003;197(2):191–197. | ||
Durante C, Haddy N, Baudin E, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91(8):2892–2899. | ||
Baudin E, Schlumberger M. New therapeutic approaches for metastatic thyroid carcinoma. Lancet Oncol. 2007;8(2):148–156. | ||
Schlumberger M, Sherman SI. Approach to the patient with advanced differentiated thyroid cancer. Eur J Endocrinol. 2012;166(1):5–11. | ||
Sherman SI. Targeted therapies for thyroid tumors. Mod Pathol. 2011;24(Suppl 2):S44–S52. | ||
Brose MS, Nutting CM, Jarzab B, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319–328. | ||
Prieto-Domínguez N, Ordóñez R, Fernández A, et al. Modulation of autophagy by sorafenib: effects on treatment response. Front Pharmacol. 2016;7:151. | ||
Mccubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–1284. | ||
Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer. 2005;12(2):245–262. | ||
Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159(3):676–690. | ||
Ranieri G, Gadaleta-Caldarola G, Goffredo V, et al. Sorafenib (BAY 43-9006) in hepatocellular carcinoma patients: from discovery to clinical development. Curr Med Chem. 2012;19(7):938–944. | ||
Zhang Z, Niu B, Chen J, et al. The use of lipid-coated nanodiamond to improve bioavailability and efficacy of sorafenib in resisting metastasis of gastric cancer. Biomaterials. 2014;35(15):4565–4572. | ||
Gadaleta-Caldarola G, Infusino S, Divella R, et al. Sorafenib: 10 years after the first pivotal trial. Future Oncol. 2015;11(13):1863–1880. | ||
Blair HA, Plosker GL. Sorafenib: a review of its use in patients with radioactive iodine-refractory, metastatic differentiated thyroid carcinoma. Target Oncol. 2015;10(1):171–178. | ||
Rosenholm JM, Peuhu E, Eriksson JE, Sahlgren C, Lindén M. Targeted intracellular delivery of hydrophobic agents using mesoporous hybrid silica nanoparticles as carrier systems. Nano Lett. 2009;9(9):3308–3311. | ||
Geng H, Zhao Y, Liu J, et al. Hollow mesoporous silica as a high drug loading carrier for regulation insoluble drug release. Int J Pharm. 2016;510(1):184–194. | ||
Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015;200:138–157. | ||
Hu Y, Wang J, Zhi Z, Jiang T, Wang S. Facile synthesis of 3D cubic mesoporous silica microspheres with a controllable pore size and their application for improved delivery of a water-insoluble drug. J Colloid Interface Sci. 2011;363(1):410–417. | ||
Lin YS, Haynes CL. Impacts of mesoporous silica nanoparticle size, pore ordering, and pore integrity on hemolytic activity. J Am Chem Soc. 2010;132(13):4834–4842. | ||
Nadrah P, Porta F, Planinšek O, Kros A, Gaberšček M. Poly(propylene imine) dendrimer caps on mesoporous silica nanoparticles for redox-responsive release: smaller is better. Phys Chem Chem Phys. 2013;15(26):10740–10748. | ||
Singh N, Karambelkar A, Gu L, et al. Bioresponsive mesoporous silica nanoparticles for triggered drug release. J Am Chem Soc. 2011;133(49):19582–19585. | ||
Souris JS, Lee CH, Cheng SH, et al. Surface charge-mediated rapid hepatobiliary excretion of mesoporous silica nanoparticles. Biomaterials. 2010;31(21):5564–5574. | ||
Lu J, Liong M, Li Z, Zink JI, Tamanoi F. Biocompatibility, biodistribution, and drug-delivery efficiency of mesoporous silica nanoparticles for cancer therapy in animals. Small. 2010;6(16):1794–1805. | ||
Li L, Guan Y, Liu H, et al. Silica nanorattle-doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS Nano. 2011;5(9):7462–7470. | ||
Patel K, Sundara Raj B, Chen Y, Lou X. Cytotoxicity of folic acid conjugated hollow silica nanoparticles toward Caco2 and 3T3 cells, with and without encapsulated DOX. Colloids Surf B Biointerf. 2016;140:213–222. | ||
Liu Y, Chen Q, Xu M, et al. Single peptide ligand-functionalized uniform hollow mesoporous silica nanoparticles achieving dual-targeting drug delivery to tumor cells and angiogenic blood vessel cells. Int J Nanomed. 2015;10:1855–1867. | ||
Cheng YJ, Luo GF, Zhu JY, et al. Enzyme-induced and tumor-targeted drug delivery system based on multifunctional mesoporous silica nanoparticles. ACS Appl Mater Interf. 2015;7(17):9078–9087. | ||
Liu S, Zhang HL, Liu TC, et al. Optimization of the methods for introduction of amine groups onto the silica nanoparticle surface. J Biomed Mater Res A. 2007;80(3):752–757. | ||
Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54(4):561–587. | ||
Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121(2):144–158. | ||
Luck AN, Mason AB. Structure and dynamics of drug carriers and their interaction with cellular receptors: focus on serum transferrin. Adv Drug Deliv Rev. 2013;65(8):1012–1019. | ||
Chitambar CR, Matthaeus WG, Antholine WE, Graff K, O’Brien WJ. Inhibition of leukemic HL60 cell growth by transferrin-gallium: effects on ribonucleotide reductase and demonstration of drug synergy with hydroxyurea. Blood. 1988;72(6):1930–1936. | ||
Deshpande P, Jhaveri A, Pattni B, Biswas S, Torchilin V. Transferrin and octaarginine modified dual-functional liposomes with improved cancer cell targeting and enhanced intracellular delivery for the treatment of ovarian cancer. Drug Deliv. 2018;25(1):517–532. | ||
van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, van Gaal EV. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65(10):1284–1298. | ||
Muddineti OS, Kumari P, Ghosh B, Biswas S. Transferrin-modified vitamin-E/lipid based polymeric micelles for improved tumor targeting and anticancer effect of curcumin. Pharm Res. 2018;35(5):97. | ||
Lu L, Yuan L, Yan J, Tang C, Wang Q. Development of core-shell nanostructures by in situ assembly of pyridine-grafted diblock copolymer and transferrin for drug delivery applications. Biomacromolecules. 2016;17(7):2321–2328. | ||
Magro G, Cataldo I, Amico P, et al. Aberrant expression of TfR1/CD71 in thyroid carcinomas identifies a novel potential diagnostic marker and therapeutic target. Thyroid. 2011;21(3):267–277. | ||
Parenti R, Salvatorelli L, Magro G. Anaplastic thyroid carcinoma: current treatments and potential new therapeutic options with emphasis on TfR1/CD71. Int J Endocrinol. 2014;2014:1–11. | ||
Green DL, Lin JS, Lam YF, Hu MZ, Schaefer DW, Harris MT. Size, volume fraction, and nucleation of Stober silica nanoparticles. J Colloid Interface Sci. 2003;266(2):346–358. | ||
Kruger NJ. The Bradford method for protein quantitation. Methods Mol Biol. 1994;32:9–15. | ||
Zhang J, Yuan ZF, Wang Y, et al. Multifunctional envelope-type mesoporous silica nanoparticles for tumor-triggered targeting drug delivery. J Am Chem Soc. 2013;135(13):5068–5073. | ||
Cui Y, Xu Q, Chow PK, Wang D, Wang CH. Transferrin-conjugated magnetic silica PLGA nanoparticles loaded with doxorubicin and paclitaxel for brain glioma treatment. Biomaterials. 2013;34(33):8511–8520. | ||
Ferris DP, Lu J, Gothard C, et al. Synthesis of biomolecule-modified mesoporous silica nanoparticles for targeted hydrophobic drug delivery to cancer cells. Small. 2011;7(13):1816–1826. | ||
Xiang C, Zhang ML, Zhao QZ, et al. LncRNA-SLC6A9-5:2: a potent sensitizer in 131I-resistant papillary thyroid carcinoma with PARP-1 induction. Oncotarget. 2017;8(14):22954–22967. | ||
Kuang Y, An S, Guo Y, et al. T7 peptide-functionalized nanoparticles utilizing RNA interference for glioma dual targeting. Int J Pharm. 2013;454(1):11–20. | ||
Qiu Y, Wu C, Jiang J, et al. Lipid-coated hollow mesoporous silica nanospheres for co-delivery of doxorubicin and paclitaxel: preparation, sustained release, cellular uptake and pharmacokinetics. Mater Sci Eng C Mater Biol Appl. 2017;71:835–843. | ||
Bernet V, Smallridge R. New therapeutic options for advanced forms of thyroid cancer. Expert Opin Emerg Drugs. 2014;19(2):225–241. | ||
Liu D, Liu Z, Jiang D, Dackiw AP, Xing M. Inhibitory effects of the mitogen-activated protein kinase kinase inhibitor CI-1040 on the proliferation and tumor growth of thyroid cancer cells with BRAF or RAS mutations. J Clin Endocrinol Metab. 2007;92(12):4686–4695. | ||
Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13(3):184–199. | ||
Broecker-Preuss M, Müller S, Britten M, et al. Sorafenib inhibits intracellular signaling pathways and induces cell cycle arrest and cell death in thyroid carcinoma cells irrespective of histological origin or BRAF mutational status. BMC Cancer. 2015;15:184. | ||
Mandal R, Becker S, Strebhardt K. Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: an emerging threat to anticancer therapy. Oncogene. 2016;35(20):2547–2561. | ||
Bible KC, Ryder M. Evolving molecularly targeted therapies for advanced-stage thyroid cancers. Nat Rev Clin Oncol. 2016;13(7):403–416. | ||
Spitzweg C, Bible KC, Hofbauer LC, Morris JC. Advanced radioiodine-refractory differentiated thyroid cancer: the sodium iodide symporter and other emerging therapeutic targets. Lancet Diabetes Endocrinol. 2014;2(10):830–842. | ||
Ho AL, Grewal RK, Leboeuf R, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368(7):623–632. |
Supplementary materials
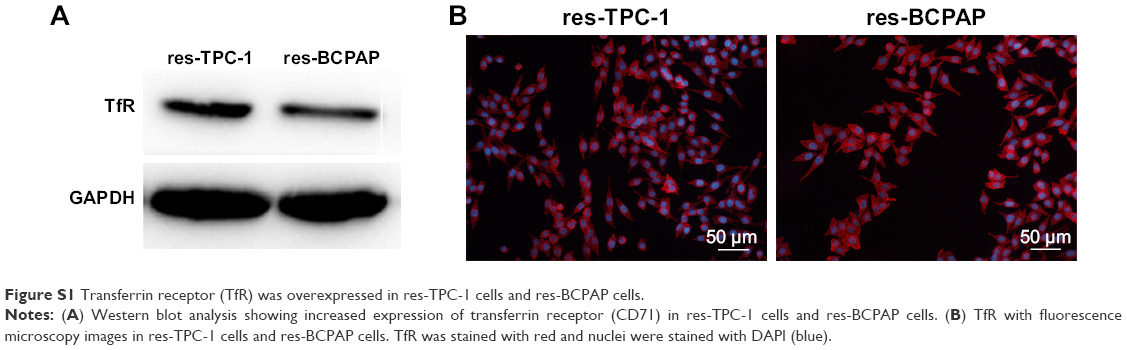 | Figure S1 Transferrin receptor (TfR) was overexpressed in res-TPC-1 cells and res-BCPAP cells. |
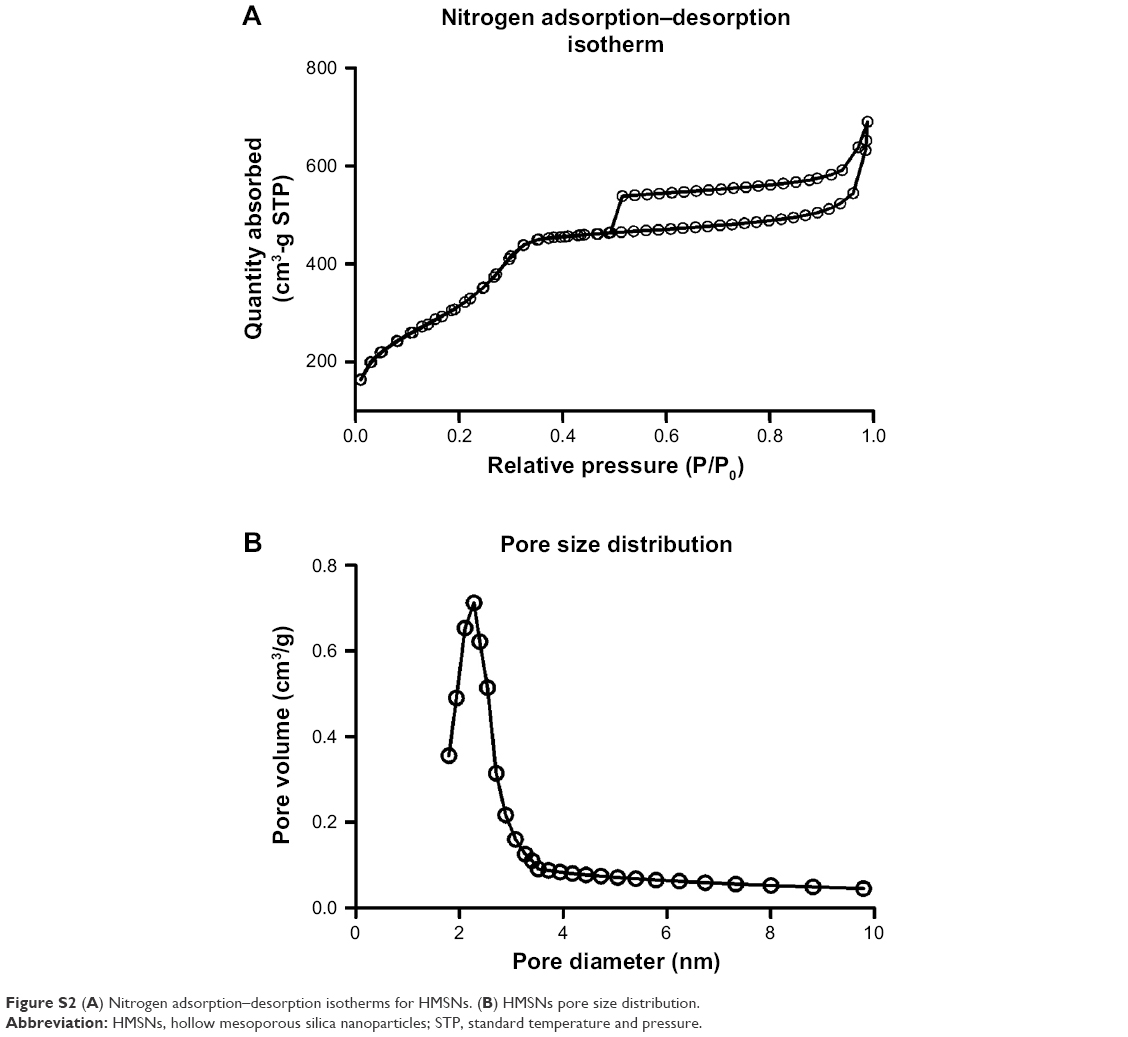 | Figure S2 (A) Nitrogen adsorption–desorption isotherms for HMSNs. (B) HMSNs pore size distribution. |
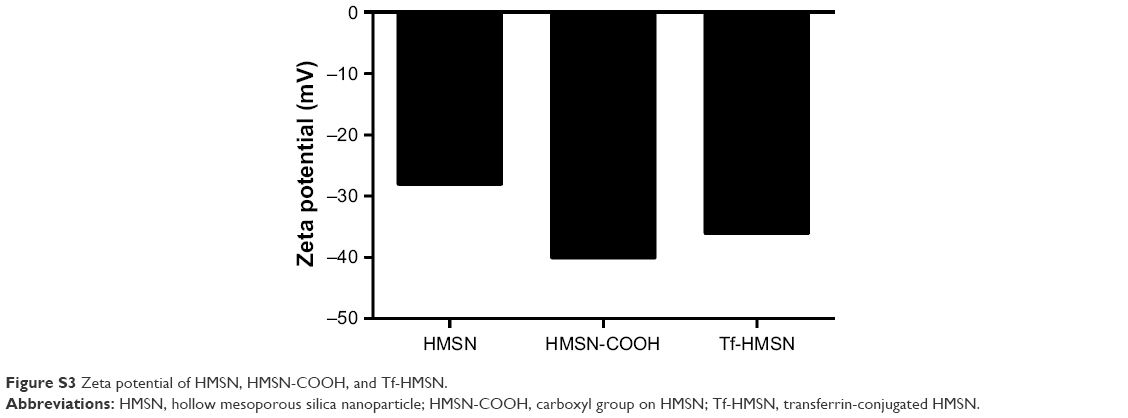 | Figure S3 Zeta potential of HMSN, HMSN-COOH, and Tf-HMSN. |
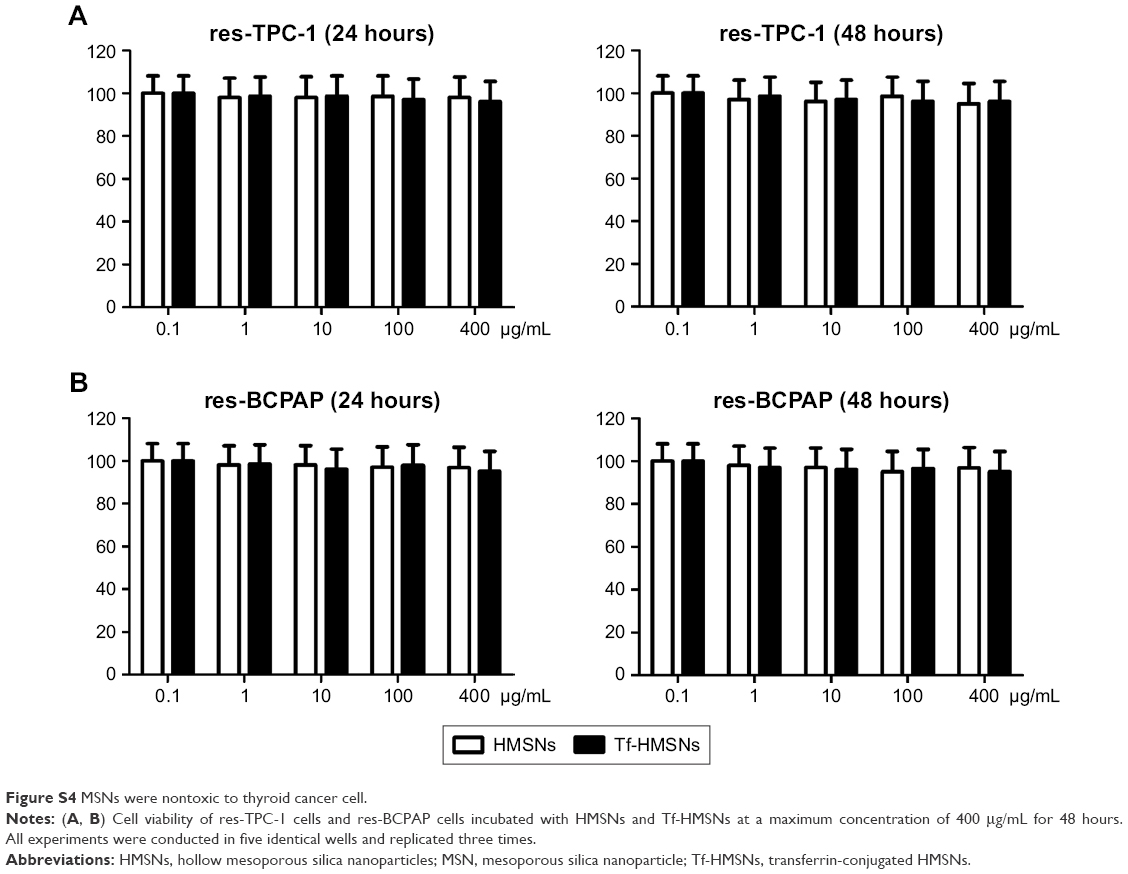 | Figure S4 MSNs were nontoxic to thyroid cancer cell. |
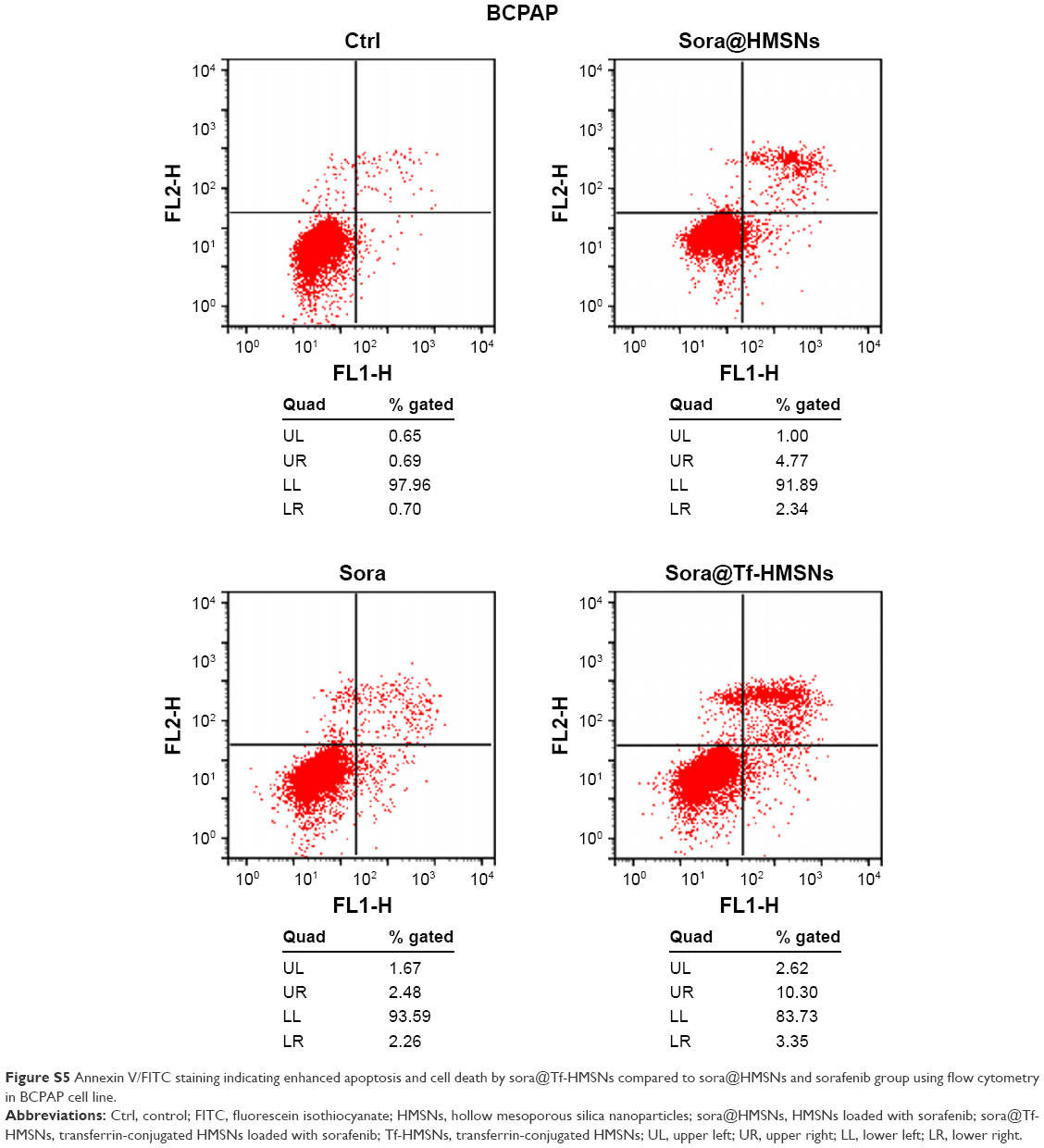 | Figure S5 Annexin V/FITC staining indicating enhanced apoptosis and cell death by sora@Tf-HMSNs compared to sora@HMSNs and sorafenib group using flow cytometry in BCPAP cell line. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.