Back to Journals » International Journal of Nanomedicine » Volume 12
Transcriptome-wide elucidation of liposomal formulations for anticancer drug delivery
Authors Li Y, Wang M, Huang B, Ping Y, You J, Gao J
Received 12 August 2017
Accepted for publication 14 October 2017
Published 29 November 2017 Volume 2017:12 Pages 8557—8572
DOI https://doi.org/10.2147/IJN.S148975
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Ying Li,1,2 Meng Wang,1,2 Bu-Wei Huang,1,2 Yuan Ping,3 Jian You,1 Jian-Qing Gao1,2
1Institute of Pharmaceutics, College of Pharmaceutical Sciences, 2Zhejiang Province Key Laboratory of Anticancer Drug Research, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, People’s Republic of China; 3School of Materials Science and Engineering, Nanyang Technological University, Singapore
Abstract: Although widely used in chemotherapy, free doxorubicin (Dox) might enhance cell malignancy undesirably. Liposomal Dox (Doxlipo) has been clinically approved for the treatment of breast cancer due to reduced systematical toxicity and increased tumor targeting, yet the transcriptome-wide elucidation of the Doxlipo formulations remains elusive. To this end, we explored the impact of two Dox liposomal formulations, Doxlipo mainly containing hydrogenated soy phosphatidylcholine or 1,2-dipalmitoyl-sn-glycero-3-phosphocholine, on the transcriptional pattern of MCF-7 cells. The two types of Dox liposomal formulations with different drug release kinetics were investigated to reveal the relationship between the formulation and tumor malignancy. Interestingly, we found that liposomal formulation significantly altered the transcriptional pattern of a wide range of genes. Under equivalent dosage of Dox, free Dox substantially changed the expression of ANK1, ACTA2, GPR87, GDF15, FZD6, and WNT4 in MCF-7 cells. Notably, free Dox induced much higher expression of ABCB1 and significantly enhanced the cell migration behavior in comparison with HSPC Doxlipo under a similar level of cytotoxicity. Finally, siRNA targeting GPR87 was codelivered with cationic Doxlipo to reduce the expression of malignancy-related genes. Our study, for the first time, provides an overview of the influence of formulation on the malignancy at transcriptional level and reveals the relationship between cytotoxicity and cell malignancy from the formulation aspect, offering valuable reference for the future formulation design for anticancer drug delivery.
Keywords: liposomes, doxorubicin, cellular uptake, cell malignancy, transcriptional profiling
Introduction
Recurrence and metastasis of tumors represent one of the major challenges after the conventional cancer chemotherapy, and an increasing number of recent studies indicate mutations and phenotypic variations of tumor cells, which derive from the selective pressure of chemotherapy, may develop the treatment-resistant clones that are believed to be the key source for such a relapse.1 Whereas many first-line chemotherapeutic drugs are demonstrated to be effective in inhibiting tumor growth, they may otherwise enhance tumor malignancy.2 These unexpected adverse effects are usually accompanied with increasing drug resistance and cell metastasis, which accounts for the main reason of failures in chemotherapies. Recent studies have indicated there is a close link between epithelial–mesenchymal transition (EMT) and chemotherapy resistance.3 As well documented, EMT is a complex process involving morphologic alteration and genetic mutations, and it is closely associated with invasion, metastasis, and even drug resistance.4,5 Tumor cells that undergo EMT usually have more aggressive phenotypes and exhibit higher degree of malignancy. Several EMT-related transcription factors or signal pathways are reported to play important roles in inducing cell metastasis and drug resistance.6,7 Unfortunately, EMT can be activated by chemotherapeutic drugs. Doxorubicin (Dox), as a first-line drug for breast cancer therapy, upregulates several EMT-related genes, including HIF1α, Twist1, Snail, and ZEB1 that can promote cancer cells to acquire more aggressive phenotypes. However, the detailed underlying mechanisms are still elusive.
As a cytotoxic drug, Dox also causes several side effects including cardiotoxicity. To this end, functional biomaterials that are exploited for loading and delivering chemotherapeutics have been widely explored to alleviate the side effects of Dox and to improve the tumor-targeting ability of free Dox. For example, liposome, one of the most explored drug delivery carriers in pharmaceutical industry, is used for encapsulating Dox, which exhibits reduced cardiotoxicity, increased stability, and better therapeutic efficiency.8 Commercially available doxorubicin, such as liposomal formulations (Doxil®; Janssen Products, Titusville, NJ, USA), has already been clinically approved.9 Recently, a thermosensitive doxorubicin liposomal formulation (ThermoDox®; Celsion Corporation, Lawrenceville, NJ, USA) is under clinical trial.10 To improve the blood circulation time, polyethylene glycol (PEG)-modified liposomes are often studied as well to augment drug accumulation at tumor tissues through enhanced permeability and retention effect, thereby minimizing the side effects. Although liposomes improve the safety and stability of chemotherapeutic drugs, their potential roles and relationships with cancer malignancy have rarely been investigated. As is well known, free Dox is internalized by cells through passive diffusion in the cell culture, which can lead to fast intracellular accumulation and instant cytotoxicity.11 Compared with free Dox, liposomal formulations usually enter cells via clathrin-mediated endocytosis,12,13 and therefore exhibit delayed intracellular accumulation. Once inside the cells, liposomes should release loaded Dox, in a fast or sustained release rate. We assume that these sharp contrasts between free drug and drug-loaded liposomal formulations might impact the cell malignancy, thus affecting tumor recurrence and metastasis.
Transcriptional profiling is an efficient approach to investigate the global gene expression pattern of cellular responses to drugs. In collaboration with microarray technology and bioinformatic tools, researchers are now fully capable of conducting high-throughput screening of genes or signal pathways. Transcriptional profiling has been applied to investigate the mechanism of Dox cytotoxicity on various cancer cell models, such as breast cancer,14 neuroblastoma,15 and hepatoma.16 The studies revealed the important function of P53-regulated genes in Dox-induced cell inhibition by regulating cell cycle-associated genes, which help elucidate the anticancer mechanism of Dox.
To further elucidate the potential relationship between liposomal formulation and tumor malignancy, we explored the transcriptional profile of MCF-7 cells treated with two types of Dox liposomal formulations exhibiting different drug release kinetics, which might affect cell tumor malignancy through cellular uptake and intracellular drug concentration. A breast cancer cell line, MCF-7, was used as a model to study the cellular uptake pattern and cytotoxicity of liposomal formulations, since MCF-7 represents the most frequently used luminal subtype with wild-type P53 protein. The global transcriptomics impacted by different liposomal formulations were examined using microarray assays, and the screened genes were further validated by quantitative real-time-PCR assay. Finally, siRNA targeting GPR87 was codelivered with Dox to check whether the inhibition of GPR87 expression can alleviate the cell malignancy induced by liposomal Dox formulations or free Dox.
Materials and methods
Reagents
Hydrogenated soy phosphatidylcholine (HSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DPSE-PEG2000) were purchased from Lipoid (Ludwigshafen, Germany). 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) were purchased from Advanced Vehicle Technology Pharmaceutical Co., LTD (Shanghai, People’s Republic of China). Cholesterol (Chol), amiloride hydrochloride hydrate, and 3-(4,5-dimethylthi-zaol-2-yl)-2,5-diphenyltetrazolum bromide (MTT) were purchased from Sigma-Aldrich (Shanghai, People’s Republic of China). Doxorubicin was obtained from Zhejiang Hisun Pharmaceutical Co. Ltd (Taizhou, People’s Republic of China). Dulbecco’s Modified Eagle’s Medium, penicillin, and streptomycin were purchased from Jinuo Biomedical Technology (Hangzhou, People’s Republic of China). Fetal bovine serum was purchased from Sijiqing Biologic Co., Ltd (Hangzhou, People’s Republic of China). Human insulin recombinant was purchased from Wisent Inc. (Saint-Jean-Baptiste, Canada). LysoTracker® Green, LysoTracker® Blue, and Hochest33342 were purchased from Life Technology (Carlsbad, CA, USA). Chlorpromazine hydrochloride (CPZ) and methyl-β-cyclodextrin (MβCD) were obtained from Aladdin (Shanghai, People’s Republic of China). RNAiso Plus and PrimeScript™ RT Master Mix were purchased from Takara (Tokyo, Japan). GoTaq® qPCR Master Mix was purchased from Promega (Madison, WI, USA).
Characterization of liposomes
Liposomes were prepared using the thin-film method. Lipid formulations are listed in Table 1. The liposomes with slow drug release mainly composed of HSPC are referred to as HSPC Doxlipo, and the thermosensitive liposomes containing DPPC are referred to as DPPC Doxlipo.
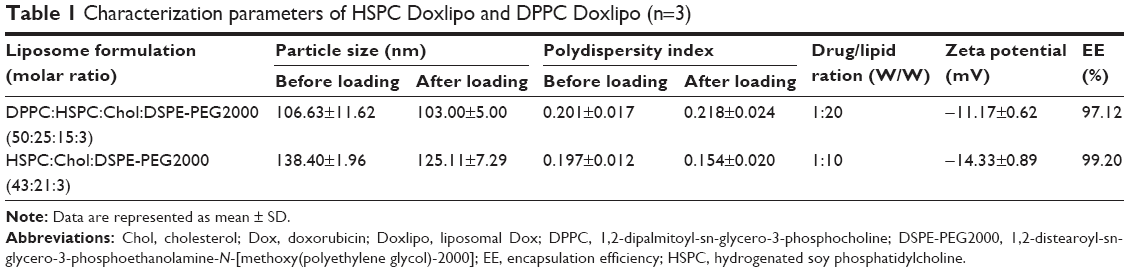 | Table 1 Characterization parameters of HSPC Doxlipo and DPPC Doxlipo (n=3) |
Preparation of thermosensitive and insensitive liposomes
All lipids were dissolved in the mixture of chloroform: methanol (3:1, v:v) and then evaporated under negative pressure at 60°C. The resulting lipid film was hydrated with ammonium sulfate (120 mM) by sonication in the water bath for 20 min, followed by probe-type sonication (200 w) for 3–5 min. The suspension was extruded successively through a 0.22 μm polycarbonate membrane (three times). External ammonium sulfate was replaced by dialysis against phosphate-buffered saline (PBS) (pH 7.4). Dox solution was added to the liposomes and incubated for 1 h at 55°C. Finally, the unencapsulated Dox was removed by dialysis against PBS.
Characterization of the Dox liposomes
The liposome particle size, zeta potential, and polydispersity were measured by dynamic light scattering analysis using a zetasizer (Nano ZS90; Malvern, Worcestershire, UK). The morphological examination of liposomes was performed via transmission electron microscope (JEM-1200EX; JEOL, Tokyo, Japan) using 2% uranyl acetate for negative staining.
The encapsulation efficiency (EE) was determined by high-speed centrifuge (Optima MAX-XP; Beckman Coulter, Inc., Indianapolis, IN, USA) at 4°C and 150,000× g for 3 h. The amount of Dox in the supernatant after centrifugation was designated as Wfree, and Dox weight in the same amount of uncentrifuged liposomes was designated as Wtotal. The Dox concentration was measured in a fluorescence spectrophotometer (Cary Eclipse; Agilent, Santa Clara, CA, USA) at Ex =488 nm, Em =590 nm. EE was calculated as the following formula:
|
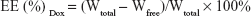
In vitro drug release
In vitro release of Dox from liposomes was investigated in PBS (pH 7.4) at both 37°C and 42°C. Briefly, 1 mL of liposome was placed in the dialysis bag (molecular weight cutoff 8,000–14,000) and immersed into 40 mL of the dialysis solution under continuous stirring at 100 rpm. At a determined time interval, 1 mL of samples were withdrawn and replaced with fresh solution. The cumulative release percentage (%) was calculated as WCumulative Dox (in dialysis medium)/WTotal Dox.
Cell culture
Human breast adenocarcinoma cell line (MCF-7) from American Type Culture Collection was kindly provided by the Center for Molecular Medicine of Zhejiang Academy of Medical Sciences. The cells were maintained in high-glucose Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 10 μg/mL insulin, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were cultured in incubators maintained at 37°C with 5% CO2 under fully humidified conditions.
In vitro cellular accumulation of Dox
Cellular Dox fluorescence intensity was determined by flow cytometry (BD FACSCalibur BD Biosciences, San Jose, CA, USA or NovoCyte, ACEA Biosciences, San Diego, CA, USA). The cells were incubated with different Dox formulations (5 μg/mL). Totally 10,000 events were collected and analyzed with Flowjo software (Ashland, OR, USA). For preheated treatment, the diluted DPPC Doxlipo was exposed to 42°C water bath for 15 min before being added into cells. The same treatments were also performed in the following experiments unless specified.
Endocytosis inhibition
To explore cellular uptake pathways, CPZ (10 μg/mL), MβCD (5 mM), and amiloride (50 μM) were pretreated for 1 h. Dox-containing medium was added and incubated for 4 h at 37°C, and then Dox fluorescence intensity was determined by flow cytometry. Cells without inhibitor treatment were regarded as control.
In vitro cellular retention of Dox
The cells were incubated with different Dox formulations (5 μg/mL); then the drug-containing medium was removed and the cells were washed with PBS. Finally fresh medium was added. Live cells were harvested after 6, 24, and 72 h. Cells without Dox treatment were assigned as blank, while cells treated with Dox formulations for the same period but not removed were assigned as control group. Cellular Dox was also measured by flow cytometry. In order to get a similar initial intracellular Dox accumulation, different Dox liposomes were incubated for a determined time (free Dox, 30 min; HSPC Doxlipo, 24 h; DPPC Doxlipo, 4 h).
In vitro cellular distribution of Dox
The cells were grown on 35 mm glass-bottom culture dishes (Nest) and coincubated with Dox formulations (5 μg/mL). 100 nM LysoTracker probe was added for 30 min, followed by nuclear staining with Hochest33342 for 20 min. The cells were washed with PBS and observed under a confocal laser scanning microscope (IX81-FV1000; Olympus, Tokyo, Japan).
In vitro cytotoxicity
In vitro cytotoxicity was measured by the MTT assay. Briefly, the cells were seeded into 96-well culture plates at 5,000/well and treated with Dox formulations. MTT (0.5 mg/mL) was added for 4 h incubation at 37°C, and the absorbance was measured using a microplate reader (Bio-Tek Instruments, Winooski, VT, USA).
Microarray analysis and quantitative RT-PCR (qRT-PCR)
Cells were treated with different Dox formulations (0.5 μg/mL) for 24 h, and total RNA was extracted by RNAiso Plus. The quality of RNA was verified by Nanodrop2000 (Thermo Fisher Scientific, Waltham, MA, USA). Microarray experiment was performed with Affymetrix Primeview Human Gene Expression Array (Affymetrix, Santa Clara, CA, USA) at Shanghai Biotechnology Corporation.
Primers for qRT-PCR was synthesized by Sangon Biotech Co., Ltd (Shanghai, People’s Republic of China) and are listed in Table S1. cDNA was converted from 0.5 μg of RNA using PrimeScript™ RT Master Mix. Then qRT-PCR was performed with GoTaq® qPCR Master Mix on the ABI7500 Fast (ABI, USA). The expression levels were normalized against the internal reference gene GAPDH and calculated using the 2−ΔΔCt method.
Cell migration
To monitor the cell migration irritated by Dox formulations, a continuous monitoring xCELLigence System was used. The cells were seeded into CIM 16 Well Device (3×105/well), and cell migration was measured by the Real-Time Cell Analyzer station (xCELLigence RTCA DP; ACEA Biosciences, Berlin, Germany). Cell migration was detected by the changes in the electrical impedance at the electrode/cell interface, when the cells moved through the microporous polyethylene terephthalate membrane of the wells. A total period of 24 h was recorded, and signal was collected every 15 min.
Preparation and characterization of cationic Dox liposomes
Based on the result of microarray analysis and qRT-PCR, a significantly upregulated gene, GPR87 was selected for codelivery with Dox using cationic liposomes. The preparation of cationic Dox liposomes was similar to the method described earlier; only the cationic lipid DOTAP was replaced in the formulation as listed in Table 2 (referred to as DOTAP Doxlipo). siRNA targeting GPR87 (siGPR87) (sense strand: 5′-GCAUCUUGCUGAAUGGUUUdTdT-3′; antisense strand: 5′-AAACCAUUCAGCAAGAUGCdTdT-3′) was synthesized by Ribobio Co., Ltd (Guangzhou, People’s Republic of China). Negative control siRNA (siNC) and FAM-labeled siNC were also obtained from Ribobio Co., Ltd. DOTAP Doxlipo was mixed with siRNA solution in serum-free medium for 20–30 min before adding into the cells.
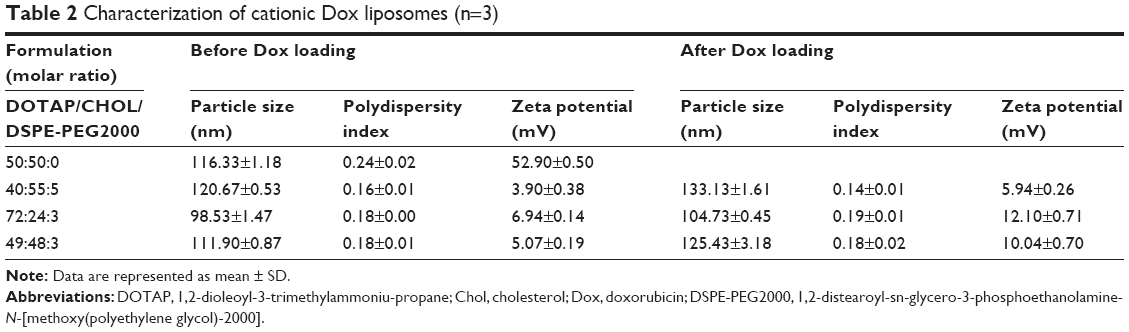 | Table 2 Characterization of cationic Dox liposomes (n=3) |
Statistical analysis
All data are represented as mean ± standard error of the mean of at least three repeats. Statistical analysis was performed using SPSS17.0 software. The statistical difference was determined by one-way analysis of variance with Tukey’s test and a P-value of <0.05.
Results
Characterization of the liposomes
The DPPC Doxlipo had a hydrodynamic diameter of ~103 nm, while the HSPC Doxlipo had a larger size of ~125 nm, and both liposomes showed similar negative zeta potential (Table 1). Dox loading did not significantly change the size and polydispersity index (PDI) of the liposomes. Both liposomal formulations showed high EE (>95%) (Table 1). Transmission electron microscopy image showed that the liposomes were generally spherical, and the bilayer of phospholipid can be observed (Figure 1A and B).
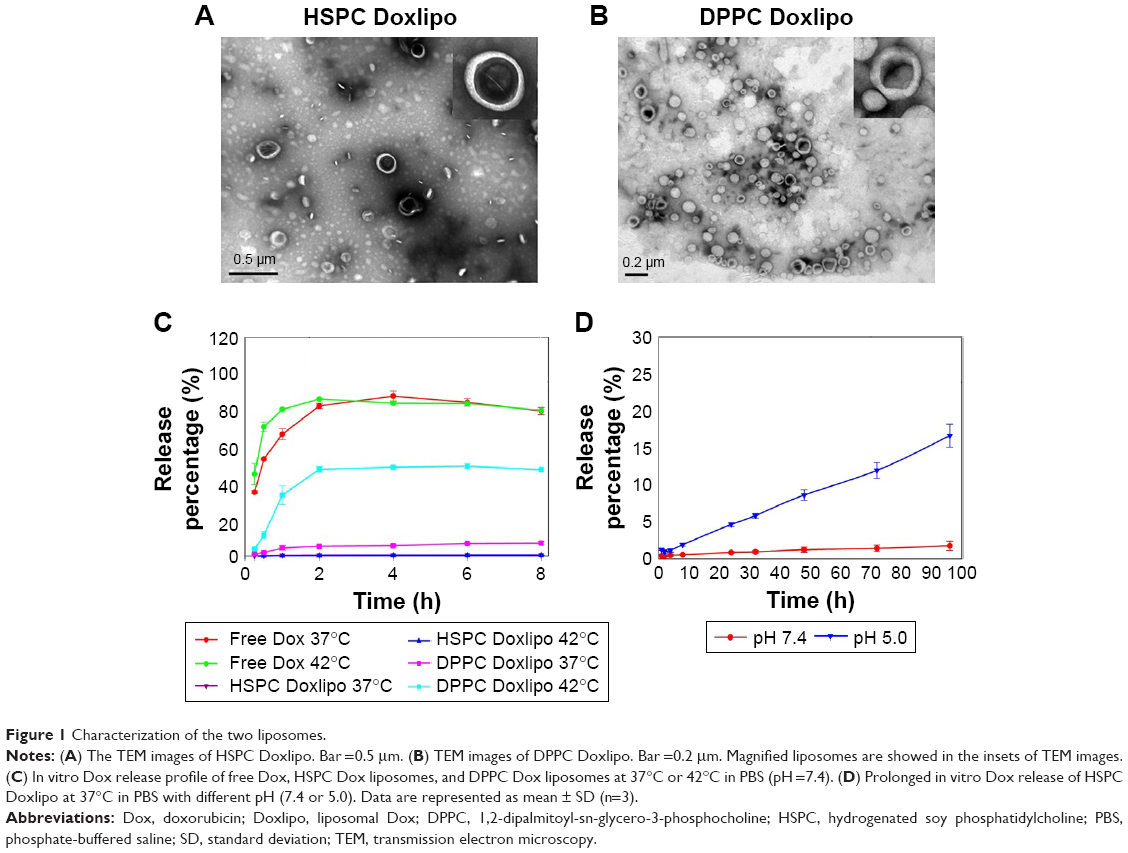 | Figure 1 Characterization of the two liposomes. |
In vitro Dox release experiment showed that the DPPC Doxlipo had a thermosensitive drug release profile, which released almost 50% Dox within 2 h under 42°C, while only 5% Dox was released under 37°C. On the other hand, Dox release from HSPC Doxlipo was minimal and unaffected by mild hyperthermia (Figure 1C). Furthermore, a longer period of Dox release (96 h) was conducted on HSPC Doxlipo in physiological pH (7.4) and acid pH (5.0), which showed that acid environment facilitated Dox release from HSPC Doxlipo (Figure 1D).
In vitro cellular Dox uptake and retention
Next, cellular Dox accumulation of the different Dox formulations was evaluated. All treatments showed time-dependent Dox accumulation (Figure 2A). Free Dox showed the highest cellular uptake, and HSPC Doxlipo showed the lowest Dox uptake. DPPC Doxlipo without preheating showed relatively higher Dox uptake than HSPC Doxlipo, but preheating enhanced Dox uptake significantly, especially at the beginning stage (2–6 h).
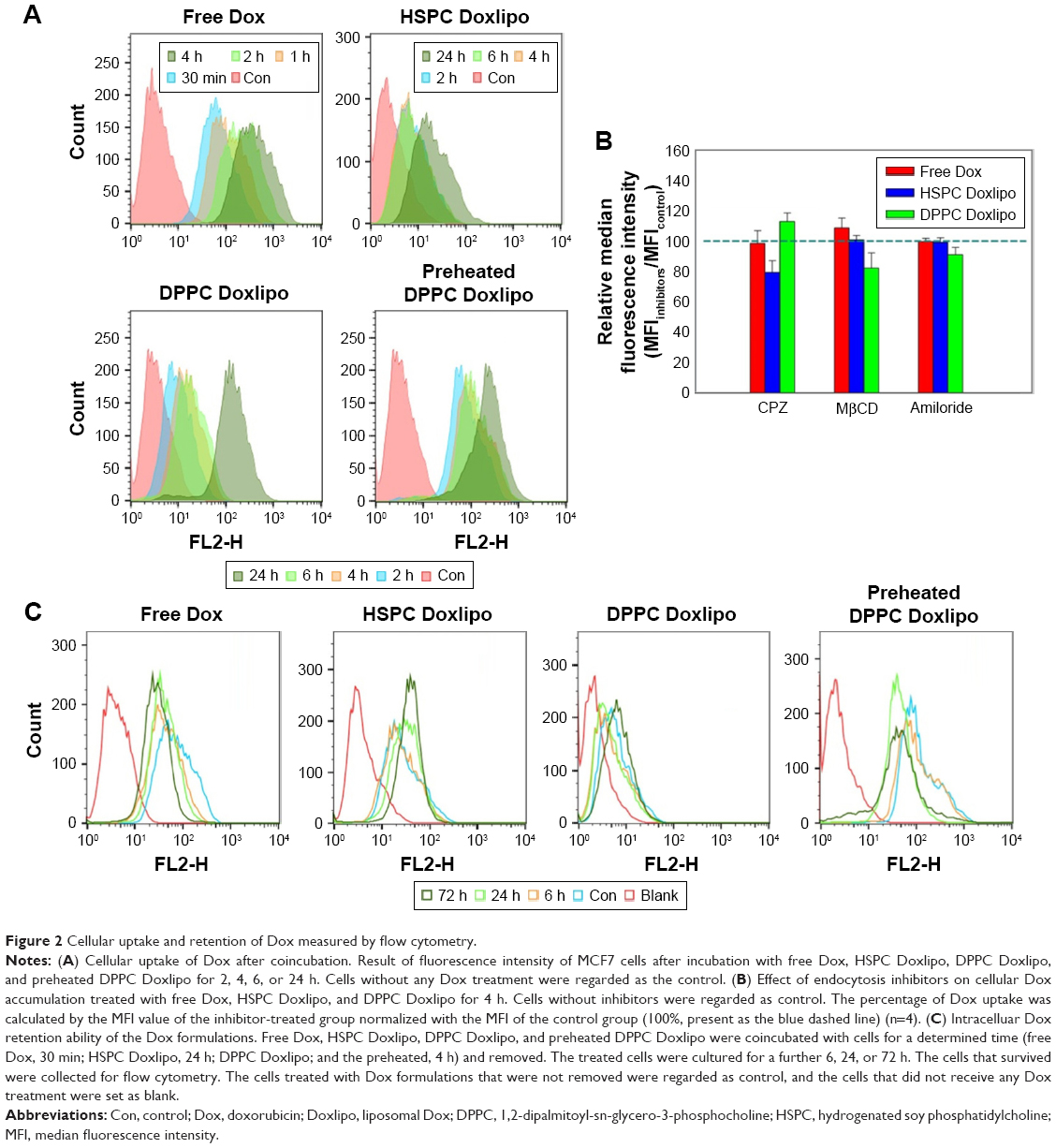 | Figure 2 Cellular uptake and retention of Dox measured by flow cytometry. |
Most liposomes enter into the cells through endocytosis; therefore, different endocytic inhibitors were employed for the mechanism study (Figure 2B). CPZ (clathrin-mediated endocytosis inhibitor) decreased the cellular uptake of HSPC Doxlipo by 21.6%, while MβCD (caveolae-mediated endocytosis inhibitor) and amiloride (macropinocytosis inhibitor) did not show a significant inhibitory effect for all treatments. The result showed that clathrin-mediated endocytosis was the main way for cells to uptake HSPC Doxlipo.
Although Dox liposomes showed slower Dox uptake, they had longer intracellular retention than free Dox. When Dox formulations were removed after incubation with cells for determined periods, free Dox showed obvious decreasing median fluorescence intensity (MFI) in a time-dependent manner, whereas HSPC Doxlipo showed increasing MFI even after the drug retreated. No significant change in MFI was observed in DPPC Doxlipo group after re-treating, but obvious decreasing MFI value was seen in preheated DPPC Doxlipo, similar as free Dox group (Figure 2C).
Intracellular Dox distribution
Distribution of Dox formulations was examined by confocal laser scanning microscopy with costaining of lysosomes and nucleus. After 4 h incubation, free Dox showed the highest Dox accumulation in the nucleus, and preheated DPPC Doxlipo showed relatively weaker staining, while HSPC Doxlipo and DPPC Doxlipo both had very weak Dox distribution in the nucleus. After 24, DPPC Doxlipo showed similar Dox distribution in the nucleus with its preheated counterpart. But Dox distribution of HSPC Doxlipo was still low (Figure 3A). As for lysosome distribution, a magnified picture showed that both HSPC Doxlipo and DPPC Doxlipo could be captured by the lysosome (yellow arrows), indicating liposomes taken by cells through endocytosis (Figure 3B and C). Besides, Dox distributed in cytoplasm was observed in HSPC Doxlipo (red triangles), which indicated the Dox released from lysosome after endocytosis (Figure 3B).
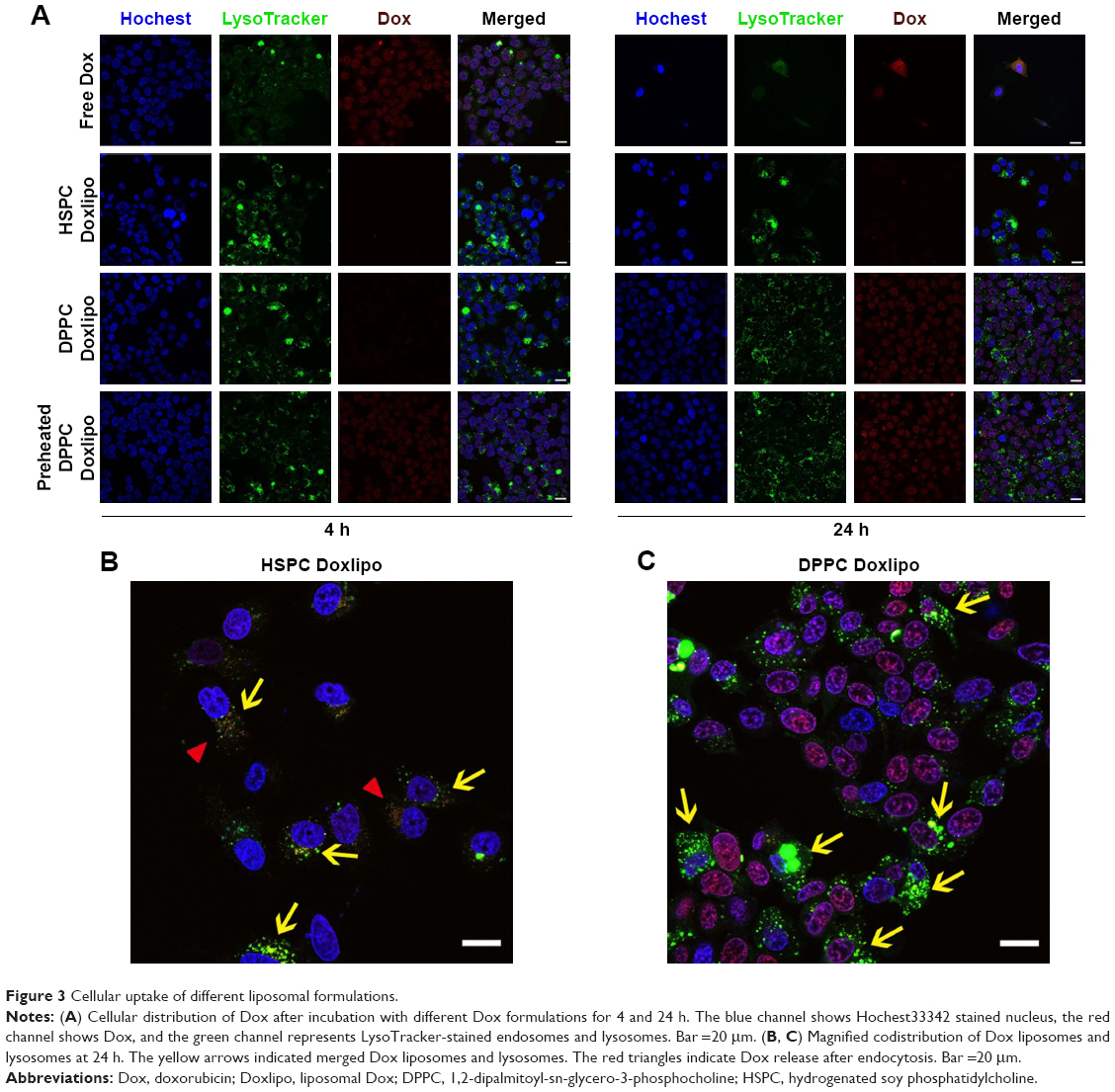 | Figure 3 Cellular uptake of different liposomal formulations. |
In vitro cytotoxicity
The in vitro cytotoxicity was determined by MTT. All treatments showed dose- and time-dependent cytotoxicity. Free Dox showed the highest cytotoxicity (Figure 4A), while HSPC Doxlipo showed the lowest cytotoxicity against MCF7 cells (Figure 4B). DPPC Doxlipo preheated displayed higher inhibitory effect at early stage (24 h) than the unheated counterpart, although the difference was not significant at 48 or 72 h (Figure 4C and D). The cytotoxicity of HSPC or DPPC liposomes without drug loading was also tested. The result showed that blank liposomes were almost not toxic (Figure 4E and F). Phospholipids and cholesterol are natural composition of cells; hence, liposomes are considered as safe drug carriers. Synthetical phospholipids HSPC and DPPC also proved to have good biocompatibility in this study.
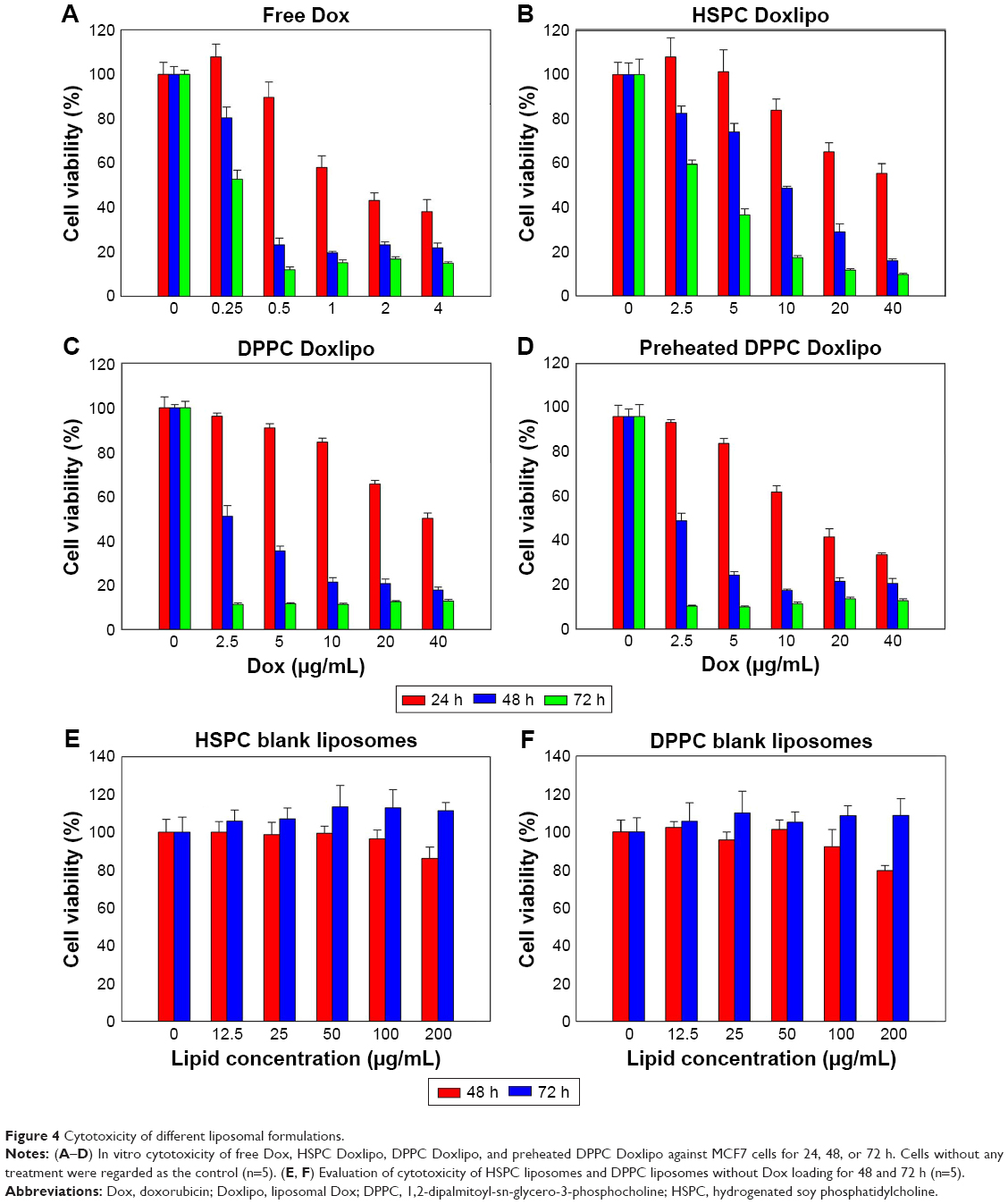 | Figure 4 Cytotoxicity of different liposomal formulations. |
Dox formulations induced global change of gene expression profile
To investigate the transcriptional profiling affected by these Dox formulations, the high-throughput screening technology microarray assay was used, so as to obtain candidate genes related to cell malignancy. RNA was isolated from MCF7 treated with Dox formulations at equal dosage (0.5 μg/mL) for 24 h. According to the previous microarray and our cytotoxicity results, this dosage did not induce significant cell death at the early stage, which minimized the impact of dead cells. The total number of differentially expressed genes was identified after normalization, as grouped by level of fold change of gene expression (Figure 5A). Free Dox triggered robust gene expression variation, and preheated DPPC Doxlipo showed mild change. But both HSPC Doxlipo and DPPC Doxlipo treatment showed fewer differentially expressed genes. The Venn diagram was used to demonstrate the differentially expressed genes in common to different treatments using Venny 2.1. Of these, 11 upregulated and 16 downregulated genes were common to all treatments with a fold change of >1.5 (Figure 5B). The heatmap of the differentially expressed genes was graphed by HemI17 to give an overall view of transcriptomics impacted by the four Dox formulations (Figure S1; Table S2). Furthermore, a selection of potential gene targets involved in tumor regression in response to the different treatments was highlighted with heatmap for further validation (Figure 5C).
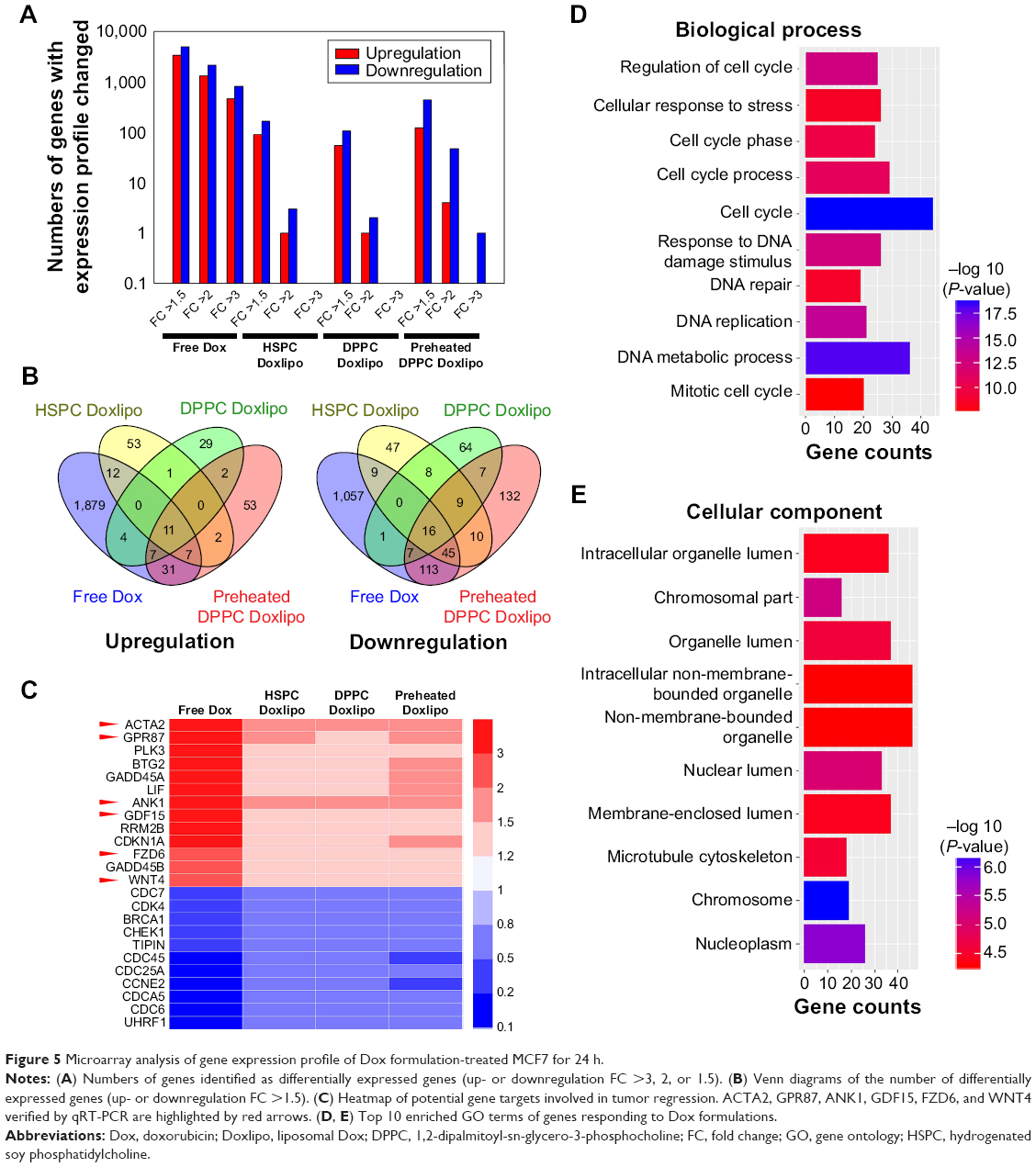 | Figure 5 Microarray analysis of gene expression profile of Dox formulation-treated MCF7 for 24 h. |
Gene Ontology (GO) was used to annotate the gene function. We performed the bioinformatics analysis using DAVID 6.718 on the genes listed in the heatmap. The top 10 enriched GO terms were illustrated by R software (Figure 5D and E). GO terms of biological process suggested all treatments induced cell cycle arrest, DNA damage, and activated DNA repair. GO terms of cellular component mainly focus on chromosome and nuclear structure change. Together, these results suggested that Dox-induced DNA damage and cell cycle arrest was an early event. Nevertheless, tumor cells quickly activated the DNA repair process to survive the stress.
Different Dox formulations stimulated the expression of EMT-related genes
Since microarray is a sensitive and high-throughput method, further verification is necessary. Eight upregulated genes were picked, which might be involved in EMT. Of these, six genes were confirmed by qRT-PCR, including ANK1, ACTA2, GPR87, GDF15, FZD6, and WNT4. Furthermore, two well-known Dox-resistant genes (ABCB1, ABCG2) were also investigated.
Total Dox (0.5 μg/mL) treatment for 24 and 48 h showed time-dependent increasing expression of these genes compared with control group (Figure 6A and B). Free Dox group induced the highest level. Especially, ANK1, ACTA2, GPR87, and WNT4 showed significantly higher expression than the other groups. However, the drug-resistant genes (ABCB1, ABCG2) were not obviously upregulated until 48 h. We speculated that drug resistance occurred as a relatively late event, which might be due to underregulation of DNA damage-related genes.
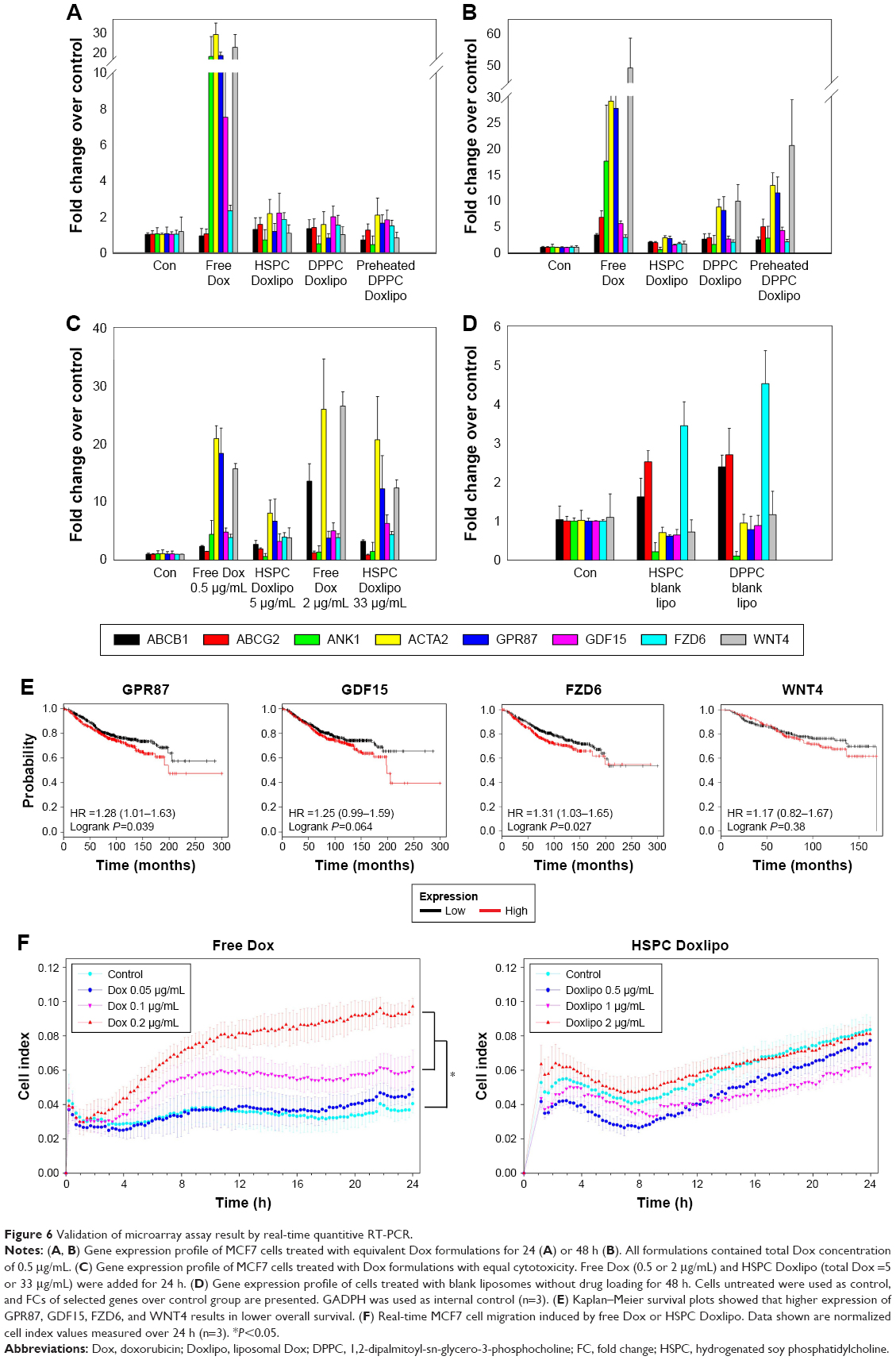 | Figure 6 Validation of microarray assay result by real-time quantitive RT-PCR. |
Since higher cellular Dox uptake led to higher cytotoxicity, the gene expression profile exhibited dose-dependent effect. We further examined the gene expression profile under dosage with similar cytotoxicity. Free Dox and HSPC Doxlipo with obviously distinctive drug release were chosen for 24 h treatment. MTT was performed with the same batch of cells and liposomes as previously to ensure similar cytotoxicity was reached. Two inhibitory dosages were used, including 15% inhibition concentration (free Dox, 0.5 μg/mL; HSPC Doxlipo, 5 μg/mL) and 50% inhibition concentration (free Dox, 2 μg/mL; HSPC Doxlipo, 33 μg/mL). Generally, free Dox still stimulated higher gene expression than HSPC Doxlipo. Especially under 50% inhibition, free Dox showed significantly higher expression of ABCB1 (Figure 6C).
Impact of blank liposomes without Dox loading was also examined, with high concentration of lipids (200 μg/mL) treating for 24 h. Surprisingly, high concentration of blank liposomes could also affect the cellular gene expression pattern, though not as significantly as Dox-encapsulated liposomes. Both HSPC and DPPC blank liposomes increased the expression of ABCB1, ABCG2, and FZD6 but decreased the expression of ANK1. This might be caused by endocytosis of liposome composition (Figure 6D).
Finally, an online tool, Kaplan Meier plotter,19 was used to assess the effect of genes on survival using clinical breast cancer sample from database. Kaplan–Meier survival plots showed that higher expression of GPR87, GDF15, WNT4, and FZD6 resulted in low overall survival, suggesting poorer prognosis (Figure 6E). Since Dox significantly upregulated the expression of these genes, their roles in cell survival or cancer relapse postchemotherapy deserve further studies.
Cell migration enhanced by Dox treatment
Enhanced cell migration is a sign of EMT and is associated with metastasis. In this study, a real-time system was used to monitor the cell movement. Free Dox and HSPC Doxlipo were tested with nontoxic dosage (within 24 h). Free Dox significantly enhanced cell migration in a dose-dependent manner, while HSPC Doxlipo did not accelerate cell migration. On the contrary, HSPC Doxlipo even slightly inhibited cell migration (Figure 6F).
Codelivery of Dox and siRNA for GPR87 using cationic Dox liposomes
To check whether inhibition of GPR87 expression can alleviate the cell malignancy induced by Dox, siRNA targeting GPR87 was codelivered using cationic Dox liposomes composed of DOTAP, Chol, and DSPE-PEG2000. DOTAP Doxlipo possessed similar size and PDI but positive zeta potential (Table 2). Agarose gel electrophoresis was used to evaluate the siRNA loading in DOTAP Doxlipo. DOTAP Doxlipo with N/P >20 (equivalent to WLipid:WsiRNA =120) could effectively entrap siRNA; hence, the combination of DOTAP:CHOL:DSPE-PEG2000 of 49:48:3 was used in the following study (Figure 7A). DOTAP Doxlipo significantly increased FAM-siRNA cellular uptake, while naked FAM-siRNA could not enter into cells. Additionally, cellular Dox uptake of DOTAP Doxlipo was also higher than HSPC or DPPC Doxlipo, probably because the positively charged liposomes facilitated internalization (Figure 7B). Cellular distribution of siRNA and Dox showed both nuclear (Figure 7C) and lysosome (Figure 7D) colocalization with DOTAP Doxlipo, though fluorescence signal in lysosomal compartments was stronger.
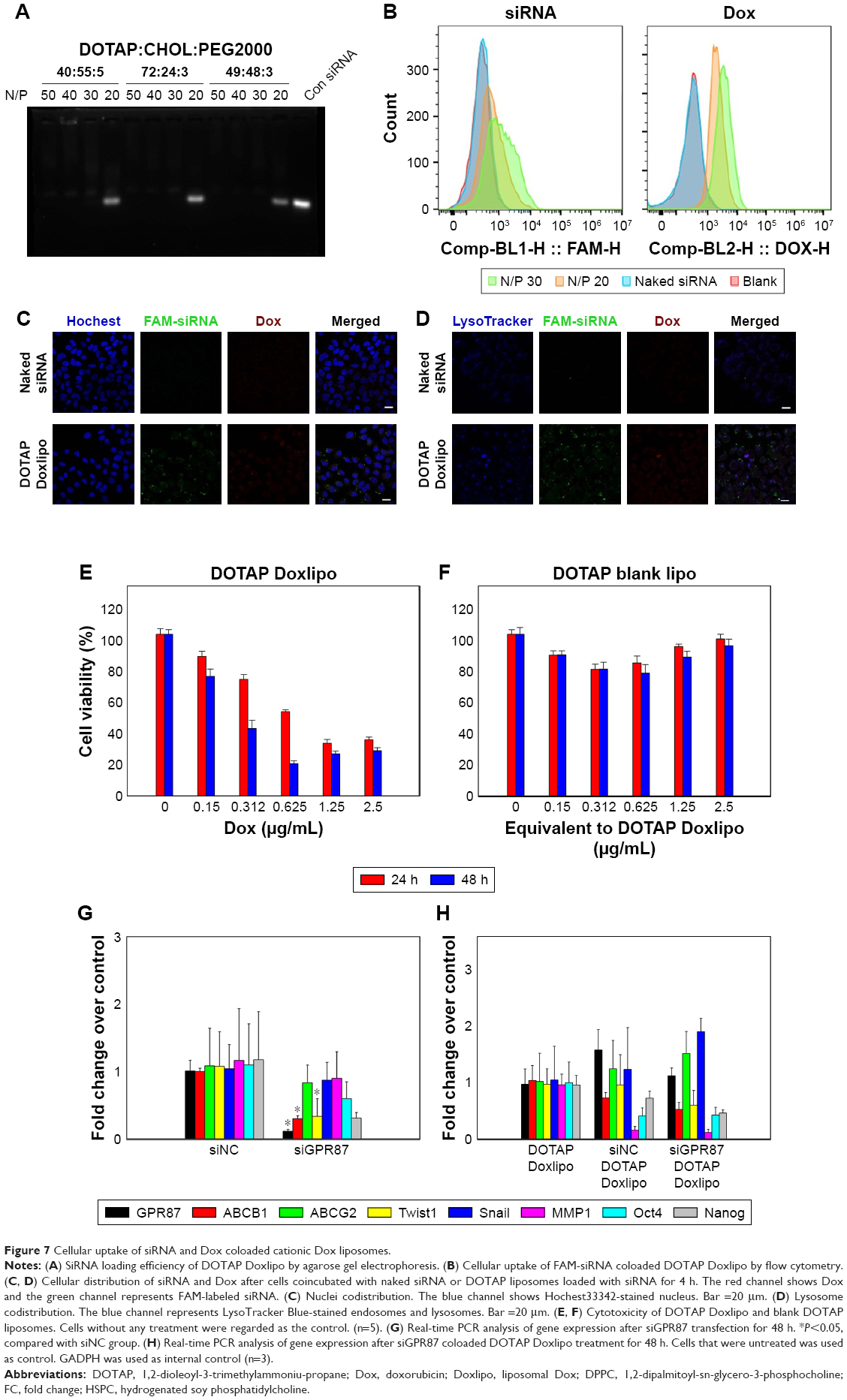 | Figure 7 Cellular uptake of siRNA and Dox coloaded cationic Dox liposomes. |
MTT results also showed higher cytotoxicity of DOTAP Doxlipo than the other Dox liposomes, which might be due to higher cellular uptake of Dox (Figure 7E). Since cationic lipids have an inhibitory effect, the cytotoxicity of blank DOTAP liposomes was also examined. The cytotoxicity of blank DOTAP liposomes was much lower than their Dox-loaded counterparts, which indicated that cytotoxicity was mainly caused by Dox (Figure 7F). However, siGPR87 and DOTAP Doxlipo codelivery did not enhance cytotoxicity (data not shown).
Although siGPR87 did not improve the cytotoxicity of Dox liposome, GPR87 knockdown could reduce the expression of some EMT-related genes. The effect of siGPR87 was first examined with commercialized transfection reagent. Compared with siNC, siGPR87 transfection knocked down the expression of GPR87 by 80% and significantly decreased the gene expression of ABCB1 and twist1. Besides, Oct4 and Nanog expression also decreased (Figure 7G). Codelivery of siGPR87 and DOTAP Doxlipo showed decreased expression of ABCB1, MMP1, Twist1, Oct4, and Nanog compared with DOTAP Doxlipo group, although ABCG2 and Snail expression was upregulated (Figure 7H).
Discussion
The aim of this study was to elucidate the potential relationship between liposomal formulation and tumor malignancy using global transcriptional profiling. Therefore, first two liposomal formulations with distinct drug release kinetics were prepared. The HSPC Doxlipo showed slow release in vitro, while DPPC Doxlipo had mild release under physical condition, but fast release under hyperthermia. DPPC with low Tm (41°C) is usually applied in the thermosensitive liposomes. By mixing DPPC with HSPC, the liposomes can quickly release the encapsulated drug under thermostimulation, while being maintained in a stable state under normal physiological temperature. On the contrary, HSPC with high Tm (55°C) is very stable, so the in vitro drug release of HSPC Doxlipo was minimal and almost unaffected by hyperthermia.
Cellular uptake of Dox is important to cancer therapy. Free Dox easily passes through the cell membrane by passive diffusion leading to fast intracellular accumulation. HSPC Doxlipo can enter into the cells through clathrin-mediated endocytosis, thus slowing Dox uptake. The acid environment in lysosome is favorable for liposome to release its cargos due to the enhanced permeability of the lipid bilayer.20 On the other side, Dox released from DPPC Doxlipo and preheated DPPC Doxlipo showed accelerated Dox uptake, while endocytosis played a less important role. Several studies also reported that most liposomes were taken up via clathrin-mediated endocytosis.12,21
Although liposomes with fast Dox release enhanced intracellular accumulation, their Dox retention ability was quite the opposite. Free Dox showed the poorest retention, and HSPC Doxlipo showed the longest cellular retention. Since free Dox can pass through the cell membrane by diffusion when the Dox concentration outside the membrane is reducing, Dox can also pass through the concentration gradient. Besides, many transporter proteins functioning as pumps will extrude drugs, such as P-glycoprotein. HSPC Doxlipo releases Dox from lysosome after endocytosis, which is a prolonged process. Also, it has been reported that some phospholipids or cholesterol can bind to the drug pumps and inhibit their function.21,22 Therefore, although HSPC Doxlipo delayed Dox uptake, it had longer retention time. Taken together, the way Dox liposomes releasing their cargos could affect cellular Dox uptake or retention.
The high-throughput screening technology was used to investigate the global transcriptional profile responding to different Dox formulations. Under equal dosage of Dox, faster drug release induced more significant variation in differentiated gene expression. These genes focused on cell cycle, DNA metabolism, DNA repair and cellular response to stress, etc., suggesting the early events caused by Dox were mainly nuclei and DNA targeted. The inhibition of the cell division cycle, cyclin E2, and cyclin-dependent kinase caused cell cycle arrest. Cell cycle arrest was an important cell adaptation to DNA damage. BRCA1 (breast cancer susceptibility gene 1), GADD45B, and GADD45A were the important molecules responding to DNA damage and activating repair, which ensured the genome integrity. All together, these results suggest that Dox formulations induced DNA damage first, and tumor cells might subsequently activate DNA repair to survive the stress.
Although DNA damage is one important mechanism of most chemodrugs, recent studies found that DNA damage otherwise might promote chemoresistance through inducing EMT-related genes.23,24 Besides, DNA repair is closely coupled with cell survival. Weyemi et al25 found that silencing H2A.X, an essential molecule involved in DNA repair, activated EMT transcription factors and increased tumor progression. However, the H2A.X-deficient cells were more vulnerable to genotoxic stresses.25,26 These results suggested the counteractive relationship between tumor inhibition and enhanced malignancy. As in our study, free Dox and DPPC Doxlipo with higher toxicity also induced more EMT-related genes.
In this study, we further validated the time- and dose-dependent expression of six genes, screened from the microarray assay, which might promote tumor malignancy. ANK1 and ACTA2 encode membrane-associated cytoskeletal proteins which are closely related to cancer metastatic risk.27,28 ANK1 responded to DNA damage agents and regulated actin cytoskeleton structure and remodeling, so as to promote cell migration. GDF15 belongs to secretory cytokines, whose high serum level correlated with poor prognosis in tumor patients. Therefore, GDF15 is suggested as a novel biomarker for cancer diagnosis.29 GPR87 is a target gene of P53, which responds to stress and promotes cell survival.30 WNT4- coded protein belongs to the WNT family, which exerts paracrine effects through activation of the transmembrane Frizzled.31 Interestingly, both WNT4 and its putative receptor FZD6 were significantly upregulated, suggesting their mutual effect after Dox treatment. Besides, ABCB1 and ABCG2 expression was also increased after Dox formulation treatment, indicating the risk of drug resistance. All together, these genes mentioned above play the roles in aggravation of malignancy, and hence deserve further study as targets for alleviation of chemodrug-induced cell malignancy.
Additionally, we found that drug release pattern might impact the global transcriptomics. When the cells were treated with free Dox and HSPC Doxlipo at dosages of similar inhibition, ABCB1 and WNT4 expression was significantly lower in HSPC Doxlipo group. This is probably because faster Dox accumulation induced DNA damage earlier. Besides, due to the short elimination time of free Dox in vivo, damaged tumor cells activated DNA repair pathway quickly. This might help cells survive stress and further enhance malignancy, thus causing tumor recurrence and metastasis. PEG-modified Dox liposomes have long circulation ability, so the drug clearance time is much longer leading to better retention in tumor site. They release drug in a constant way; thus, they might induce DNA damage and repair in a mild way. Moreover, Dox has severe cardiotoxicity, and thus, it is much safer to use Dox liposomes than free Dox of equal dosage. Although both free Dox and Dox liposomes triggered EMT, free Dox might have higher risk of acquiring more aggressive phenotypes. This was validated in our real-time cell migration results, as liposomal Dox did not enhance cell migration as free Dox did.
High expression of GPR87 decreases the overall survival of breast cancer patients. Recent studies also found that GPR87 was involved in cancer survival and metastasis. Overexpression of GPR87 promotes proliferation of lung cancer and bladder cancer cells.32,33 It was found that GPR87 as a downstream target gene of P53 signal pathway was necessary for P53-dependent cell survival after genotoxic stress.30 Recently, Park et al,34 suggested activated GPR87 transcription could induce lung cancer cell migration and invasion. Furthermore, GPR87 is found to be correlated to the maintenance of CD133+ cancer stem-like cell population in hepatocellular carcinoma.35 Our study also found codelivery of siGPR87 with Dox could downregulate the parts of EMT-related genes, although siGPR87 could not improve the cytotoxicity. These results suggest GPR87 could be used as a potential target for drug development and an adjuvant agent for current chemodrugs so as to alleviate cell malignancy induced by chemotherapy.
Conclusion
In summary, two types of Doxlipo formulations with different drug release kinetics were prepared, and their impact over a wide range of genes using global transcription profiling was investigated. In comparison with liposomal formulation, free Dox significantly changed the transcriptional pattern of a wide range of genes, especially those correlated with drug resistance and cell metastasis. Free Dox also considerably enhanced the cell migration behavior in comparison with HSPC Doxlipo under a similar level of cytotoxicity. Similarly, DPPC Doxlipo with faster drug release triggered more aggressive transcriptional profile in tumor cells than HSPC Doxlipo, suggesting more attention should be paid on balance of toxicity enhancement and tumor malignancy inhibition. The current study reveals the impact of pharmaceutical formulation on the malignancy at transcriptional level and illustrates the relationship between formulation-induced cytotoxicity and accompanied cell malignancy, which may facilitate the future design of anticancer formulations.
Acknowledgments
This research was supported by National Natural Science Foundation of China (81620108028), the Fundamental Research Funds for the Central Universities (2017XZZX011-04) and Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents. The authors thank Prof Xin Chen from College of Pharmaceutical Sciences, Zhejiang University, for assistance with bioinformatics analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang Q, Feng Y, Kennedy D. Multidrug-resistant cancer cells and cancer stem cells hijack cellular systems to circumvent systemic therapies, can natural products reverse this? Cell Mol Life Sci. 2017;74(5):777–801. | ||
Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6(13):10697–10711. | ||
Li QQ, Chen ZQ, Cao XX, et al. Involvement of NF-kappaB/miR-448 regulatory feedback loop in chemotherapy-induced epithelial-mesenchymal transition of breast cancer cells. Cell Death Differ. 2011;18(1):16–25. | ||
Li QQ, Xu JD, Wang WJ, et al. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. 2009;15(8):2657–2665. | ||
Du B, Shim JS. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21(7). | ||
Wang C, Jin H, Wang N, et al. Gas6/Axl axis contributes to chemoresistance and metastasis in breast cancer through Akt/GSK-3beta/beta-catenin signaling. Theranostics. 2016;6(8):1205–1219. | ||
Xu T, Zhang J, Chen W, et al. ARK5 promotes doxorubicin resistance in hepatocellular carcinoma via epithelial-mesenchymal transition. Cancer Lett. 2016;377(2):140–148. | ||
Tahover E, Patil YP, Gabizon AA. Emerging delivery systems to reduce doxorubicin cardiotoxicity and improve therapeutic index: focus on liposomes. Anti-cancer Drugs. 2015;26(3):241–258. | ||
Shibata H, Izutsu KI, Yomota C, Okuda H, Goda Y. Investigation of factors affecting in vitro doxorubicin release from PEGylated liposomal doxorubicin for the development of in vitro release testing conditions. Drug Dev Ind Pharm. 2015;41(8):1376–1386. | ||
Kokuryo D, Nakashima S, Ozaki F, et al. Evaluation of thermo-triggered drug release in intramuscular-transplanted tumors using thermosensitive polymer-modified liposomes and MRI. Nanomedicine. 2015;11(1):229–238. | ||
Siegfried JM, Burke TG, Tritton TR. Cellular transport of anthracyclines by passive diffusion. Implications for drug resistance. Biochem Pharmacol. 1985;34(5):593–598. | ||
Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. | ||
Ziello JE, Huang Y, Jovin IS. Cellular endocytosis and gene delivery. Molecular Med. 2010;16(5–6):222–229. | ||
Bisio A, Zamborszky J, Zaccara S, et al. Cooperative interactions between p53 and NFkappaB enhance cell plasticity. Oncotarget. 2014;5(23):12111–12125. | ||
Horwacik I, Durbas M, Boratyn E, et al. Analysis of genes involved in response to doxorubicin and a GD2 ganglioside-specific 14G2a monoclonal antibody in IMR-32 human neuroblastoma cells. Acta Biochim Pol. 2015;62(3):423–433. | ||
Wang J, Chan JY, Fong CC, Tzang CH, Fung KP, Yang M. Transcriptional analysis of doxorubicin-induced cytotoxicity and resistance in human hepatocellular carcinoma cell lines. Liver Int. 2009;29(9):1338–1347. | ||
Deng W, Wang Y, Liu Z, Cheng H, Xue Y. HemI: a toolkit for illustrating heatmaps. PLoS One. 2014;9(11):e111988. | ||
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. | ||
Szasz AM, Lanczky A, Nagy A, et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016;7(31):49322–49333. | ||
Karve S, Alaouie A, Zhou Y, Rotolo J, Sofou S. The use of pH-triggered leaky heterogeneities on rigid lipid bilayers to improve intracellular trafficking and therapeutic potential of targeted liposomal immunochemotherapy. Biomaterials. 2009;30(30):6055–6064. | ||
Un K, Sakai-Kato K, Oshima Y, Kawanishi T, Okuda H. Intracellular trafficking mechanism, from intracellular uptake to extracellular efflux, for phospholipid/cholesterol liposomes. Biomaterials. 2012;33(32):8131–8141. | ||
Kang DI, Kang HK, Gwak HS, Han HK, Lim SJ. Liposome composition is important for retention of liposomal rhodamine in P-glycoprotein-overexpressing cancer cells. Drug Deliv. 2009;16(5):261–267. | ||
Lee HJ, Li CF, Ruan D, et al. The DNA damage transducer RNF8 facilitates cancer chemoresistance and progression through twist activation. Mol Cell. 2016;63(6):1021–1033. | ||
Alam SK, Yadav VK, Bajaj S, et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 2016;23(4):707–722. | ||
Weyemi U, Redon CE, Choudhuri R, et al. The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition. Nat Commun. 2016;7:10711. | ||
Celeste A, Difilippantonio S, Difilippantonio MJ, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114(3):371–383. | ||
Lee HW, Park YM, Lee SJ, et al. Alpha-smooth muscle actin (ACTA2) is required for metastatic potential of human lung adenocarcinoma. Clinical Cancer Res. 2013;19(21):5879–5889. | ||
Omura N, Mizuma M, MacGregor A, et al. Overexpression of ankyrin1 promotes pancreatic cancer cell growth. Oncotarget. 2016;7(23):34977–34987. | ||
Zhang Y, Hua W, Niu LC, et al. Elevated growth differentiation factor 15 expression predicts poor prognosis in epithelial ovarian cancer patients. Tumour Biol. 2016;37(7):9423–9431. | ||
Zhang Y, Qian Y, Lu W, Chen X. The G protein-coupled receptor 87 is necessary for p53-dependent cell survival in response to genotoxic stress. Cancer Res. 2009;69(15):6049–6056. | ||
Vouyovitch CM, Perry JK, Liu DX, et al. WNT4 mediates the autocrine effects of growth hormone in mammary carcinoma cells. Endocr Relat Cancer. 2016;23(7):571–585. | ||
Nii K, Tokunaga Y, Liu D, et al. Overexpression of G protein-coupled receptor 87 correlates with poorer tumor differentiation and higher tumor proliferation in non-small-cell lung cancer. Mol Clin Oncol. 2014;2(4):539–544. | ||
Zhang X, Liu D, Hayashida Y, et al. G Protein-Coupled Receptor 87 (GPR87) promotes cell proliferation in human bladder cancer cells. Int J Mol Sci. 2015;16(10):24319–24331. | ||
Park SM, Choi EY, Bae M, et al. Histone variant H3F3A promotes lung cancer cell migration through intronic regulation. Nat Commun. 2016;7:12914. | ||
Yan M, Li H, Zhu M, et al. G protein-coupled receptor 87 (GPR87) promotes the growth and metastasis of CD133(+) cancer stem-like cells in hepatocellular carcinoma. PLoS One. 2013;8(4):e61056. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.