Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
TPMT Genetic Variability and Its Association with Hematotoxicity in Indonesian Children with Acute Lymphoblastic Leukemia in Maintenance Therapy
Authors Rosdiana DS, Setiabudy R, Andalusia R, Gatot D, Louisa M ![]() , Bardosono S, Instiaty I
, Bardosono S, Instiaty I
Received 31 October 2020
Accepted for publication 29 December 2020
Published 3 February 2021 Volume 2021:14 Pages 199—210
DOI https://doi.org/10.2147/PGPM.S288988
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Dewi Selvina Rosdiana,1 Rianto Setiabudy,1 Rizka Andalusia,2,3 Djajadiman Gatot,4 Melva Louisa,1 Saptawati Bardosono,5 Instiaty Instiaty1
1Department of Pharmacology and Therapeutics, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia; 2Dharmais National Cancer Hospital, Jakarta, Indonesia; 3Drug Registration Directorate, National Agency for Drug and Food Control, Jakarta, Indonesia; 4Division of Hematology-Oncology, Department of Pediatrics, Faculty of Medicine, Universitas Indonesia/Cipto Mangunkusumo Hospital, Jakarta, Indonesia; 5Department of Nutrition, Faculty of Medicine, University of Indonesia, Jakarta, Indonesia
Correspondence: Dewi Selvina Rosdiana
Department of Pharmacology and Therapeutics, Faculty of Medicine, Universitas Indonesia, Jl. Salemba Raya No. 6, Jakarta, 10430, Indonesia
Tel +62 21 31930481
Fax +62 21 3920947
Email [email protected]
Purpose: Hematotoxicity monitoring in children with acute lymphoblastic leukemia (ALL) is critical to preventing life-threatening infections and drug discontinuation. The primary drug that causes hematotoxicity in ALL children is 6-mercaptopurine (6-MP). Genetic variability of the drug-metabolizing enzymes of 6-MP, thiopurine S-methyltransferase (TPMT), is one factor that might increase the susceptibility of children to hematotoxicity. The present study aimed to determine the variability in TPMT genotypes and phenotypes and its association with the occurrence of hematotoxicity in ALL children in maintenance therapy.
Patients and Methods: A cross-sectional study was conducted at Cipto Mangunkusumo and Dharmais National Cancer Hospital, Jakarta, Indonesia, from June 2017 to October 2018. We included ALL patients, 1– 18 years, who were receiving at least one month of 6-MP during maintenance therapy according to the Indonesian protocol for ALL 2013. Direct sequencing was used to determine TPMT*3A, *3B, and *3C genotypes, and LC-MS/MS analysis was performed to measure the plasma concentrations of 6-MP and its metabolites. Association analysis between the TPMT genotype and hematotoxicity was evaluated using the unpaired t-test or Mann–Whitney’s test.
Results: The prevalence of neutropenia, anemia, and thrombocytopenia in ALL children during maintenance therapy was 51.9%, 44.3%, and 6.6%, respectively. We found a low frequency of TPMT*3C, which is 0.95%. No association was found between hematotoxicity and TPMT genotypes or age, nutritional status, serum albumin levels, risk stratification, the daily dose of 6-MP, and cotrimoxazole co-administration. However, hematotoxicity was associated with 6-methylmercaptopurine (6-MeMP) plasma concentrations and the ratio 6-MeMP/6-thioguanine (6-TGN). We also found no association between TPMT genotypes and TPMT phenotypes.
Conclusion: The 6-MeMP/6-TGN ratio is associated with hematotoxicity in ALL children during maintenance therapy but is not strong enough to predict hematotoxicity.
Keywords: thiopurine methyltransferase, mercaptopurine, methylmercaptopurine, thioguanine, neutropenia
Introduction
Acute lymphoblastic leukemia (ALL) is the most common pediatric malignancy, comprising 30% of cancers occurring before 15. It accounts for 80% of all leukemia cases in children. Despite steady improvements in outcome, ALL remains the leading cause of childhood cancer death.1,2 A complete remission rate is more than 90% after induction therapy, but in developed countries, around 11% of children relapse while receiving maintenance therapy,3 whereas in Cipto Mangunkusumo Hospital, Indonesia, it is 28.7%.4
During the maintenance phase, pediatric ALL patients received a treatment regimen containing 6-mercaptopurine (6-MP). However, 6-MP can cause life-threatening side effects such as myelosuppression, which may lead to treatment interruptions, and subsequently contribute to an increased relapse incidence.5
Studies have shown that the polymorphism of 6-MP drug-metabolizing enzymes, thiopurine S-methyltransferase (TPMT), has an essential role in patients’ susceptibility to myelosuppression from 6-MP. Individuals with TPMT polymorphisms represent approximately 10% of the population. Numerous variant alleles of TPMT polymorphisms have been identified and associated with decreased TPMT enzyme activity levels. Reduced enzyme activity will enhance the hematotoxicity of 6-MP by increased concentrations of 6-thioguanine nucleotide (6-TGN) levels, an active metabolite of 6-MP. Consequently, 6-MP dose reduction is needed to avoid the hematotoxicity of 6-MP. Based on population studies, three alleles account for more than 95% of TPMT variants: TPMT*3A, TPMT*3C, and TPMT*2.6 The Clinical Pharmacogenetics Implementation of the Consortium (CPIC) Guidelines recommended adjusting the starting dose of 6-MP based on the TPMT genotype to avoid hematotoxicity.7 However, some studies in Asian population indicate the relationship between TPMT genotypes and hematotoxicity, with conflicting results. In one study, patients with TPMT heterozygotes received a less 6-MP dose compared to those with wild-type alleles. The above study concluded that the identification of the TPMT genotype might be significant.8 In another study in an Indian population, TPMT heterozygous patients received a similar 6-MP dose, and the study showed that TPMT genotyping might not be necessary.9
To date, there are no data on TPMT polymorphism and its relationship with hematotoxicity in Indonesian pediatric ALL. Thus, dose adjustment for 6-MP maintenance therapy based on TPMT genotypes, which reflects ethnic allele distribution, needs to be evaluated in Indonesian pediatric ALL.
Patients and Methods
Study Design
This cross-sectional study was conducted from June 2017 to October 2018 at Cipto Mangunkusumo Hospital and Dharmais Cancer Hospital to recruit pediatric ALL patients undergoing maintenance therapy. The study was conducted following the Declaration of Helsinki. The Ethics Committee of Medical Faculty, Universitas Indonesia, approved this study. Written informed consent/assent was obtained from patients and/or parents/legal guardians.
We included children diagnosed with ALL, aged 1–18 years, who underwent 6-MP treatment for at least one month according to the Indonesian protocol for ALL 2013.10 Patients were excluded if they had a severe infection, received colony-stimulating factors, allopurinol, mesalazine, olsalazine, or sulfasalazine on their treatment regimen. The sample size estimation for the polymorphism study was determined based on reference, and Hong et al stated that the minimal sample size in the polymorphism study was 100.11
The blood samples of each patient who underwent 6-MP 50 mg/m2/day for at least one month were drawn once, in the morning (a day before receiving vincristine intravenously), to evaluate TPMT genotyping, to quantify 6-TGN and 6-methylmercaptopurine (6-MeMP) concentrations, as well as for hematology data. We recorded the demography of all eligible patients. Hematology data were categorized according to the Common Terminology Criteria for Adverse Events v3.0.12 Later, we correlated the metabolite levels with the hematological data obtained simultaneously.
Drug Regimen in Maintenance Therapy
Maintenance therapy was initiated at treatment week 13 for standard risk (SR) ALL or 18 for high risk (HR) ALL and continued until 2.5 years after diagnosis. The 6-MP starting dose was 50 mg/m2/day. Some patients received breaking tablets (the same daily dose of 6-MP), and others were given different doses on alternating days to achieve specific doses (modified daily dose of 6-MP). During maintenance therapy, patients also received low-dose oral (20 mg/m2/week) pulse methotrexate (MTX) and vincristine intravenously every fifth and sixth week with dexamethasone orally 6 mg/m2/day (HR ALL) or 4 mg/m2/day (SR ALL), for two weeks. During the first year of the maintenance phase, patients received pulses of intrathecal MTX at seven-week intervals until six courses.10
TPMT Genotyping
Genomic DNA was isolated from 200 µL of buffy coat from peripheral blood using the Genomic DNA Mini Kit (Geneaid Biotech Ltd, Taipei, Taiwan) according to the manufacturer’s instructions. Genotyping was performed using polymerase chain reaction on the LightCycler® 480 Real-Time PCR System (Roche, Basel, Switzerland) for mutations in exon 7 (G460A) and exon 10 (A719G) (TPMT*3A, *3B, and *3C) followed by direct DNA sequencing using the primers as previously described by Dervieux et al13.
Phenotyping
The blood sample of each patient who underwent 6-MP treatment for at least one month was tested for 6-TGN and 6-MeMP concentrations. LC-MS/MS was used to quantify the levels of 6-TGN and 6-MeMP, according to the method previously published.14 The blood sample was drawn in patients who underwent 6-MP treatment for at least one month because the steady-state level of mercaptopurine metabolites was reached after 2–4 weeks of administration.15
Statistical Analysis
We used SPSS 20 software (New York, USA) for statistical analysis. The Kolmogorov–Smirnov test was used to evaluate the normality of data distribution. Association analysis between the TPMT genotype and hematotoxicity and subgroup analysis were assessed using the unpaired t-test, Mann–Whitney’s test, the chi-square test, or Fisher’s exact test, as appropriate. Receiver operating characteristic (ROC) curve analysis was used to determine the predictive 6-MP metabolite levels and hematotoxicity risk.
Results
Patient Characteristics and Prevalence of Hematotoxicity
A total of 106 ALL patients were included in our study. The baseline characteristics of these subjects are shown in Table 1. The median age at study participation was 6.21 years. In all, 53% of the subjects were male, and 54.7% presented with high-risk disease. More than 95% of the subjects had normal to obese nutritional status and normal serum albumin levels. Subjects with concomitant cotrimoxazole were 46.2%, and 52.8% received the same daily dose of 6-MP.
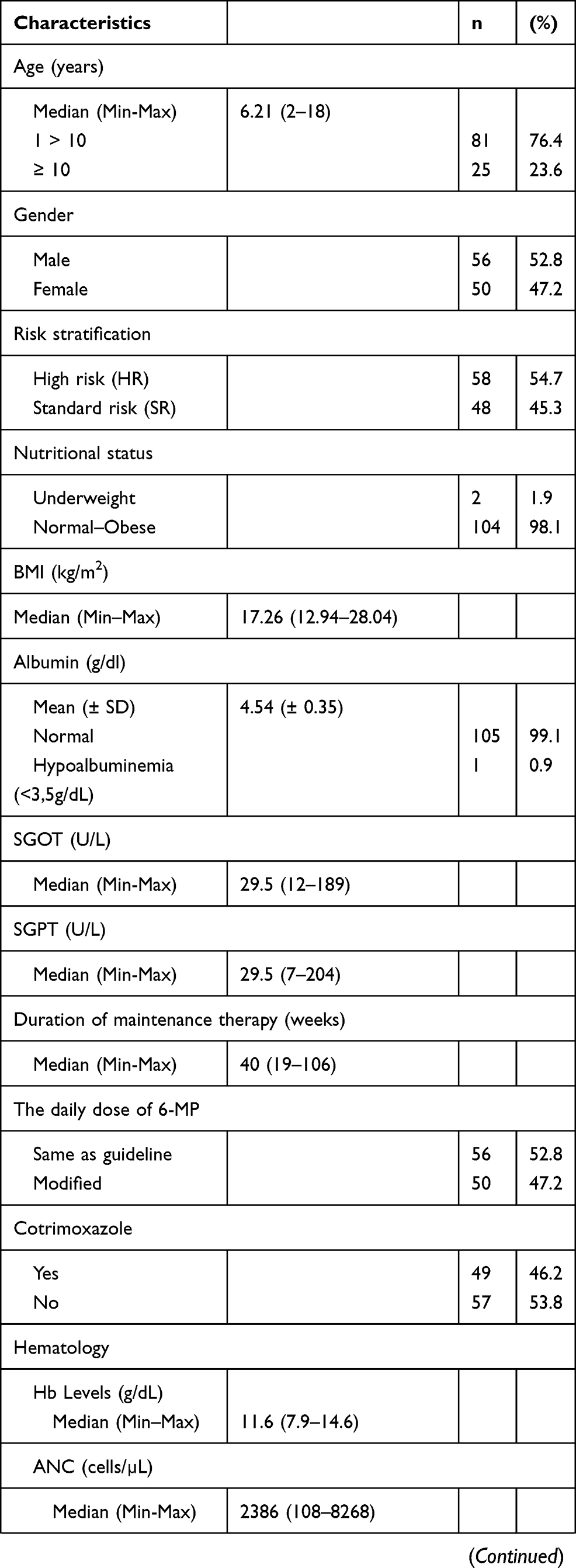 | 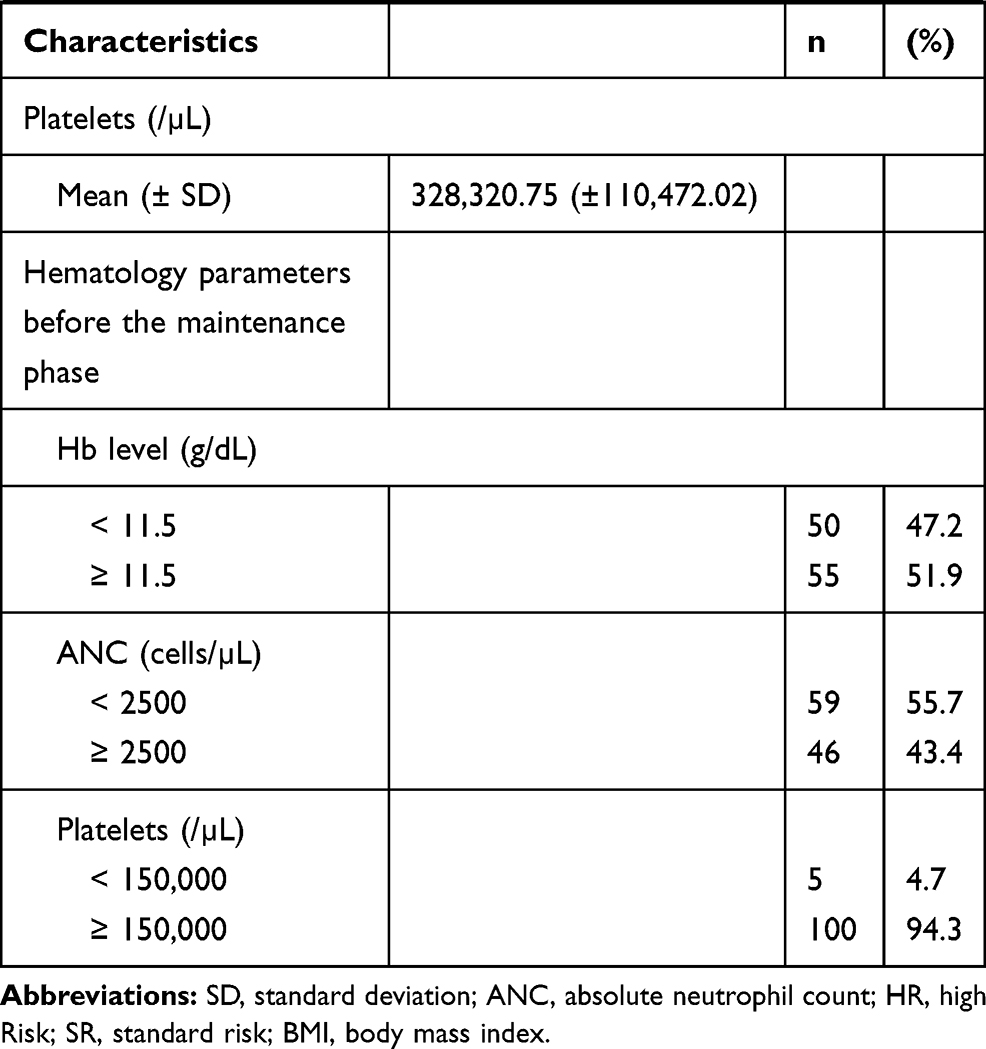 |  |
Table 1 Patients Characteristics |
The prevalence of hematotoxicity was 71.7%, the majority in grade 1 (Table 2). The prevalence of neutropenia, anemia, and thrombocytopenia was 51.9%, 44.3%, and 6.6%, respectively. Ten subjects (9.4%) presented with grade 3–4 neutropenia (absolute neutrophil count [ANC] < 1000 cells/mm3).
 |
Table 2 Hematotoxicity Grading According to CTCAE |
TPMT Genotyping and Phenotyping
TPMT genotyping is shown in Table 3. The result of the TPMT genotype was that 104 subjects (98.1%) were *1/*1 (wild type), and two subjects (1.9%) were *1/*3C. The frequency of TPMT*3C was 0.95%. TPMT*3A and *3B were not found.
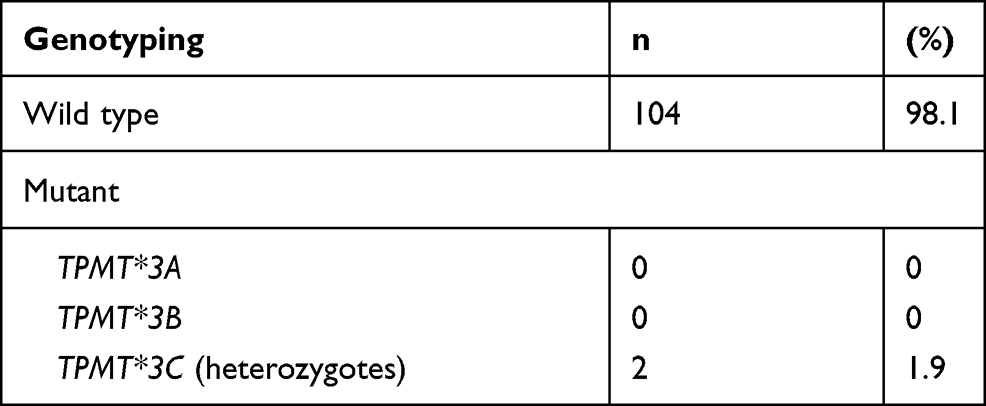 |
Table 3 Genotyping of Subjects |
The median 6-TGN levels, 6-MeMP levels, and ratio 6-MeMP/6-TGN levels were 19.12 (6–234.04), 44.54 (3.5–3167.01), and 3.1 (0.06–100.64) pmol/8×108 RBC, respectively. The association of hematotoxicity with genotypes and several factors are shown in Table 4. There was no association between TPMT genotypes and hematotoxicity (p = 1.000). There were no statistical differences in 6-TGN levels between the group with hematotoxicity and that without hematotoxicity (p = 0.938; Figure 1). In contrast, there were statistically significant differences in 6-MeMP levels (p = 0.004; Figure 2) and ratio 6-MeMP/6-TGN levels (p = 0.022; Figure 3) with hematotoxicity. We tried to predict the risk of hematotoxicity based on the ratio of 6-MeMP/6-TGN levels using ROC curve analysis. The area under the curve was 0.66. The 6-MeMP/6-TGN ratio threshold at 0.6 was shown to have the best sensitivity (71.1%) and specificity (53.3%).
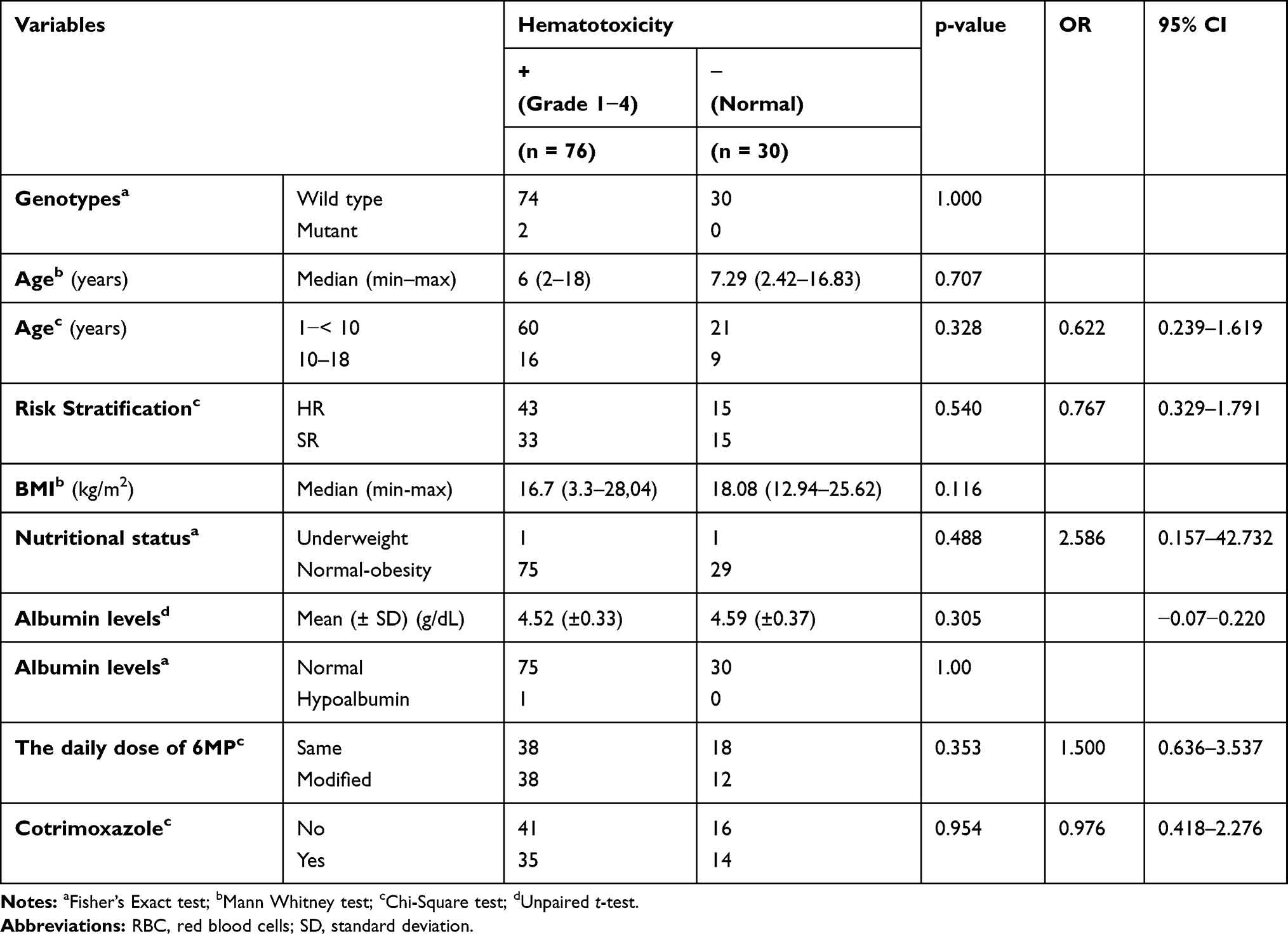 |
Table 4 Association Between Hematotoxicity with TPMT Genotypes, Phenotypes, and Several Variables |
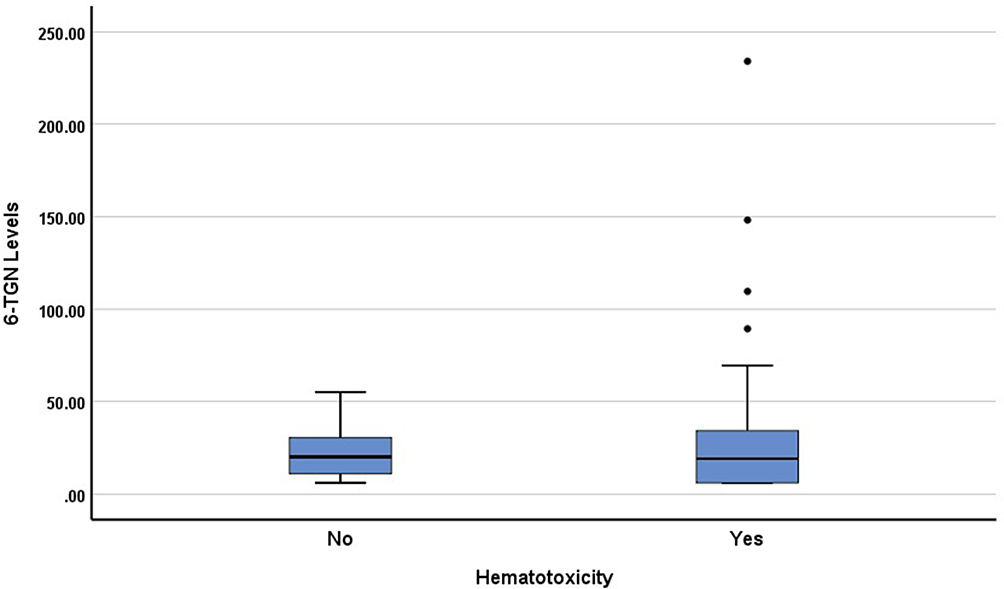 |
Figure 1 Box plots of 6-TGN levels in patients with and without hematotoxicity. The 6-TGN levels were not significantly different in both groups (p=0.938). |
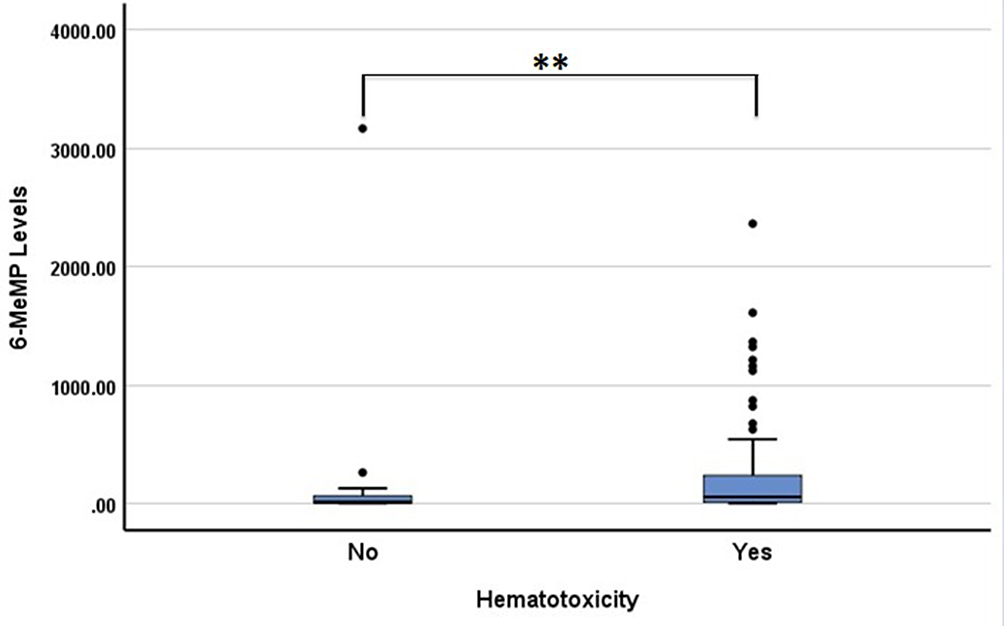 |
Figure 2 Box plots of 6-MeMP levels in patients with and without hematotoxicity. The 6-MeMP levels were higher in patients with hematotoxicity (**p<0.01). |
 |
Figure 3 Box plots of 6-MeMP/6-TGN ratio in patients with and without hematotoxicity. The 6-MeMP/6-TGN ratio was higher in patients with hematotoxicity (*p<0.05). |
Subgroup analysis was carried out based on the occurrence of anemia, grade 3–4 neutropenia, and thrombocytopenia (Table 5). Based on subgroup analysis, there was a statistical difference of 6-MeMP levels between subjects with anemia and anemia. A ROC curve analysis to determine the predictive 6-MeMP levels and the risk of anemia resulted in an area under the ROC curve of 0.61. The best sensitivity and specificity were defined for the 6-MeMP level threshold at 44.54 pmol/8 × 108 erythrocytes, 61%, and 60%. There was also a significant difference in 6-MeMP levels between subjects with and without grade 3–4 neutropenia. Based on the ROC curve analysis, the area under the curve was 0.77. The 6-MeMP level threshold at 60 pmol/8 × 108 erythrocytes was shown to have the best sensitivity (90%) and specificity (62%). There was also a significant difference in the 6-MeMP/6-TGN ratio between subjects with and without grade 3–4 neutropenia (Figure 4). Based on the ROC curve analysis, the area under the curve was 0.75. The 6-MeMP/6-TGN ratio threshold at 3.1 was shown to have the best sensitivity (90%) and specificity (54%).
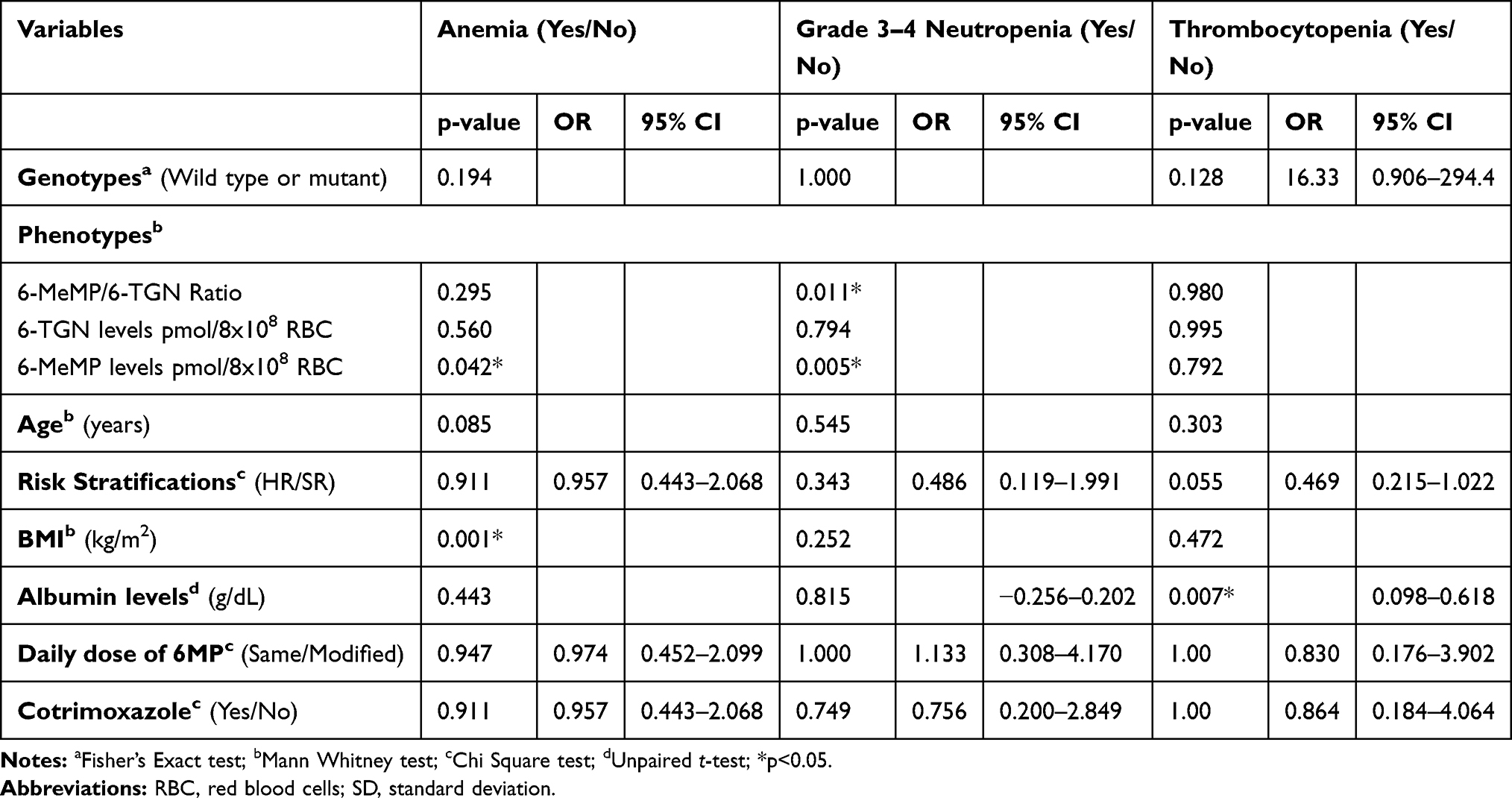 |
Table 5 Association Between Anemia, Neutropenia Grade 3–4, Thrombocytopenia, and Other Variables |
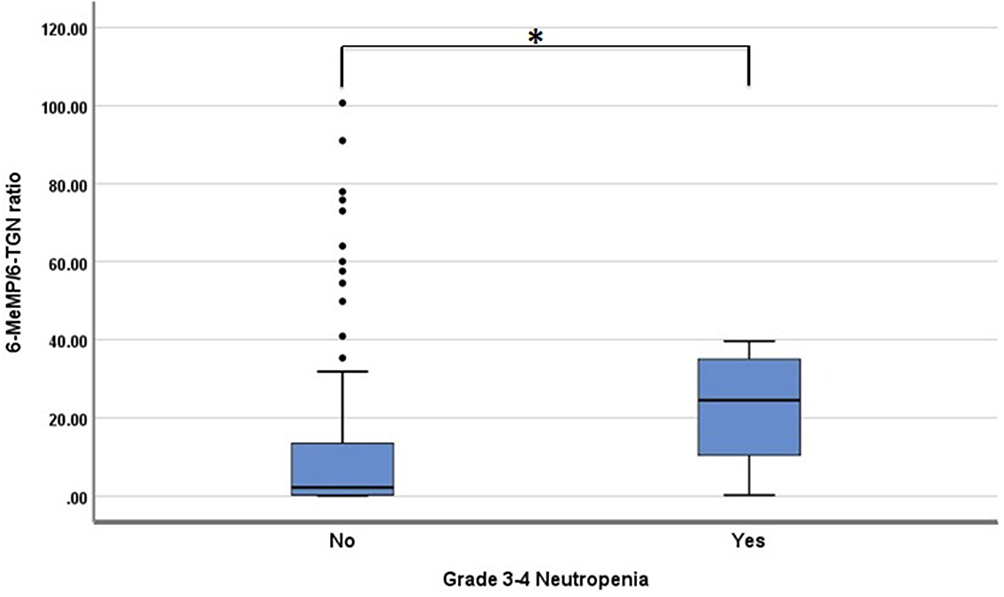 |
Figure 4 Box plots of 6-MeMP/6-TGN ratio in patients with and without grade 3–4 neutropenia. The 6-MeMP/6-TGN ratio was higher in patients with grade 3–4 neutropenia (*p<0.05). |
There was no association in hematotoxicity prevalence analyzed based on patients’ characteristics (age, nutritional status, serum albumin levels, risk stratification, a daily dose of 6-MP, and co-administration cotrimoxazole). Multivariate logistic regression analysis was not carried out because none of the bivariate analysis was significant (Table 4). No relationship between TPMT genotypes and phenotypes was found. Analysis using Mann–Whitney’s test did not show statistically significant differences between both metabolites (6-TGN levels and 6-MeMP levels) and the TPMT genotype, p = 0.075 and p = 0.935, respectively (Figure 5).
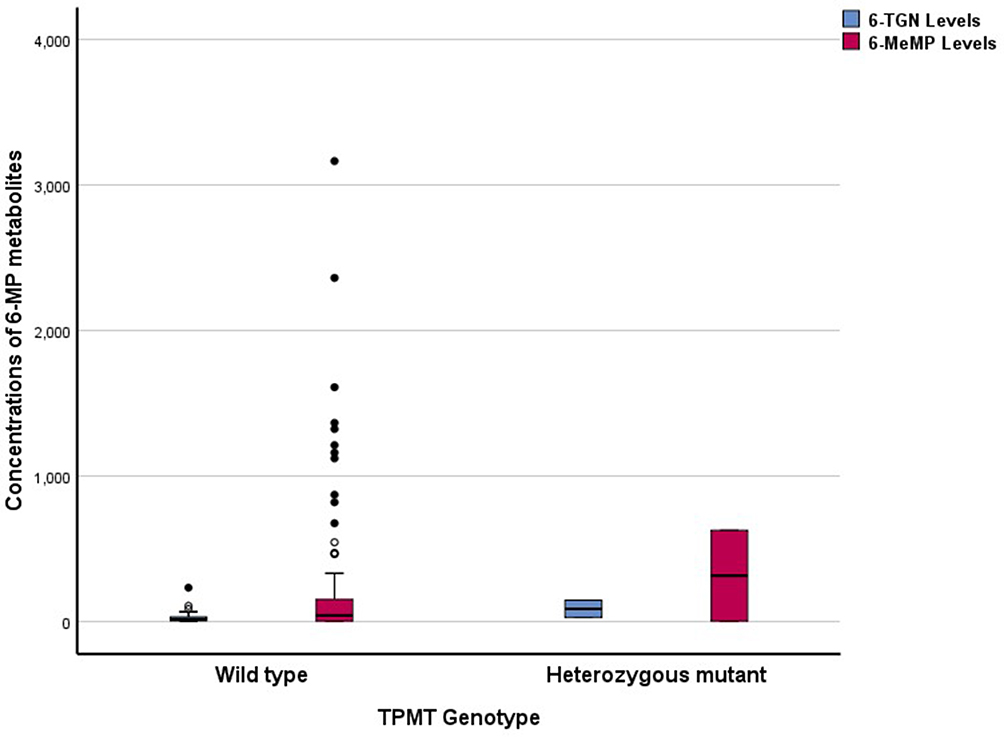 |
Figure 5 Erythrocyte 6-TGN and 6-MeMP concentrations (pmol/8x108 RBC) by TPMT genotype (wild type, n=104; mutant, n=2). |
Discussion
Several studies have shown that genetic variability in TPMT is essential to determine the susceptibility of ALL patients to hematotoxicity due to 6-MP.7 Yet, studies in Asian populations have been inconclusive. In the present study, we found the lack of a relationship between TPMT *3A, *3B, and *3C and the occurrence of hematotoxicity. However, we found strong associations between the TPMT phenotypes and grade 3–4 neutropenia prevalence.
The present study results showed that the allele frequencies among patients were similar to those found in other Asian populations, with TPMT*3C as the primary variant allele (0.95%). In Asian populations, the overall prevalence of TPMT mutant alleles is lower (~1.7%) and is predominantly represented by the TPMT*3C variant (1.6%).7
No association was found between the three TPMT variants studied (*3A, *3B, and *3C) and hematotoxicity. The 97.4% of subjects with hematotoxicity were of wild-type TPMT genotypes. Similarly, in subgroup analysis, the TPMT genotype showed no significant association with the incidence of anemia, grade 3–4 neutropenia, or thrombocytopenia. All the patients who experienced grade 3–4 neutropenia (ANC<1000 cells/m3) in this study had wild-type genotypes. However, both subjects with heterozygous TPMT*3C (TPMT *1/*3C) alleles belong to the group of subjects who are experiencing hematotoxicity. The results of this study are in agreement with those of Levinsen et al16. who also found no difference in myelotoxicity in subjects with high TPMT activity (wild type) compared with those with intermediate TPMT activity (heterozygous mutants). These results are in accordance with other studies in Asian populations.17–22 No TPMT mutant alleles were even found in patients experiencing hematotoxicity.19 The result of our study indicates that the TPMT genotype alone could not explain the high incidence of hematotoxicity in ALL children in Indonesia.
However, in Caucasian pediatric ALL, Relling et al found that hematotoxicity was higher in heterozygous mutant patients than wild-type patients. In that study, the most frequent allele found was TPMT*3A and not TPMT*3C.23
TPMT enzyme activity differs between alleles. The TPMT*3A allele is the most common mutant allele responsible for low TPMT activity in the Caucasian, Mediterranean, American, Middle Eastern, and Mexican populations. TPMT*3A encodes a protein with two single-nucleotide polymorphisms (SNPs), ie, G460A in exon 7 and A719G in exon 10, leading to amino acid substitutions at codon 154 (Ala>Thr) and codon 240 (Tyr>Cys), resulting in low activity. TPMT*3A had more than 200-fold reduced enzyme activity than the wild type. While the TPMT*3C mutant allele, the most common mutant allele in Asia and Africa, contains an SNP (A719G in exon 10), it produces intermediate enzyme activity compared with the wild type.24–26 The variation of TPMT enzyme activity in mutant alleles caused a significant difference in the level of 6-TGN in TPMT genotypes *1/*3A, which was significantly higher than TPMT genotype patients *1/*3C.27 In our study, the proportion of subjects with heterozygous mutant alleles was low, 1.9% (2/106). We only found TPMT*3C mutant alleles, which caused a slight decrease in TPMT activity. The allele is one reason for the difference in hematotoxicity based on the TPMT genotype in our study.
In the Asian population, the frequency of the TPMT mutant allele is low and dominated by TPMT*3C.7 However, similar to our results, hematotoxicity associated with mercaptopurine administration in Asian patients is common.18–21 TPMT deficiency cannot explain the high hematotoxicity in patients in Asia.28 TPMT genotyping cannot predict hematotoxicity in 6-MP administration in Asian patients.29
In this study, 106 patients who received 6-MP 50 mg/m2/day for at least one month showed highly varied metabolite levels of 6-TGN and 6-MeMP. Our result is in line with various studies conducted previously.5,30,31 The study of Relling et al23. in 180 ALL pediatric patients who received 6-MP therapy during the maintenance phase, gave highly variable 6-TGN levels, with an inter-patient variation coefficient of 78% and an intra-patient variation coefficient of 37.9%.
The therapeutic level range of 6-TGN in inflammatory bowel disease (IBD) patients is around 235–450 pmol/8 × 108 erythrocytes.32 In pediatric ALL patients, a range of 6-TGN therapeutic levels has not been determined. In our study, the 6-TGN levels varied significantly in a wide range, as in other studies. Nevertheless, the levels obtained in this study were lower, 6–234.04 pmol/8 × 108 erythrocytes, compared with the Chrzanowska et al study,30 <60–833 pmol/8 × 108 erythrocytes, despite the patient receiving the same 6-MP dose of 50 mg/m2/day. In the Bhatia et al study,5 patients received a higher dose of 6-MP, 75 mg/m2/day. Still, the 6-TGN levels in the study were 0.3–714.1 pmol/8 × 108 erythrocytes, similar to the study of Chrzanowska et al30. Several factors can contribute to inter-individual variations in 6-MP metabolite levels: differences in 6-MP absorption; a variety of TPMT enzyme activities, which are influenced by genetic polymorphisms, interactions with other drugs, and patient compliance.6,33–36 Although the 6-TGN levels in this study were relatively low, the myelosuppression effect, as indicated by the ANC values, was in the range of 108–8268 cells/mm3. Our study’s ANC value was not much different from that reported by Bhatia et al5; with higher 6-TGN levels, ANC ranged from 600 to 8100 cells/mm3.
We found higher 6-MeMP levels and a higher ratio of 6-MeMP/6-TGN levels in patients with hematotoxicity and grade 3–4 neutropenia. However, the sensitivity and specificity values were not strong enough to predict the risk of hematotoxicity and grade 3–4 neutropenia.
To date, no studies have been done to assess the relationship between the ratio of 6-MeMP/6-TGN levels and hematotoxicity in pediatric ALL patients. Nevertheless, there were studies in patients with IBD. They found no difference in the ratio 6-MeMP/6-TGN levels, 6-TGN levels, and 6-MeMP levels in IBD patients with or without leukopenia.37 Another study showed that the 6-MeMP/6-TGN level ratio >11 were included in the thiopurine hyper-methylator and for those patients necessary to reduce 25–33% 6-MP dose.36
TPMT phenotyping to predict hematotoxicity is associated with some limitations, ie, the therapeutic range of 6-TGN levels and 6-MeMP levels for ALL is not yet determined due to inter-and intra-individual variations, higher cost, and because the measurement results are affected by patient compliance.
We found no association between TPMT genotypes and TPMT phenotypes. In this study, 6-TGN levels tended to be higher, and 6-MeMP levels tended to be lower in heterozygous mutant patients, which was in line with another study.13 However, the difference did not reach statistical significance due to the small number of patients with heterozygous mutants found in this study.
This study found no association of patients’ characteristics with 6-MP hematotoxicity. The genetic factor, such as polymorphism, contributed to 6-MP hematotoxicity. TPMT polymorphism plays an essential role in the metabolism of 6-MP and affects the efficacy and safety of the treatment. However, the frequencies of TPMT variants in most Asian populations, including Indonesia, are low. More recently, the CPIC Guidelines recommended testing the genotype of the nudix enzyme (nucleoside diphosphate-linked moiety X)-type motif 15 (NUDT15) for 6-MP initial dose adjustment to avoid hematotoxicity in Asian patients.38 The frequency of NUDT15 mutant alleles in Asia was more than 10%.29
Based on this study’s results, routine TPMT genotyping for adjusting the initial 6-MP dose in Indonesian pediatric ALL patients provides limited clinical benefits. Further studies are needed to confirm the role of NUDT15 in pediatric ALL patients in Indonesia.
Conclusion
In the present study, we found no association between TPMT*3A, *3B, and *3C genotype variations and hematotoxicity in Indonesian pediatric ALL patients in maintenance therapy containing 6-mercaptopurine. Yet, there was an association between the ratio of 6-MeMP/6-TGN and the occurrence of grade 3–4 neutropenia.
Abbreviations
ALL, acute lymphoblastic leukemia; CTCAE, Common Terminology Criteria for Adverse Events; 6MP, 6-mercaptopurine; 6MeMP, 6-methylmercaptopurine; 6TGN, 6-thioguanine nucleotide; TPMT, thiopurine S-methyltransferase; SD, standard deviation; ANC, absolute neutrophil count; HR, high risk; SR, standard risk; RBC, red blood cells; BMI, body mass index.
Acknowledgments
This study was supported by Research Grant from the University of Indonesia. We thank Dr. Pustika Amalia W, dr. Sp.A(K), head of the Hemato-oncology Division of Department of Pediatrics, Cipto Mangunkusumo Hospital, and Haridini Intan, dr. Sp.A(K), head of the Pediatric Oncology Polyclinic, Dharmais National Cancer Hospital, allows us to access patient and medical records. The Authors would like to thank Enago (www.enago.com) for the English language review.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hunger SP, Loh ML, Withlock JA, et al. Children’s oncology group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer. 2013;60(6):957–963. doi:10.1002/pbc.24420
2. Cancer Facts & Figures. American cancer society; 2015. Available from: http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf.
3. Bhatia S, Landier W, Shangguan M, et al. Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and Non-Hispanic white children with acute lymphoblastic leukemia: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(17):2094–2101. doi:10.1200/JCO.2011.38.9924
4. Windiastuti E Childhood acute leukemia. Cipto Mangunkusumo Hospital experiences. Dipresentasikan pada Pertemuan Perhimpunan Hematologi dan Transfusi Darah Indonesia. Medan. 2011.
5. Bhatia S, Landier W, Hageman L, et al. Systemic exposure to thiopurines and risk of relapse in children with acute lymphoblastic leukemia: a children’s oncology group study. JAMA Oncol. 2015;1(3):287–295. doi:10.1001/jamaoncol.2015.0245
6. Fotoohi AK, Coulthard SA, Albertioni F. Thiopurines: factors influencing toxicity and response. Biochem Pharmacol. 2010;79:1211–1220. doi:10.1016/j.bcp.2010.01.006
7. Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther. 2013;89(3):1–5.
8. Kapoor G, Sinha R, Naithani R, et al. Thiopurine S-methyltransferase gene polymorphism and 6-mercaptopurine dose intensity in Indian children with acute lymphoblastic leukemia. Leuk Res. 2010;34(8):1023–1026. doi:10.1016/j.leukres.2010.01.029
9. Linga VG, Patchva DB, Mallavarapu KM. Thiopurine methyltransferase polymorphisms in children with acute lymphoblastic leukemia. Indian J Med Paediatr Oncol. 2014;35(4):276–280. doi:10.4103/0971-5851.144989
10. Ikatan Dokter Anak Indonesia. Indonesian protocol of acute lymphoblastic leukemia; 2013. Available from: https://kupdf.com/download/panduan-protokol-indonesia-all-2013_58e9fef1dc0d600d38da981c_pdf.
11. Hong EP, Park JW. Sample size and statistical power calculation in genetic association studies. Genome Inform. 2012;10(2):117–122.
12. Cancer therapy evaluation program, common terminology criteria for adverse events, version 3.0, DCTD, NCI, NIH, DHHS; 2003 Available from: http://ctep.cancer.gov.
13. Dervieux T, Meyer G, Barham R, et al. Liquid Chromatography–tandem mass spectrometry analysis of erythrocyte thiopurine nucleotides and effect of thiopurine methyltransferase gene variants on these metabolites in patients receiving azathioprine/6-mercaptopurine therapy. Clin Chem. 2005;51(11):2074–2084. doi:10.1373/clinchem.2005.050831
14. Rosdiana DS, Setiabudy R, Louisa M, et al. Development and method validation of 6-thioguanine and 6-methylmercaptopurine in erythrocytes using LC-MS/MS. J Pharm Sci Res. 2018;November(S):56–63.
15. Sahasranaman S, Howard D, Roy S. Clinical pharmacology and pharmacogenetics of thiopurines. Eur J Clin Pharmacol. 2008;64:753–767. doi:10.1007/s00228-008-0478-6
16. Levinsen M, Rosthøj S, Nygaard U, et al. Myelotoxicity after high-dose methotrexate in childhood acute leukemia is influenced by 6-mercaptopurine dosing but not by intermediate thiopurine methyltransferase activity. Cancer Chemother Pharmacol. 2015;75(1):59–66. doi:10.1007/s00280-014-2613-7
17. Tanaka Y, Kato M, Hasegawa D, et al. susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol. 2015;171:109–115. doi:10.1111/bjh.13518
18. Takatsu N, Matsui T, Murakami Y, et al. Adverse reactions to azathioprine cannot be predicted by thiopurine S-methyltransferase genotype in Japanese patients with inflammatory bowel disease. J Gastroenterol Hepatol. 2009;24:1258–1264. doi:10.1111/j.1440-1746.2009.05917.x
19. Uchiyama K, Nakamura M, Kubota T, et al. Thiopurine S-methyltransferase and inosine triphosphate pyrophosphohydrolase genes Japanese patients with inflammatory bowel disease in whom adverse drug reactions were induced by azathioprine/6mercaptopurine treatment. J Gastroenterol. 2009;44:197–203. doi:10.1007/s00535-008-2307-1
20. Zhu Q, Cao Q. Thiopurine methyltransferase gene polymorphisms and activity in Chinese patients with inflammatory bowel disease treated with azathioprine. Chin Med J (Engl). 2012;125:3665–3670.
21. Cao Q, Zhu Q, Shang Y, et al. Thiopurine methyltransferase gene polymorphisms in Chinese patients with inflammatory bowel disease. Digestion. 2009;79:58–63. doi:10.1159/000205268
22. Lee JH, Kim TJ, Kim ER, et al. Measurements of 6-thioguanine nucleotide levels with TPMT and NUDT15 genotyping in patients with Chron’s disease. PLoS One. 2017;12(12):1–13. doi:10.1371/journal.pone.0188925
23. Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine s-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi:10.1093/jnci/91.23.2001
24. Kubota T, Chiba K. Frequencies of thiopurine S-methyltransferase mutant alleles (TPMT*2, *3A, *3B and *3C) in 151 healthy Japanese subjects and the inheritance of TPMT*3C in the family of a propositus. Br J Clin Pharmacol. 2001;51(5):475–477. doi:10.1046/j.1365-2125.2001.01371.x
25. Lennard L, Cartwright CS, Wade R, et al. Thiopurine methyltransferase genotype-phenotype discordance and thiopurine active metabolite formation in childhood acute lymphoblastic leukaemia. Br J Clin Pharmacol. 2012;7(1):125–136.
26. Ladislava B, Michal K, Katerina W, et al. Gene polymorphisms and inflammatory bowel diseases. In: Szabo I, editor. Inflammatory Bowel Disease. Chapter 2. 2012.
27. Lennard L, Cartwight S, Wade R, et al. Thiopurine methyltransferase genotype-phenotype discordance and thiopurine active metabolite formation in childhood acute lymphoblastic leukemia. Br J Clin Pharmacol. 2012;76(1):125–136. doi:10.1111/bcp.12066
28. Kakuta Y, Kinouchi Y, Shimosegawa T. Pharmacogenetics of thiopurines for inflammatory bowel disease in east asia: prospects for clinical application fo NUDT15 genotyping. J Gastroenterol. 2018;53:172–180. doi:10.1007/s00535-017-1416-0
29. Ho CC, Fong WY, Lee YH, et al. Novel tetra-primer ARMS-PCR assays for thiopurine intolerance susceptibility mutations NUDT15 c.415C>T and TPMT c.719A>G (TPMT*3C) in East Asians. Genes. 2017;285(8):1–8.
30. Chrzanowska M, Kolecki P, Cichocka BD, et al. Metabolites of mercaptopurine in red blood cell: a relationship between 6-thioguanine nucleotides and 6-methylmercaptopurine metabolite concentrations in children with lymphoblastic leukemia. Eur J Pharm Sci. 1999;8:329–334. doi:10.1016/S0928-0987(99)00027-5
31. Lennard L, Lilleyman JS. Variable mercaptopurine metabolism and treatment outcome in childhood lymphoblastic leukemia. J Clin Oncol. 1990;8(3):567–574.
32. Sheffield LJ, Irving P, Gupta A, et al. Thiopurine methyltransferase and thiopurine metabolite testing in patients with inflammatory bowel disease who are taking thiopurine drugs. Pharmacogenomics. 2009;10(7):1091–1099. doi:10.2217/pgs.09.60
33. Kuzelicki NK, Rascan IM. Individualization of thiopurine therapy: thiopurine S-methyltransferase and beyond. Pharmacogenomics. 2009;10(8):1309–1322.
34. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) assessment report of mercaptopurine. 2011.
35. Dervieux T, Hancock M, Evans W, et al. Effect of methotrexate on thioguanine nucleotide concentrations during continuation therapy of acute lymphoblastic leukemia with mercaptopurine. Leukemia. 2002;16:209–212. doi:10.1038/sj.leu.2402373
36. Goel RM, Blaker P, Mentzer A, et al. Optimizing the use of thiopurines in inflammatory bowel disease. Ther Adv Chronic Dis. 2015;6(3):138–146. doi:10.1177/2040622315579063
37. Broekman MMTJ, Coenen MJH, Wanten GJ, et al. Risk factors for thiopurine-induced myelosuppression and infections in inflammatory bowel disease patients with a normal TPMT genotype. Aliment Pharmacol Ther. 2017;46:953–963. doi:10.1111/apt.14323
38. Relling MV, Schwab M, Carrillo MW, et al. Clinical pharmacogenetics implementation consortium guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin Pharmacol Ther. 2019;1–11.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.