Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
Townes-Brocks Syndrome as Familial Isolated Bilateral Cup Ear Deformity: A Case Report
Authors Chang Y ![]() , Yang L, Zhang Z, Zhang H, Xue F
, Yang L, Zhang Z, Zhang H, Xue F
Received 13 February 2026
Accepted for publication 26 May 2026
Published 18 June 2026 Volume 2026:19 603309
DOI https://doi.org/10.2147/CCID.S603309
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Anne-Claire Fougerousse
Yanbin Chang,1,2,* Lishuo Yang,1,2,* Zihan Zhang,3 Huayu Zhang,3 Feng Xue3
1Graduate Department of Shandong First Medical University, Jinan, Shandong, People’s Republic of China; 2Department of Burns, Plastic and Aesthetic Surgery, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, People’s Republic of China; 3Department of Auricular Reconstruction and Plastic Surgery, The Second Qilu Hospital of Shandong University, Jinan, Shandong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Feng Xue, Department of Auricular Reconstruction and Plastic Surgery, The Second Qilu Hospital of Shandong University, Jinan, Shandong, People’s Republic of China, Email [email protected]
Abstract: Townes-Brocks syndrome (TBS) is a rare autosomal dominant disorder caused by pathogenic variants in SALL1. Although classically characterized by external ear anomalies, anorectal malformations, and thumb/radial defects, its expressivity is highly variable and mild presentations may be overlooked. We report a 7-year-old boy presenting for cosmetic correction of bilateral cup ear deformities. Initial hearing screening and renal ultrasonography showed no abnormalities, and no other anomalies were identified on clinical examination. Detailed history-taking and construction of a three-generation pedigree revealed that the patient’s father and paternal grandfather had similar bilateral auricular deformities, suggesting autosomal dominant familial transmission. Genetic testing of the proband and father demonstrated a heterozygous frameshift variant in exon 2 of SALL1 (c.1949del; p.Gly650AlafsTer46). The proband and his father underwent corrective otoplasty and achieved satisfactory early cosmetic outcomes. This family illustrates that SALL1-related TBS can present as an apparently isolated auricular phenotype. Familial congenital cup ear deformity, even when encountered in a cosmetic setting, should prompt careful family history-taking, consideration of genetic evaluation, counseling, and longitudinal surveillance.
Keywords: Townes-Brocks syndrome, SALL1, cup ear, genetic counseling
Introduction
Townes–Brocks syndrome (TBS; OMIM 107480) is an autosomal dominant developmental disorder traditionally defined by the triad of external ear anomalies, thumb/radial malformations, and anorectal abnormalities.1,2 The phenotype is highly variable, even within the same family, and additional manifestations can include hearing impairment, renal anomalies and dysfunction, congenital heart disease, genitourinary anomalies, and neurodevelopmental differences.3
SALL1 encodes a C2H2 zinc-finger transcriptional regulator involved in developmental programs across multiple organ systems.4 Most individuals with TBS carry heterozygous pathogenic variants in SALL1, and reported variants are predominantly truncating (nonsense/frameshift) or splice-site changes consistent with a loss-of-function mechanism.5 Because SALL1-related TBS shows broad phenotypic variability, the presence of a pathogenic variant alone cannot reliably predict the specific manifestations or clinical severity in an affected individual.6
Here, we report a three-generation family in which a bilateral constricted (cup) ear deformity was the predominant and, at presentation, isolated manifestation. This case underscores the importance of obtaining a detailed family history and conducting a genetic assessment in individuals presenting for cosmetic repair of congenital auricular malformations.
Case Presentation
Patient Information
A 7-year-old male patient was evaluated for the correction of bilateral congenital cup ear deformities. The patient’s guardian reported a history of normal growth and development, with no prior anorectal surgery, chronic constipation, urinary symptoms, recurrent infections, or other congenital anomalies.
Clinical Findings
Physical examination revealed bilateral constricted auricles with superior helical rim folding and reduced upper auricular height. A left preauricular accessory tag was present (Figure 1). No obvious thumb/radial anomalies or other dysmorphic features were identified on examination.
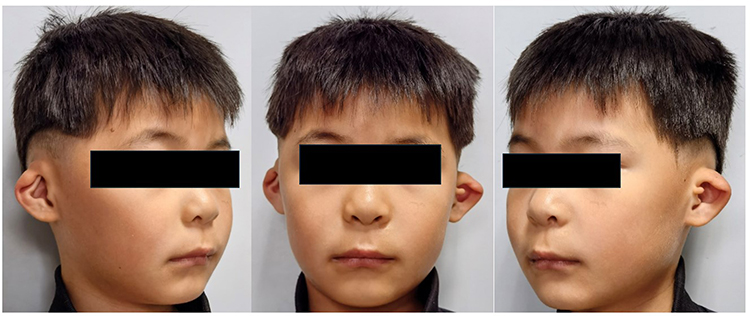 |
Figure 1 Preoperative photographs showing bilateral cup auricles in the proband, with a left preauricular accessory tag. |
Family History
The patient’s father had a similar bilateral auricular phenotype (Figure 2), and the paternal grandfather reportedly had the same congenital deformity. The mother and younger sister had normal auricular morphology. The family denied a history of anorectal malformations, limb deformities, or known renal disease among affected relatives.
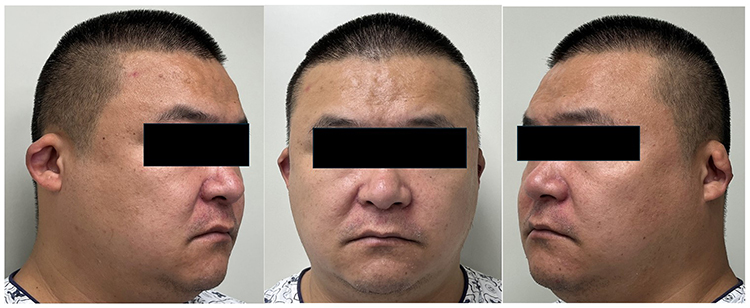 |
Figure 2 Auricular deformity in the proband’s father, showing a similar congenital cup ear phenotype. |
Diagnostic Assessment
Given the observed familial clustering and suspected hereditary predisposition, a three-generation pedigree was constructed (Figure 3). Genetic analysis of the proband and his father revealed a heterozygous frameshift variant in exon 2 of SALL1 (NM_002968.3:c.1949del; p.Gly650AlafsTer46), which was classified as pathogenic and was identified in both individuals. Initial clinical assessments, including office-based hearing screening and renal ultrasonography, revealed no abnormalities at the time of diagnosis. Genetic counseling was provided, emphasizing that hearing loss and renal impairment in TBS may exhibit delayed onset or progressive deterioration, and longitudinal surveillance was recommended.
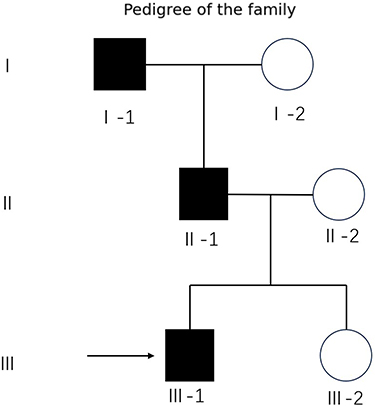 |
Figure 3 Pedigree of the family with Townes–Brocks syndrome. |
Therapeutic Intervention and Outcome
The proband underwent one-stage bilateral correction of the constricted auricles via posterior auricular incisions. The skin was elevated to expose the deformed cartilage. After release of the constricted segments, cartilage scoring and suture techniques were used to recreate a stable antihelical fold and improve upper auricular projection. Excess skin was tailored and the incisions were closed in layers. The preauricular tag was excised. Early follow-up demonstrated improved auricular contour and symmetry without recurrent helical rim collapse (Figure 4). The father underwent a similar corrective procedure and reported satisfaction with the cosmetic outcome.
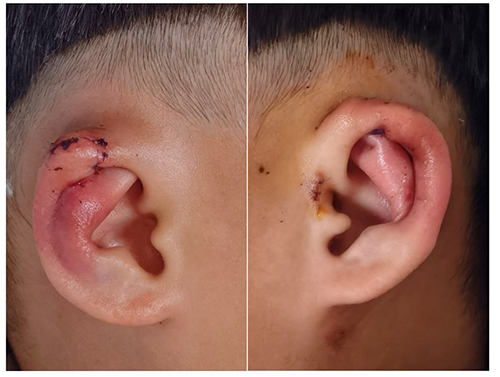 |
Figure 4 Postoperative photographs at showing improved auricular contour and symmetry after one-stage bilateral otoplasty. |
Discussion
TBS is typically recognized by the triad of anorectal malformation, thumb/radial anomalies, and dysplastic ears. However, many affected individuals do not manifest the complete triad, and mild phenotypes can be overlooked, particularly when patients present with a single apparently isolated feature such as an external ear deformity.
Recent cohort-level studies provide context for the present family. In a large Chinese hearing-loss cohort, Yan et al identified genetically confirmed TBS cases and emphasized that the disorder may be underdiagnosed when the full classical triad is absent.7 Our case further confirms this observation. Leduc et al reported a series of 49 patients and reviewed 207 previously reported SALL1-related TBS patients; only 25% of their series and 49.7% of evaluable literature cases had the complete triad, while dysplastic ears were among the most frequent findings.8 Stein et al similarly showed that kidney gene panel testing can reveal SALL1-related TBS in individuals whose initial presentation is renal rather than dysmorphic.9 Compared with these hearing- and renal-ascertained cohorts, our family is unusual because the first medical contact was for cosmetic auricular correction and because hearing and renal screening were normal at presentation. Thus, the key diagnostic clues were bilateral cup ear deformity and an autosomal dominant family history.
The frameshift variant identified in this family (NM_002968.3:c.1949del; p.Gly650AlafsTer46) is predicted to introduce a premature termination codon. Truncating SALL1 variants are a common molecular mechanism in TBS. Mechanistically, SALL1 disruption can alter transcriptional programs during organogenesis, and truncated SALL1 proteins may perturb downstream gene expression.10 Nonetheless, current genotype-phenotype correlations remain incomplete; therefore, the mild auricular-predominant presentation in this family should not be interpreted as evidence that future hearing, renal, or other systemic manifestations are impossible.
In the present family, the phenotype was dominated by congenital external ear anomalies, with normal baseline hearing screening and renal ultrasonography in the proband. Although reassuring, normal early screening does not exclude future complications. Hearing impairment and renal dysfunction in TBS may be delayed or progressive; therefore, periodic audiologic evaluation and renal surveillance, including renal ultrasonography and renal function testing when clinically appropriate, should be considered even in individuals with mild phenotypes.11
Congenital cup ear is characterized by deficiency and inward folding of the upper third of the auricle.12 Aesthetic correction may involve cartilage release, scoring, and suture-based reshaping, with or without cartilage grafting, depending on severity.13 In our patient, a one-stage posterior approach achieved satisfactory early cosmetic results. Importantly, identifying an underlying syndromic diagnosis may not alter the basic surgical principles of auricular reconstruction, but it expands the clinical significance of treatment by prompting broader perioperative assessment, genetic counseling, screening for associated complications, and long-term follow-up.
When external ear anomalies cluster in families, TBS should be considered alongside other genetic conditions with overlapping otologic phenotypes (eg, branchio-oto-renal syndrome and oculo-auriculo-vertebral spectrum) Confirmatory genetic testing can clarify diagnosis, guide counseling regarding autosomal dominant transmission, and prompt appropriate screening of at-risk relatives.
This report has several limitations. First, as a single-family case report, it cannot determine the frequency of isolated cup ear deformity among individuals with SALL1-related TBS. Second, genetic testing was performed in the proband and father, but not in the paternal grandfather or unaffected relatives; therefore, segregation in the wider pedigree remains partly inferred from phenotype. Third, postoperative follow-up was short, so long-term auricular stability and patient-reported outcomes require continued observation. Finally, baseline hearing and renal findings were normal, but delayed-onset manifestations remain possible and require surveillance.
Conclusion
This three-generation familial case demonstrates that SALL1-related Townes–Brocks syndrome may present predominantly as an apparently isolated bilateral cup ear deformity. For patients seeking cosmetic correction of congenital auricular anomalies, especially when familial clustering is present, a detailed family history and appropriate genetic evaluation can facilitate early recognition of mild syndromic disease, inform counseling regarding autosomal dominant inheritance, guide screening of at-risk relatives, and support long-term audiologic, renal, and postoperative follow-up.
Ethics Approval
This report complies with the Helsinki Declaration of Ethical Principles. The Ethics Committee of The Second Qilu Hospital of Shandong University, has approved the publication of the case details.
Consent for Publication
Written informed consent for publication of the clinical details and accompanying images was obtained from the legal guardian of the minor participant and from the adult participant (father).
Funding
This work was supported by the Wu Jieping Medical Foundation Clinical Research Special Grant (Grant No. 320.6750.2025-25-5).
Disclosure
Yanbin Chang and Lishuo Yang are co-first authors for this study. The authors report no conflicts of interest in this work.
References
1. Townes PL, Brocks ER. Hereditary syndrome of imperforate anus with hand, foot, and ear anomalies. J Pediatr. 1972;81(2):321–5. doi:10.1016/s0022-3476(72)80302-0
2. Powell CM, Michaelis RC. Townes-Brocks syndrome. J Med Genet. 1999;36(2):89–93.
3. Wang Z, Sun Z, Diao Y, et al. Identification of two novel SALL1 mutations in Chinese families with townes-brocks syndrome and literature review. Orphanet J Rare Dis. 2023;18(1):250. doi:10.1186/s13023-023-02874-4
4. Asagai Y, Tanaka Y, Hanafusa H, et al. Clinical characteristics of patients with SALL1-related disorder. Pediatr Nephrol. 2025;40(11):3407–3414. doi:10.1007/s00467-025-06878-z
5. Kohlhase J, Taschner PE, Burfeind P, et al. Molecular analysis of SALL1 mutations in Townes-Brocks syndrome. Am J Hum Genet. 1999;64(2):435–445. doi:10.1086/302238
6. Botzenhart EM, Green A, Ilyina H, et al. SALL1 mutation analysis in Townes-Brocks syndrome: twelve novel mutations and expansion of the phenotype. Hum Mutat. 2005;26(3):282. doi:10.1002/humu.9362
7. Yan X, Wang J, Yang W, et al. Molecular diagnosis, clinical evaluation and phenotypic spectrum of Townes-Brocks syndrome: insights from a large Chinese hearing loss cohort. J Med Genet. 2024;61(5):459–468. doi:10.1136/jmg-2023-109579
8. Leduc F, Brunelle P, Escande F, et al. Townes-Brocks syndrome: genotype-phenotype correlations of SALL1 variants in our series and the literature. Eur J Hum Genet. 2025;33(11):1442–1450. doi:10.1038/s41431-025-01855-4
9. Stein Q, Vostrizansky A, Magay Y, et al. Townes-Brocks syndrome revealed by kidney gene panel testing. Kidney Int Rep. 2024;9(6):1810–1816. doi:10.1016/j.ekir.2024.03.030
10. Kiefer SM, Robbins L, Barina A, Zhang Z, Rauchman M. SALL1 truncated protein expression in Townes-Brocks syndrome leads to ectopic expression of downstream genes. Hum Mutat. 2008;29(9):1133–1140. doi:10.1002/humu.20759
11. Graziano C, Olivucci G. SALL1-Related Townes-Brocks Syndrome. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews®. Seattle: University of Washington, Seattle; 2007.
12. Tanzer RC. The constricted (cup and lop) ear. Plast Reconstr Surg. 1975;55(4):406–415.
13. Huang X, Ma C, Chang J, et al. Classification and surgical strategies of constricted ears in a chinese specialty clinic: a retrospective study. Aesthetic Plast Surg. 2022;46(5):2194–2207. doi:10.1007/s00266-021-02699-1
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.