Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
Towards Precision Medicine in Systemic Lupus Erythematosus
Authors Lever E ![]() , Alves MR, Isenberg DA
, Alves MR, Isenberg DA
Received 29 October 2019
Accepted for publication 14 January 2020
Published 4 February 2020 Volume 2020:13 Pages 39—49
DOI https://doi.org/10.2147/PGPM.S205079
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Elliott Lever,1 Marta R Alves,2 David A Isenberg1
1Centre for Rheumatology, Division of Medicine, University College Hospital London, London, UK; 2Internal Medicine, Department of Medicine, Centro Hospitalar do Porto, Porto, Portugal
Correspondence: David A Isenberg
Centre for Rheumatology, Division of Medicine, University College Hospital London, Room 424, the Rayne Building, 5 University Street, London WC1E 6JF, UK
Email [email protected]
Abstract: Systemic lupus erythematosus (SLE) is a remarkable condition characterised by diversity amongst its clinical features and immunological abnormalities. In this review, we attempt to capture the major immunological changes linked to the pathophysiology of lupus and discuss the challenge it presents in moving towards the concept of precision medicine. Currently broadly similar types of drugs, e.g., steroids, immunosuppressives, hydroxychloroquine are used to treat many of the diverse clinical features of SLE. We suspect that, as the precise immunopathological abnormalities differ between the various organs/systems in lupus patients, it will be some time before precision medicine can be fully applied to SLE.
Keywords: systemic lupus erythematosus, anti-DNA antibodies, lupus nephritis, lupus genetics, immunosuppression
Introduction
Systemic lupus erythematosus (SLE) is a chronic, complex, auto-immune disease of unknown origin with multiorgan involvement. Knowledge of its epidemiology, genetic susceptibility and pathophysiologic mechanisms has evolved, but it remains a disease with a highly variable course and considerable inter-individual variability. It has an unpredictable prognosis, depending mainly on the severity of the disease activity, organ damage and response to treatment. SLE is characterized by loss of self-immune tolerance, production of self-reacting antibodies and formation of immune complexes (IC) that precipitate in tissues, causing chronic systemic inflammation and organ damage.1 It affects primarily women (90%) during childbearing years and is more prevalent in non-white populations. It is a disease with an important genetic linkage, as data suggest that concordance in monozygotic twins is 10 times higher than in dizygotic twins, an important epigenetic role is likely in disease pathogenesis. Beside the endogenous factors (sex, age, hormones), it is also known that environmental factors (psychological stress, viral infections, smoking, chemicals, nutrition) influence the disease course and may trigger it.2–7
Several sets of classification criteria have been developed. In 2012 Systemic Lupus International Collaborating Clinics (SLICC) developed a set of criteria8 shown to have a higher sensitivity than the American College of Rheumatology’s revised criteria (1997),9 although with slighter lower specificity. In 2017 European League against Rheumatism (EULAR) and American College of Rheumatology (ARC) developed the ACR-EULAR classification criteria.10 Studies have compared the performance of SLICC criteria (sensitivity between 85% and 96.7%; specificity 76% and 83.6%) and ARC-EULAR (sensitivity between 87% and 96.3%; specificity 74% and 93.4%).11–13 Both criteria performed worse in one of the studies, this, according to the authors, being due to a more selected population with several confounding factors. Thus, in more challenging situations, when classification criteria may be more useful, the performance can be sub-optimal.
Treatment of SLE is based on the use of antimalarials, shown to be beneficial in most SLE patients, glucocorticoids (GC) and immunosuppressants (IS). Treatment is individualized to some extent, according to organ involvement and having as a primary objective, remission of disease signs and symptoms in order to prevent organ damage. Complete clinical and immunological remission (no clinical activity with normal ds-DNA antibody and C3 levels, on no corticosteroid or immunosuppressive treatment) is limited. In a study of more than 600 SLE patients, followed up for more than 30 years only 14% achieved full remission for 3 years and 20% relapsed thereafter.14 Therefore, according to the most recent EULAR guidelines,13 another treatment target is now a low-disease activity state, defined by a SLE disease activity index (SLEDAI) score ≤3, on antimalarials, or alternatively [Systemic Lupus Erythematosus Disease Index (SLEDAI) Score] ≤4, physician global assessment (PGA) ≤1 with glucocorticoid therapy ≤7.5 mg of prednisone and well-tolerated IS agents.
Precision medicine consists of a tailored approach to each patient, based on genetic and epigenetic singularities, which influence disease pathophysiology and drug response. Precision medicine in SLE is trying to address the need to assess SLE patients optimally, predict disease course and treatment response at diagnosis. Ideally every patient would undergo an initial evaluation that would profile his/her disease, assessing the main pathophysiologic pathway through biomarkers, therefore predicting risk of specific organ damage, most adequate treatment, and would allow better follow-up and flare prediction.
In this review, we will outline the pathological processes in lupus in general terms with particular emphasis on neuropsychiatric and renal involvement. Epigenetic, microbiome and environmental aspects are also considered. The current treatment approach is addressed and a perspective to evolve precision with respect to clinical appraisal of disease activity and management is envisaged. There is exciting scope towards improving precision in lupus care, but there is unlikely to be a short-term universal achieving of this goal.
SLE Immunobiology
As stated above, SLE is complex disease whose precise pathophysiology remains uncertain. It results from an interaction of genetic susceptibility, epigenetic modification of the genome, endocrine factors and one or more triggers. One central concept in SLE pathogenesis is an imbalance between apoptotic cell production and inefficient apoptotic material disposal, through increased production and/or decreased clearance.1 Known risk factors, such as exposure to ultraviolet light, toxins and infections, lead to increased apoptotic burden, supporting this hypothesis.15 Microparticles are small membrane vesicles circulating in blood, that detach from apoptotic cells. These particles contain cellular constituents derived mainly from activated platelets. Microparticles from nucleated cells contain chromatin that can react with anti-DNA antibodies to form immune complexes (IC). Microparticles also exist in healthy individuals, although differences have been noticed, compared to SLE patients. These particles may provide the nuclear autoantigens that induce anti-nuclear antibodies (ANA) and support the theory of imbalance in apoptotic material in the genesis of SLE. Neutrophils are also involved in SLE pathophysiology.16 These cells, in SLE patients, were shown to have defective phagocytosis capacity which might contribute to the increased susceptibility to infection in these patients. Neutrophils have a short lifespan and contribute largely to the apoptotic cell burden. Defective production of reactive oxygen species (ROS) may contribute to SLE, and one study correlated their production with disease severity and organ damage.17 SLE patients have an abnormal subset of neutrophils that exhibit increased NETosis.18,19 This mechanism of cell death occurs in response to several stimuli and consists of extrusion of cytoplasmatic and nuclear material, then called neutrophil extracellular trap (NET). NETosis contributes to the type 1 INF response, stimulating its production by pDC, via TLR9.20 Type 1 INF cytokines also induce NETosis, suggesting a positive feedback loop. Extruded nuclear material provides antigens that may result in antibody production.
One of the most important advances in SLE knowledge was the discovery of the innate immune system participation.21–23 It has been recognised that nucleic acids and IC containing nucleic acids occur in the intracellular medium, outside the nucleus, via both TLR and TLR-independent nucleic acid sensors and can trigger the production of type 1 INF and other cytokines.24
Several studies have implicated the Toll-like receptor (TLR) family involvement in SLE pathogenesis.25 TLR are expressed by both immune cells (macrophages, B and T cells, dendritic cells) and non-immune (epithelial cells and fibroblasts). TRL3, TLR7, TLR8, TLR9 are stored in the endoplasmic reticulum and can travel to endosomes where they exercise their recognition function. Both TLR9 and TLR7 trigger a strong interferon (INF) response. TLR7 recognises single-stranded RNA and was shown to be implicated in RNA-reactive antibodies production and glomerulonephritis (GN) development in a mouse model. TLR9 recognises DNA containing unmethylated CpG sequence motifs. It has been demonstrated that active SLE patients develop a higher number of TLR9 expressing B-cells and macrophages, in contrast to low activity SLE patients, and levels of these cells correlated with ds-DNA antibody levels.25
Several cells of the immune system are thought to be involved in SLE pathogenesis, and some of them belong to the innate immune system. Dendritic cells (DC) are effective antigen presenting cells and malfunction of this step in the immune response may cause the loss of self-tolerance of B and T-cells in auto-immune diseases.26 In SLE patients the DC population has well-established abnormalities thus there is an increased population of plasmacytoid DC (pDC), which produce type 1 INF in response to nucleic acid via TLR7 and TLR 9 activation, a reduced conventional DC population which, in SLE, promote autoreactivity instead of self-tolerance.27
The complement system also participates in SLE.28 Complement consumption is one of the markers for SLE activity as inflammation leads to complement system activation and, among other functions, opsonization of IC. Genetic complement deficits, notably C1q deficit, are linked to a lupus-like disease. This complement molecule is responsible for the initiation of classical pathway and apoptotic cell clearance, without initiating an inflammatory response. Autoantibodies against complement have also been linked to SLE. Complement was shown to interact with B-cells, during their development, negatively selecting those who react with autoantigens. Much of the complement-B cell interaction occurs via complement receptor 2 (CR2), expressed on B cells. Polymorphisms in this receptor can alter self-tolerance. This receptor expression is lower in SLE patients. Furthermore, pDC production of INF type 1 responses is lower when the IC are coated with C1q. In addition, C1q also helps in NET clearance by macrophages. The evidence thus points to a possible role for complement in SLE pathophysiology. Other Complement protein deficits linked to SLE, include C2 and C4.
With respect to the adaptive immune system, T lymphocytes are closely related to the major histocompatibility complex (MHC) proteins and are, therefore, considered central to SLE pathophysiology.29 Loss of self-tolerance by T cells is pivotal in all auto-immune diseases and may occur in the thymus or peripherally. Defects in lymphocyte signalling may alter thymic education. T cells in SLE patients have an aberrant T cell receptor (TCR) signalling that is not cell intrinsic, it can be induced by SLE serum IgG.30 Lupus T cells express CD40 ligand after activation (an important co-stimulatory molecule) and maintain this expression longer than control T cells. These hyperactive T-cells are Th17 polarised. IL-2 production is also impaired and Treg cells, which are IL-2 dependent, are deficient or defective.31,32 An expansion of follicular helper T cells (ThF) that produce IL-21, a cytokine that promotes B-cell differentiation has been noticed. ThF cells can be found within lymphoid aggregates in kidney biopsy samples from active lupus GN, and antibody titres, in these patients, correlate with activated ThF cells.33 Interactions between T and B cells were also found altered in SLE patients.34 These encounters occur outside their usual locations, in secondary lymphoid organs.35
B cells, which are responsible for antigen response, the regulation of other cells, antibody and cytokine production, also have a central role in SLE.36 Targeting B cells, with belimumab through BLyS/BAFF blockage, or B cell depletion with rituximab, are known to be an effective way to treat SLE patients.37 Studies that target SLE B-cells found that their regulation is impaired. Both baseline activation status and response to antigen stimulation is altered, when compared with healthy controls. Activated SLE B cells, are capable of promoting self-antigen presentation to T cells. Early immature B cells show increased autoreactivity in SLE patients, raising the question of whether loss of tolerance happens in central B-cells. Some cytokines, particularly BLyS, may also induce loss of tolerance. Furthermore, B-cells that secrete IL-10, a regulatory cytokine, are functionally impaired. Loss of tolerance is not an all-or-nothing phenomenon as it was shown that “lupus antibodies” may develop up to 10 years prior to SLE clinical manifestations.38 These antibodies have fairly constant levels and are refractory to immunosuppressive therapies, including B cell depletion. Anti-dsDNA and anti-phospholipid antibodies are believed to be produced by circulating plasma cells and plasmablasts. These antibodies fluctuate over time and, in most patients, the anti-dsDNA antibodies relate to disease activity. B-cell depletion reduces their levels efficiently.
However, the precise pathophysiology of lupus is not fully established and is varies from organ to organ. In the next section, we will focus on two organs to discuss representative pathophysiology.
Lupus Nephritis Pathophysiology
Clinically significant glomerulonephritis (GN) develops in up to 50% of SLE patients and is a major cause of morbidity and mortality. Afro-Caribbean patients are more likely to get lupus nephritis (LN).39
The International Society of Nephrology/Renal Pathology Society (ISN/RPS) classification of GN focuses primarily on glomerular lesions.40 These classes are associated with prognosis and dictate the treatment to some extent. Class I and II refer to mesangial nephritis. Due to the high-regenerative capacity of mesangial cells, these GN classes have better prognosis than type III and IV.41 Proliferative GN is class III (focal) and IV (diffuse), occurs with subendothelial deposition of ICs of the glomerular capillaries, leading to endothelial activation. There is also germinal centre formation. In class V, the membranous IC deposition in the subepithelial space, causes local inflammation and, ultimately, podocyte damage. Tubulointerstitial inflammation does not always co-exist with glomerular lesions. The severity of tubulointerstitial inflammation correlated better with renal outcome. Several studies have addressed tubulointerstitial injuries in LN patients.42
There is a complex network of processes leading to LN. Endothelial activation through direct damage or reactive oxygen species (ROS) causes complement activation and cellular recruitment. Recent single-cell transcriptome analysis shows multiple lineages of innate and adaptive cells are active locally in the renal parenchyma.43 B cells are activated locally and undergo somatic hypermutation, clonal expansion, and intrarenal production of autoantibodies. Costimulation through DC, macrophage, B and T cell interactions are essential in the LN pathogenic network. Type 1 interferons are produced by resident and recruited cells in renal lupus and associated with damage. Immune complexes and anti-DNA antibodies aggregate along the tubular basement membrane and bind podocytes. It is thought that free DNA may be involved in the binding of the antibody from defective NETosis.44 More recently microRNAs have been implicated. Low level of a microRNA in LN was found to cause dysregulation of the podocyte cytoskeleton and another causing deficiency in E-cadherin and intercellular adhesion.45
Chronic inflammation leads to cell death. Cellular regeneration and laying down of fibrotic material, leads to glomerulosclerosis. In LN any cell in the kidney may be damaged including the podocytes. Several studies of podocyte injury in LN found that podocyte injury correlated with the degree of proteinuria, and renal outcome.46 Morphologically these podocyte injuries are similar to minimal change disease or focal segmental glomerulosclerosis (FSGS). These biopsies show extensive podocyte injury, without evidence of IC deposition.
Concomitant with glomerular inflammation, vascular rarefaction and tubular infarction also contribute to glomerulosclerosis. This process eventually leads to end-stage renal disease. Several vascular lesions are commonly observed in LN biopsies: vascular immune IC deposits, arteriosclerosis, thrombotic microangiopathy (TMA), non-inflammatory necrotizing vasculopathy and true renal vasculitis. Studies comparing vascular damage in renal biopsies found that the type of vascular lesion correlated with renal outcome and that vasculopathy correlated with disease activity.47
Neuropsychiatric SLE Pathophysiology
Neuropsychiatric SLE (NPSLE) encompasses a group of central and peripheral nervous system syndromes, and its diagnosis may pose a clinical challenge.48 NPSLE manifestations vary from nonspecific common symptoms (headaches, mood disorders, cognitive dysfunctions) that may be difficult to attribute to NPSLE, to rare syndromes (such as autonomic dysfunction). Seizures and psychosis are the most serious CNS manifestations often occurring in the presence of activity in other organs/systems.
NPSLE pathophysiology is poorly understood. Several possible pathophysiologic mechanisms including vasculitis and cross-reactive antibodies contribute to nervous system damage. In general, NPSE may be divided in central nervous system (CNS) damage, which may be focal or diffuse, and peripheral nervous system (PNS) damage. Focal damage is frequently secondary to cerebrovascular disease, mainly due to accelerated atherosclerosis and/or anti-phospholipid syndrome (APS). Diffuse CNS damage thought to be mainly due to IC deposition/formation in situ. The entrance of (auto)antibodies, activated immune cells and cytokine in the CNS is thought to imply a blood-brain barrier (BBB) dysfunction or loss of integrity, as it is typically antibody impermeable. However, both meninges and choroid plexus, where cerebrospinal fluid (CSF) is produced, are richly vascularized and may be another point of immune interaction.
Anti-phospholipid antibodies correlate with cerebrovascular disease and focal NPSLE. A subset of anti-dsDNA antibodies cross reacts with the N-methyl-D-asparte receptor (NDMAR) and its presence in the CSF is frequently associated with diffuse CNS damage. Anti-ribosomal P protein antibodies have also been found to relate to NPSLE in some, but not all, reports. Anti-Sm antibodies have also been shown to cross-react with ribosomal P protein.49
Cellular-induced inflammation might also contribute to NPSLE.50 CNS resident microglia are potent INF type 1 cytokine producers and were found aberrant in mouse models.
Several cytokines that participate in inflammation (as TNF, IL1, IL4, IL17, INFγ) also have roles of synaptic modulation, in normal homeostatic conditions, and are involved in high neurological superior functions (such as memory acquisition, social skills).51 Some studies demonstrate that pathological elevation of these cytokines induce neurological dysfunctions (mainly in mouse models). NPLE CSF studies indicate INFα and IL6 as possibly related to CNS diffuse damage52,53.
SLE Genetics and Epigenetics
Although a strong genetic association was always evident, little was known of SLE genetics until the development of the genome wide association studies (GWAS).54,55 These studies are hypothesis-free genome screening that link gene loci with disease phenotype in multifactorial diseases, as SLE. Prior to GWAS, a few genes were known to be associated to SLE, as is the case of the human leukocyte antigen (HLA) haplotypes. The most ancient gene to be a known associate with SLE is the HLA, specifically the DRB1 and BQA1 loci, that encode part of the MHC class II proteins. Several studies demonstrated positive haplotype association with SLE (HLA-DR3; DR9; DR15; DQA1*0101), with LN (DQA1*0101; DR3; DR15) and possible negative associations, therefore possible protective haplotypes (HLA-DR4, DR11, DR14).
Epigenetic events seem likely to play an important role in the pathophysiology of lupus. In particular (CpG DNA methylation, histone modifications and non-coding RNAs are likely to be critically involved.56,57
Most of the genes that are associated with SLE, with known function, can be sub-divided according to one of four-key molecular pathways: genes that affect lymphocyte activation; genes related to innate immune activation and signalling; genes related to handling of apoptotic debris, chromatin and IC; genes related to a specific organ damage in SLE. Some genetic variants may fall within more than one category.
Flares in lupus are probably linked to epigenetic disequilibrium in a multifaceted and multi-hit fashion. Studies of DNA methylation have found reduced CpG island methylation in lupus at type 1 interferon genes and GWAS loci including MHC genes.58,59 In contrast, there is increased methylation of IL-2 and FOXP3 required for regulatory T cell maturation. DNA methylation has a distinct tissue signature. In lupus where selective targeting of organ systems occurs, it can be inferred that differential methylation, whether local at the tissue or as de novo methylation cause organ selectivity in lupus. MicroRNAs are intimately linked to DNA methylation. They are mechanistically linked to de novo methylation and differentially expressed in lupus active, inactive and healthy individuals. An example is miR-146a which is linked to type-1 interferon gene expression. Histone modification and other architectural changes to chromatin have an established association with the disease state of lupus.60 The conformation of DNA and accessibility of transcription factors determines cell and tissue level gene regulation. In patients with SLE, CD4+ cells are globally H3 and H4 hypo-acetylated. Hypo-acetylation is associated with active chromatin. Specific histone modifications are also found at important immunoregulatory genes.
The environment has been proposed to interact at the epigenetic level in lupus. Ultraviolet exposure is associated with reduced DNTM1 expression from SLE patients.61 There are extensive associations with vitamin D both correlating serum levels and vitamin D-receptor genomic binding sites.62 Epstein-Barr Virus viraemia occurs in lupus patients.63 The proliferation of B cells driven by this virus can induce a stimulated autoinflammatory response and some attribute this to the clinical response and often remission of the disease following treatment with rituximab or Belimumab. Oxidative stress may have a role in stimulating cellular proliferation via the mechanistic target of rapamycin (mTOR) pathway. mTOR a key player in T cell survival, proliferation and activation.64 Cyclophosphamide has been shown to increase DNA methylation through induction of DNMT1,65 although its main action is through alkylation and cell cycle arrest.
Intestinal Microbiota and SLE
There have been recent advances in understanding the microbiome in lupus. SLE and Sjogrens patients have been found to have differential gut microbiota.66,67 Work on mice have suggested the gut is leaky in lupus like disease models.68 An enterococcus was found translocated to the liver and triggering interferon expression and anti-dsDNA antibodies.68 Hyper-responsiveness to gut flora occurs in a mouse model that has overexpression of TLR4 and develops lupus. Oestrogen receptor deficiency impairs TLR4 signaling and reduces activity in lupus prone mice.68 In MRL/lpr mice lactobacillus species are reduced and supplementation with lactobacillus caused increased gut mucosal barrier, reduced gut inflammation and nephritis.69 Bacterial gut commensals have been found to produce a variant of Ro60 antigen which stimulates immune cells.70 Diet modifies the gut with fish oils reducing proinflammatory cytokines. Polyunsaturated fatty acids and polyphenols ingestion are associated with beneficial changes to microorganisms present in the gastrointestinal tract.71 Septrin, tetracylines and pencillins can trigger lupus. The suspicion remains that the gut flora changes can interplay with immune regulatory cells such as Tregs.
Current Treatment of Lupus
The clinical assessment of patients dictates the management. Diagnostic tools help to define the affected organ system. The condition is heterogeneous and can present in unusual ways. Most patients are given hydroxychloroquine and prednisolone accompanied by immunosuppressives. In Table 1 we indicate the general outline for the management of SLE in the treatment of different organ and systems. This is adapted from BSR guideline on SLE.72
 |
Table 1 SLE Treatment Based on Severity of the Disease |
Use of the British Isles Lupus Assessment Group (BILAG) scoring system provides a comprehensive approach to managing patients with lupus72 The assessor needs to define activity accurately distinguishing clinical features due to activity, distinct from previous damage or complications of therapy to capture active disease correctly. The multimodal facets of the BILAG allow classification of mild, moderate and severe disease. Mild disease includes non-life threatening organ involvement, equivalent to SLEDAI <6 with one or more grade C scores or a single grade B. Treatment may include topical steroid, low dose oral steroid and/or hydroxychloroquine. In contrast, patients with moderate disease require stronger suppression of the immune system and would consistently have BILAG scores of 2 Bs or more. The most severe disease BILAG A implies high dose steroid (≥20mg/day; intravenous or oral, and other immunosuppressive agents. Other therapies outlined in Table 2 (which designates treatments according to disease severity) include anticoagulation or antiplatelet therapy in antiphospholipid patients and the use of rituximab or cyclophosphamide as second-line treatments to induce remission.
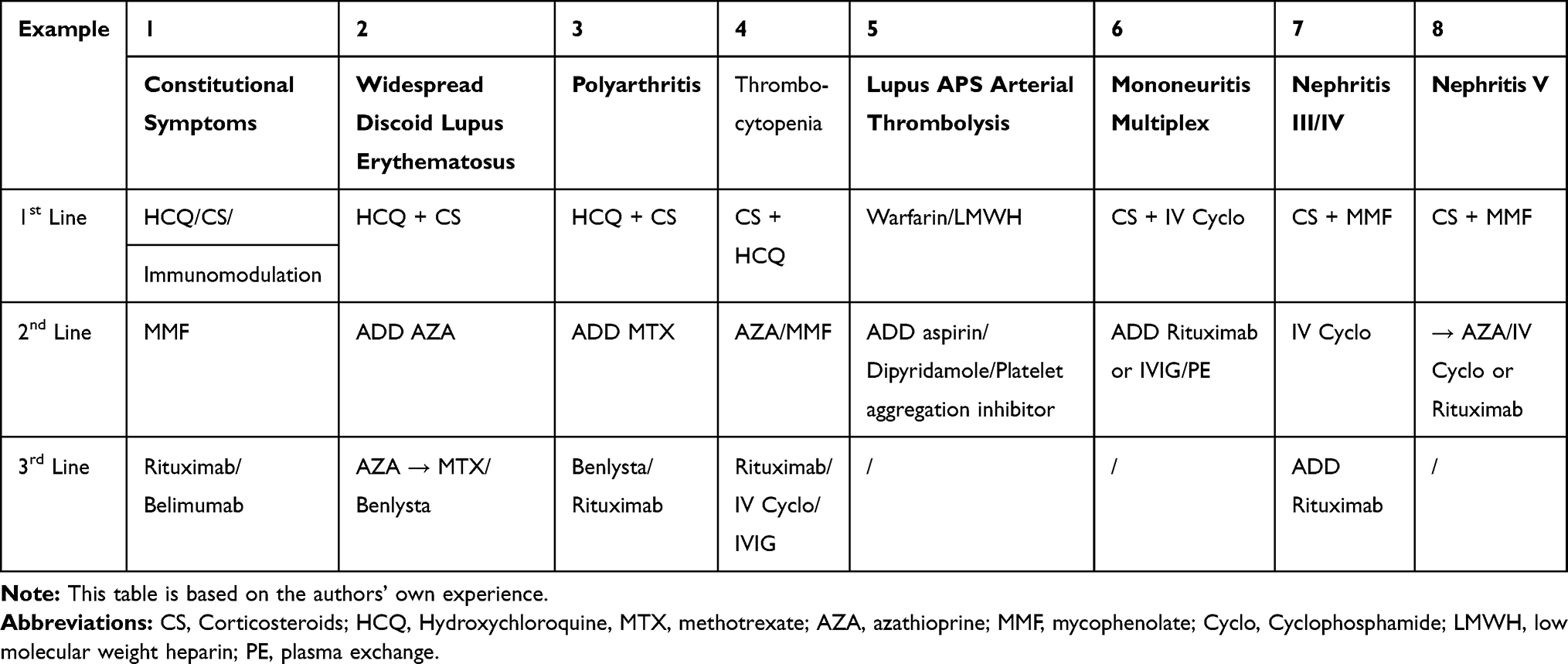 |
Table 2 Treatment Based on the Organ/Systemic Involved |
Patients with skin-limited disease usually respond to nonaggressive treatment including topical and oral steroids and hydroxychloroquine. We recently reported on the use of rituximab in severe cutaneous lupus73 and showed that some types of subcutaneous lupus [e.g., acute cutaneous LE] respond better than other forms [e.g., subacute and chronic]. Severe life-threatening lupus with renal or neuropsychiatric involvement is treated with potent agents notably cyclophosphamide. BILAG grading and following the pattern over time assists in determining the degree of immunosuppression. Use of blood markers notably anti-dsDNA antibody levels [which increase with active disease] or C3 levels [which fall when the disease is active] provide help to the clinician when assessing disease activity. Scoring is determined based on new, improving, worse and similar categories. The art of the physician is to identify the activity features clinically and apply the appropriate management. Renal lupus can, rarely, be active without the presence of other SLE disease involvement. Cerebral lupus is scored in 20 separate sections from seizures to demyelination. In patients with subacute presentations, it may be difficult to assess and treat. Anti-dsDNA antibodies and complement can be normal in a proportion of cases.
SLEDAI scoring systems are more limited in representation of the features of lupus that are present. It provides a global score rather than distinguishing the organ systems and does not distinguish patients who are partly responding, from those whose activity is unchanged or worse.
Both SLEDAI and BILAG have been validated74,75 Disease activity is more comprehensively assessed by BILAG and shown to be reliable and sensitive to change Manifestations such as gastrointestinal disease and haemolytic anaemia are not captured at all in SLEDAI.
The precision of lupus care in the twenty-first century is focused on minimizing the use of potent medication and the side-effects that they cause, while proactively treating the autoimmunity and preventing damage. Rituximab use is increasing but Belimumab (an anti-BAFF monoclonal) is currently only allowed by the National Institute of Clinical Excellence (NICE) for skin and joint disease only in the UK in BILAG centres. We recently reported on predictors of response.76 These include the BILAG score and renal involvement. More severe disease had greater benefit from B-cell depletion. Patients need a careful balance of immunosuppression that controls the disease without rendering them too susceptible to infection and long-term risks such as increased cardiovascular risk. Our approach as detailed in Table 1 does not help in distinguishing subgroups of patients for methotrexate or azathioprine. The molecular tools for this are on their way to the clinic. In rheumatoid arthritis gene signature has been shown to assist response prediction.77
In the current treatment of lupus longterm clinical and immunological remission is only achieved in a proportion of patients. The inherent pathophysiological complexity of lupus described at the cellular and molecular level cannot be overstated. Using targeted drug therapy and improving clinical assessment remains vital to improving disease control and patient outcomes.
The Challenge of Moving Towards Precision Medicine in SLE
As the foregoing text has confirmed the pathophysiological aspects of SLE are highly diverse and the combination of factors leading to say skin, kidney and renal disease are very different. In spite of these differences, a rather restricted set of drugs steroids, immunosuppressives and Hydroxychloroquine [see Tables 1 & 2] are used with some success to treat these “diversely induced” clinical features. Given that the mortality figures for lupus have improved from 50% 4-year survival in 195078 to approximately 90% 10-year survival now79 these standard drugs have been beneficial. Some modest clues to precision targeting do exist with conventional drugs. Thus it has been shown that black lupus patients do not respond as well to Cyclophosphamide as Caucasians.80 There is a debate about whether a seemingly well patient with a rising dsDNA antibody level and falling C3 should be treated prophylactically as these markers anticipate a flare.81 However, as we have argued elsewhere the improvement in survival has largely stalled82 and it is likely that biologic drugs capable of targeting individual molecules will be needed to provide the kind of precise approach that precision medicine warrants. Sadly SLE lags far behind the precision approaches available for the treatment of rheumatoid arthritis where currently five-biologic/small molecule drugs [anti-TNF, anti-CD20, blocking the link between the antigen-presenting cell and the T-cell, anti-IL6/IL6 receptor and JAK-STAT inhibition] are approved for treatment.81 In SLE the approval by the Federal Drug Administration of Benlysta [anti-BAFF] in 2012 remains the only biologic to be allowed in the disease in the USA.82 In the UK NHS England permits the use of Rituximab but both the American College of Rheumatology83 and the European League Against Rheumatism84 recommend its use in renal lupus. Our [unpublished] observations are that it often works quickly to correct haematological problems [thrombocytopenia or haemolytic anaemia] but is much slower to benefit renal disease.
The prospects of other approaches such as the use of Anifrolumab85 which blocks interferon α and Atacicept86 which blocks two B-cell activation factors [BAFF and APRIL] also open up the possibilities for a more precise approach to the treatment of SLE.
Predicting response to treatment could be improved in the future. In the case of belimumab high SLEDAI score and polyarthritis.87 More precise information was gleaned with BAFF or APRIL although this is preclinical in utility.88,89 For rituximab, the type of interferon signature helps to distinguish response.90 Vital and colleagues found two signatures; A and B. Signature B is a set of nonclassical interferon genes which were better at predicting response. TNF-like weak inducer of apoptosis (TWEAK) is a proinflammatory cytokine from the TNF superfamily that binds monogamously to its receptor Fn14. In a study of 110 patients, it was a useful predictor of response to induction therapy in lupus nephritis.91 For mycophenolate, there is no clear marker of response from a systematic review.92 These studies encourage the view that ultimately SLE will be susceptible to a precision medicine approach.
Conclusions
We have analysed recent facets of the pathological and immunogenetic basis of lupus. The network of factors that drives organ selectivity and how the precise within individuals conspires to cause the diverse clinical features remains a mystery. Current treatment can be approached in a more precise and systematic fashion as suggested here. Future care will involve molecular diagnostics throughout the patient timecourse to drive the least toxic combination of therapies. Recent evidence suggests a paradigm shift is on the way but it is hard to predict how fast it will come.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lisnevskaia L, Murphy G, Isenberg DA. Systemic lupus erythematosus. Lancet. 2014;384:1878–1888. doi:10.1016/S0140-6736(14)60128-8
2. Moulton VR. Sex hormones in acquired immunity and autoimmune disease. Front Immunol. 2018;9:2279. doi:10.3389/fimmu.2018.02279
3. Sharif K, Watad A, Coplan L, et al. The role of stress in the mosaic of autoimmunity: an overlooked association. Autoimmun Rev. 2018;17:967–983. doi:10.1016/j.autrev.2018.04.005
4. Smatti MK, Cyprian FS, Nasrallah GK, et al. Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses. 2019;11(8):762. doi:10.3390/v11080762
5. Barbhaiya M, Tedeschi SK, Lu B, et al. Cigarette smoking and the risk of systemic lupus erythematosus, overall and by anti-double stranded DNA antibody subtype, in the nurses’ health study cohorts. Ann Rheum Dis. 2018;77:196–202. doi:10.1136/annrheumdis-2017-211675
6. Mak A, Tay SH. Environmental factors, toxicants and systemic lupus erythematosus. Int J Mol Sci. 2014;15(9):16043–16056. doi:10.3390/ijms150916043
7. Mu Q, Zhang H, Luo XM. SLE: another autoimmune disorder influenced by microbes and diet? Front Immunol. 2015;6:608. doi:10.3389/fimmu.2015.00608
8. Petri M, Orbai AM, Alarcon GS, et al. Derivation and validation of the systemic lupus erythematosus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677–2686. doi:10.1002/art.34473
9. Hochberg MC. Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi:10.1002/(ISSN)1529-0131
10. Aringer M, Costenbader KH, Daikh D, et al. European league against rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78:1151–1159. doi:10.1136/annrheumdis-2018-214819
11. Aringer M, Costenbader KH, Brinks R, et al. Validation of new systemic lupus erythematosus classification criteria. Ann Rheum Dis. 2018;77:60–62. doi:10.1136/annrheumdis-2017-212463
12. Gegenava M, Beaart HJL, Monahan RC, et al. Performance of the proposed ACR-EULAR classification criteria for systemic lupus erythematosus (SLE) in a cohort of patients with SLE with neuropsychiatric symptoms. RMD Open. 2019;5:e000895. doi:10.1136/rmdopen-2019-000895
13. Fanouriakis A, Kostopoulou M, Alunno A, et al. update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78:736–745. doi:10.1136/annrheumdis-2019-215089
14. Medina-Quinones CV, Ramos-Merino L, Ruiz-Sada P, Isenberg DA. Analysis of complete remission in systemic lupus erythematosus patients over a 32-year period. Arthritis Care Res. 2016;68:981–987. doi:10.1002/acr.v68.7
15. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. 2018;36:489–517. doi:10.1146/annurev-immunol-042617-053010
16. Smith CK, Kaplan MJ. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr Opinion Rheum. 2015;5:448–453. doi:10.1097/BOR.0000000000000197
17. Urbonaviciute V, Luo H, Sjöwall C, Bengtsson A, Holmdahl R. Low production of reactive oxygen species drives systemic lupus erythematosus. Trends Mol Med. 2019;25:826–835. doi:10.1016/j.molmed.2019.06.001
18. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. 2011;7:691–699. doi:10.1038/nrrheum.2011.132
19. Frangou E, Vassilopoulos D, Boletis J, Boumpas DT, Boletis J. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): implications for the pathogenesis and treatment. Autoimmun Rev. 2019;18:751–760. doi:10.1016/j.autrev.2019.06.011
20. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, et al. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. 2017;8(1–20):81. doi:10.3389/fimmu.2017.00081
21. Herrada AA, Escobedo N, Iruretagoyena M, et al. Innate immune cells’ contribution to systemic lupus erythematosus. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.00772
22. Shin JI, Lee KH, Joo YH, et al. Inflammasomes and autoimmune and rheumatic diseases: A comprehensive review. J Autoimmun. 2019;103:102299. doi:10.1016/j.jaut.2019.06.010
23. Ma C, Xia Y, Yang Q, et al. The contribution of macrophages to systemic lupus erythematosus. Clin Immunol. 2019;207:1–9. doi:10.1016/j.clim.2019.06.009
24. Thorlacius GE, Wahren-Herlenius M, Rönnblom L. An update on the role of type I interferons in systemic lupus erythematosus and Sjögren’s syndrome. Curr Opin Rheumatol. 2018;30:471–478. doi:10.1097/BOR.0000000000000524
25. Celhar T, Fairhurst A. Toll-like receptors in systemic lupus erythematosus: potential for personalized treatment. Frontiers Pharmacol. 2014;5(265):1–8. doi:10.3389/fphar.2014.00265
26. Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017;17:30–48. doi:10.1038/nri.2016.116
27. Klarquist J, Zhou Z, Shen N, Janssen EM. Dendritic cells in systemic lupus erythematosus: from pathogenic players to therapeutic tools. Mediators Inflamm. 2016;2016:5045248. doi:10.1155/2016/5045248
28. Sandhu V, Quan M. SLE and serum complement: causative, concomitant or coincidental. Open Rheumatol. 2017;11:113–122. doi:10.2174/1874312901711010113
29. Zhou H, Li B, Li J, et al. Dysregulated T-cell activation and aberrant cytokine expression profile in systemic lupus erythematosus. Mediators of Inflamm. 2019;2019. doi:10.1155/2019/8450947
30. SL P. Altered T and B lymphocyte signaling pathways in lupus. Autoimmun Rev. 2009;8:179–183. doi:10.1016/j.autrev.2008.07.040
31. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. 2018;87:1–15. doi:10.1016/j.jaut.2017.12.007
32. Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. 2015;45:344–355. doi:10.1002/eji.201344280
33. Blanco P, Ueno H, Schmitt N. T follicular helper (Tfh) cells in lupus: activation and involvement in SLE pathogenesis. Eur J Immunol. 2016;46:281–290. doi:10.1002/eji.201545760
34. Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol. 2017;13:141–154. doi:10.1038/nrrheum.2016.217
35. Costa S, Bevilacqua D, Cassatella MA. Recent advances on the crosstalk between neutrophils and B or T lymphocytes. Immunology. 2019;156:23–32. doi:10.1111/imm.13005
36. Karrar S, Cunningham-Graham D. Abnormal B-cell development in systemic lupus erythematosus. Arthritis Rheumatol. 2018;70:496–502. doi:10.1002/art.40396
37. Murphy G, Isenberg DA. New therapies for systemic lupus erythematosus: past, imperfect, future tense? Nat Rev Rheum. 2019;15:403–412. doi:10.1038/s41584-019-0235-5
38. Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi:10.1056/NEJMoa021933
39. Flower C, Hennis A, Hambleton IR, Nicholson G. Lupus nephritis in an Afro-Caribbean population: renal indices and clinical outcomes. Lupus. 2006;15(10):689–694. doi:10.1177/0961203306072415
40. Mavragani CP, Fragoulis GE, Somarakis G, Drosos A, Tzioufas AG, Moutsopoulos HM. Clinical and laboratory predictors of distinct histopathological features of lupus nephritis. Medicine. 2015;94:1–8. doi:10.1097/MD.0000000000000829
41. Yu F, Haas M, Glassock R, et al. Redefining lupus nephritis: clinical implications of pathophysiologic subtypes. Nat Rev Nephrol. 2017;13:483–495. doi:10.1038/nrneph.2017.85
42. Londoño Jiminez A, Mowrey WB, Putternam C, Buyon J, Goilav B, Broder A. Brief report: tubulointerstitial damage in lupus nephritis: A comparison of the factors associated with tubulointerstitial inflammation and renal scarring. Arthritis Rheumatol. 2018;70:1801–1806. doi:10.1002/art.40575
43. Arazi A, Rao DA, Berthier CC etal,. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. 2019;20:902–914. doi:10.1038/s41590-019-0398-x
44. Yung S, Chan TM. Anti-dsDNA antibodies and resident renal cells - Their putative roles in pathogenesis of renal lesions in lupus nephritis. Clin Immunol. 2017;185:40–50. doi:10.1016/j.clim.2016.09.002
45. Nalewajska M, Gurazda K, Styczyńska-Kowalska E, et al. The role of MicroRNAs in selected forms of glomerulonephritis. Int J Mol Sci. 2019;20:1–9. doi:10.3390/ijms20205050
46. Sakhi H, Moktefi A, Bouachi K, et al. Podocyte injury in lupus nephritis. J Clin Med. 2019;8:1340. doi:10.3390/jcm8091340
47. Mejía-Vilet JM, Córdova-Sánchez BM, Uribe-Uribe NO, Correa-Rotter R, Morales-Buenrostro LE. Prognostic significance of renal vascular pathology in lupus nephritis. Lupus. 2017;26:1042–1050. doi:10.1177/0961203317692419
48. Jafri K, Patterson SL, Lanata C. Central nervous system manifestations of systemic lupus erythematosus. Rheumatic Dis Clin N Am. 2017;43:531–548. doi:10.1016/j.rdc.2017.06.003
49. Hanly J, Urowitz MB, Su L, et al. Autoantibodies as biomarkers for the prediction of neuropsychiatric events in systemic lupus erythematosus. Ann Rheum Dis. 2011;10:1726–1732. doi:10.1136/ard.2010.148502
50. McGlasson S, Wiseman S, Wardlaw J, Dhaun N, Hunt DPJ. Neurological disease in lupus: toward a personalized medicine approach. Front Immunol. 2018;9(1146):1–12. doi:10.3389/fimmu.2018.01146
51. Tchessalova D, Posillico CK, Tronson NC. Neuroimmune activation drives multiple brain states. Front Syst Neurosci. 2018;12:39. doi:10.3389/fnsys.2018.00039
52. Schwartz N, Stock AD, Putterman C. Neuropsychiatric lupus: new mechanistic insights and future treatment directions. Nat Rev Rheumatol. 2019;15:137–152. doi:10.1038/s41584-018-0156-8
53. Stock AD, Ben-Zui A, Putterman C. Pathogenesis of neuropsychiatric lupus. In: Wallace DJ, Hahn BH, editors. Dubois Lupus Erythematosus and Related Symptoms.
54. Julià A, López-Longo FJ, Pérez Venegas JJ, et al. Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Ther Res. 2018;1(100):1–10. doi:10.1186/s13075-018-1604-1
55. Amlöf JC, Alexsson A, Imgenberg-Kreuz J, et al. Novel risk genes for systemic lupus erythematosus predicted by random forest classification. Sci Rep. 2017;1(6236):1–11. doi:10.1038/s41598-017-06516-1
56. Catalina MD, Owen KA, Labonte AC, Grammer AC, Lipsky PE, Labonte AC. The pathogenesis of systemic lupus erythematosus: harnessing big data to understand the molecular basis of lupus. J Autoimmun. 2019;2:102359. doi:10.1016/j.jaut.2019.102359
57. Surace AEA, Hedrich CM. The role of epigenetics in autoimmune/inflammatory disease. Front Immunol. 2019. doi:10.3389/fimmu.2019:01525
58. Imgenberg-Kreuz J, Carlsson Almlöf J, Leonard D, et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis. 2018;77(5):736–743. doi:10.1136/annrheumdis-2017-212379
59. Hedrich CM. Epigenetics in SLE. Curr Rheumatol Rep. 2017;19(58):1–13. doi:10.1007/s11926-017-0685-1
60. Wang Z, Yin H, Lau CS, Lu Q. Histone posttranslational modifications of CD4+ T cell in autoimmune diseases. Int J Mol Sci. 2016;17(1547):1–16. doi:10.3390/ijms18010001
61. Wu Z, Mei X, Ying Z, Sun Y, Song J, Shi W. Ultraviolet B inhibition of DNMT1 activity via AhR activation dependent SIRT1 suppression in CD4+ T cells from systemic lupus erythematosus patients. J Dermatol Sci. 2017;86:230–237. doi:10.1016/j.jdermsci.2017.03.006
62. Mak A. The impact of vitamin D on the immunopathophysiology, disease activity, and extra-musculoskeletal manifestations of systemic lupus erythematosus. Int J Mol Sci. 2018;19:
63. Draborg A, Izarzugaza JM, Houen G. How compelling are the data for Epstein-Barr virus being a trigger for systemic lupus and other autoimmune diseases? Curr Opin Rheumatol. 2016;28:398–404. doi:10.1097/BOR.0000000000000289
64. Myers DR, Wheeler B, Roose JP. mTOR and other effector kinase signals that impact T cell function and activity. Immunol Rev. 2019;291:134–153. doi:10.1111/imr.v291.1
65. Zhang J1, Yuan B, Zhang F, et al. Cyclophosphamide perturbs cytosine methylation in Jurkat-T cells through LSD1-mediated stabilization of DNMT1 protein. Chem Res Toxicol. 2011;24:2040–2043. doi:10.1021/tx2003849
66. Silverman GJ. The microbiome in SLE pathogenesis. Nat Rev Rheumatol. 2019;15(2):72–74.
67. Kim JW, Kwok SK, Choe JY, et al. Recent advances in our understanding of the link between the intestinal microbiota and systemic lupus erythematosus. Int J Mol Sci. 2019;20(19). doi:10.3390/ijms20194871.
68. Manfredo Vieira S, Hiltensperger M, Kumar V, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359(6380):1156–1161. doi:10.1126/science.aar7201
69. Mu Q, Zhang H, Liao X, et al. Control of lupus nephritis by changes of gut microbiota. Microbiome. 2017;5(1):73. doi:10.1186/s40168-017-0300-8
70. Greiling TM, Dehner C, Chen X, et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci Transl Med. 2018;10(434). doi:10.1126/scitranslmed.aan2306.
71. Cuervo A, Hevia A, López P, et al. Association of polyphenols from oranges and apples with specific intestinal microorganisms in systemic lupus erythematosus patients. Nutrients. 2015;7(2):1301–1317. doi:10.3390/nu7021301
72. Gordon C, Amissah-Arthur MB, Gayed M, et al. The British Society for Rheumatology guideline for the management of systemic lupus erythematosus in adults. Rheumatology (Oxford). 2018;57:e1–45. doi:10.1093/rheumatology/kex286
73. Da Costa R, Aguirre-Alastuey E, Isenberg DA, Saracino A. Assessment of response to B-cell depletion using Rituximab in cutaneous lupus erythematosus. JAMA Dermatol. 2018;154(12):1432–1440. doi:10.1001/jamadermatol.2018.379
74. Romero-Diaz J, Isenberg D, Ramsey-Goldman R. Measures of adult systemic lupus erythematosus. Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S37–46. doi:10.1002/acr.20572
75. Yee CS, Isenberg DA, Prabu A, et al. BILAG 2004 index captures systemic lupus erythematosus disease activity better than SLEDAI-2000. Ann Rheum Dis. 2008;67(21):873–876. doi:10.1136/ard.2007.070847
76. Freitas S, Mozo Ruiz M, Costa Carneiro A, Isenberg DA. Why do some patients with systemic lupus erythematosus fail to respond to B-cell depletion using rituximab?Clin Exp Rheumatol. 2019;58(Supplement_3):kez107–055 In Press.
77. Plant D, Maciejewski M, Smith S, Nair N. Maximising therapeutic utility in rheumatoid arthritis consortium, the RAMS study group, Hyrich K1, Ziemek D2, Barton A1, Verstappen S1 Profiling of gene expression biomarkers as a classifier of methotrexate nonresponse in patients with rheumatoid arthritis. Arthritis Rheumatol. 2019;71:678–684. doi:10.1002/art.40810
78. Merrell M, Shulman LE. Determination of prognosis in chronic disease illustrated by systemic lupus erythematosus. J Chronic Dis. 1955;1:12–32. doi:10.1016/0021-9681(55)90018-7
79. Mak A, Cheung MW-L, Chiew KJ, et al. Global trend of survival and damage of systemic lupus erythematosus: meta-analysis and meta-regression of observational studies from the 1950s to 2000s. Semin Arthritis Rheum. 2012;41:830–839. doi:10.1016/j.semarthrit.2011.11.002
80. Dooley MA, Hogan S, Falk R. Cyclophosphamide therapy for lupus nephritis: poor survival in black Americans. glomerular disease collaborative network. Kidn Int. 1997;51:1188–1195. doi:10.1038/ki.1997.162
81. Ciurtin C, Isenberg DA, Eds. Biologics in Rheumatology. New York: Nova Biomedical; 2016.
82. Van Vollenhoven RF, Petri MA, Cervera R, et al. Belimumab in the treatment of systemic lupus erythematosus: high disease activity predictions of response. Ann Rheum Dis. 2012;71:1343–1349. doi:10.1136/annrheumdis-2011-200937
83. Hahn BH, McMahon MA, Wilkinson A, et al. American college of rheumatology guidelines for screening, treatment and management of lupus nephritis. Arthritis Care Res. 2012;64:797–808. doi:10.1002/acr.21664
84. Bertsios G, Tektonidou M, Amura Z, et al. Joint European League Against Rheumatism and European Renal Association – European Dialysis and Transplant Association [EULAR/ERA-EDTA] recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis. 2012;71:177–182.
85. Felten R, Scher F, Sagez F, Chasset F, Arnaud L. Spotlight on anifrolumab and its potential for the treatment of moderate-to-severe systemic lupus erythematosus: evidence to date. Drug Des Devel Ther. 2019;13:1535–1543. doi:10.2147/DDDT.S170969
86. Merrill JT, Wallace DJ, Wax S, et al. Efficacy and safety of Atacicept in patients with systemic lupus erythematosus. Arthritis Rheum. 2018;70:266–276. doi:10.1002/art.40360
87. Laccarino L, Andreoli L, Bocci EB, et al. Clinical predictors of response and discontinuation of belimumab in patients with systemic lupus erythematosus in real life setting. Results of a large, multicentric, nationwide study. J Autoimmun. 2018;86:1–8. doi:10.1016/j.jaut.2017.09.004
88. Piantoni S, Andreoli L, Lowin T. Baseline serum levels of baff or april are independent predictors of sledai response after 12 months of treatment with belimumab in patients with refractory systemic lupus erythematosus. doi:10.1136/lupus-2018-abstract.43
89. Parodis I, Johansson P, Gomez A, Soukka S, Emamikia S, Chatzidionysiou K, Gomez A. Predictors of low disease activity and clinical remission following belimumab treatment in systemic lupus erythematosus. Rheumatology (Oxford). 2019;58(12):2170–2176. doi:10.1093/rheumatology/kez191
90. Alase A, Wigston Z, Burska A. Prediction of response to rituximab in SLE using a validated two-score system for interferon status. doi:10.1136/lupus-2019-lsm.71
91. Suttichet TB, Kittanamongkolchai W, Phromjeen C. Urine TWEAK level as a biomarker for early response to treatment in active lupus nephritis: a prospective multicentre study. Lupus Sci Med. 2019. eCollection 2019. doi:10.1136/lupus-2018-000298
92. Mendoza-Pinto C, Pirone C, van der Windt DA. Can we identify who gets benefit or harm from mycophenolate mofetil in systemic lupus erythematosus? Syst Rev Semin Arthritis Rheum. 2017;47:65–76.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.