Back to Journals » OncoTargets and Therapy » Volume 12
Toward fully exploiting the therapeutic potential of marketed pharmaceuticals: the use of octreotide and chloroquine in oncology
Authors Papanagnou P, Papadopoulos GE ![]() , Stivarou T, Pappas A
, Stivarou T, Pappas A
Received 5 August 2018
Accepted for publication 20 November 2018
Published 31 December 2018 Volume 2019:12 Pages 319—339
DOI https://doi.org/10.2147/OTT.S182685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Panagiota Papanagnou,1,* Georgios E Papadopoulos,2,* Theodora Stivarou,3 Anastasios Pappas1
1Department of Urology, Agios Savvas Cancer Hospital, Athens 11522, Greece; 2Department of Biochemistry and Biotechnology, University of Thessaly, Larissa 41500, Greece; 3Immunology Laboratory, Immunology Department, Hellenic Pasteur Institute, Athens, Greece
*These authors contributed equally to this work
Abstract: Pleiotropy in biological systems and their targeting allows many pharmaceuticals to be used for multiple therapeutic purposes. Fully exploiting the therapeutic properties of drugs that are already marketed would be highly advantageous. This is especially the case in the field of oncology, where the ineffectiveness of typical anticancer agents is a common issue, while the development of novel anticancer agents is a costly and particularly time-consuming process. Octreotide and chloroquine are two pharmaceuticals that exhibit profound antitumorigenic activities. However, the current therapeutic use of octreotide is restricted primarily to the management of acromegaly and neuroendocrine tumors, both of which are rare medical conditions. Similarly, chloroquine is used mainly for the treatment of malaria, which is designated as a rare disease in Western countries. This limited exploitation contradicts the experimental findings of numerous studies outlining the possible expansion of the use of octreotide to include the treatment of common human malignancies and the repositioning of chloroquine in oncology. Herein, we review the current knowledge on the antitumor function of these two agents stemming from preclinical or clinical experimentation. In addition, we present in silico evidence on octreotide potentially binding to multiple Wnt-pathway components. This will hopefully aid in the design of new efficacious anticancer therapeutic regimens with minimal toxicity, which represents an enormous unmet demand in oncology.
Keywords: drug repositioning, pleiotropy, neuroendocrine tumors, lysosomotropic agent, cancer, docking
Introduction
Several marketed pharmaceuticals are being experimentally characterized as pleiotropic agents whose therapeutic potency is currently rather underrated.1–6 As such, they could be repurposed for use in therapeutic fields that are different to their current therapeutic field. In addition, the use of certain commercially available pharmacological agents could be expanded for use in a wider range of histological (sub)types of human cancer. This reflects the admirable pleiotropy that is found in biological systems. In particular, chloroquine has been reported to be a possible neurorestorative agent in animal models of Parkinson’s disease7 and has been used in rheumatology,8 treatment of amebic liver abscess,9 and management of systemic lupus erythematosus, due to its immunomodulatory effects.10 Harnessing the well-described repositionability of certain marketed pharmacological agents and/or fully exploiting the therapeutic potential of agents that are currently underexploited may save time (related to conducting new research from bench to bedside) and would take advantage of the years of accumulated clinical experience of their use. In this way, clinicians could avoid getting into uncharted waters in their efforts to fight cancer via using newly launched antitumor drugs.
Repositionability is relevant not only for pharmaceuticals that are currently used for the management of common pathological conditions, such as acute/chronic pain,11 diabetes mellitus type 2,12 dyslipidemia,13 and asthma,14 but also for drugs that are currently used only for the management of less commonly occurring diseases. This is the case for octreotide, a drug that is currently used for the treatment of midgut carcinoids (which are a dozen times less common than colon adenocarcinomas) or acromegaly, as well as for chloroquine. The latter is an antimalarial drug that blocks autophagy, and it is not commonly prescribed in most Western countries, where malaria has been eliminated. In fact, the autophagy signaling pathway encompasses several druggable components and can be targeted at multiple levels not only by chloroquine, which is commercially available, but also by other agents currently used only in experimental settings. These agents include the small molecule spautin 1 and SAR405, which both selectively inhibit Vps34 kinase, a key regulator of the autophagic PI3K complex.15 However, in striking contrast to chloroquine, there is no clinical experience with regard to these newly discovered agents.
Hydroxychloroquine, a chloroquine derivative, is also an antimalarial that exhibits a well-known anticancer function, which has been extensively reviewed elsewhere.16 The chloroquine derivative cymanquine also exhibits anticancer potency.17 However, in this review, we focus on chloroquine, which is the prototype antimalarial agent and drug of choice for malaria chemotherapy that is currently under consideration for repositioning in oncology.18–20 The subsequent paragraphs review the literature that supports the possible repurposing of chloroquine in the field of oncology, as well as the literature that supports the expansion of the oncological use of octreotide beyond its current use in cancer.
In silico data further support the notion that the signaling properties of octreotide are not fully exploited in cancer therapeutics. Other synthetic somatostatin analogues, such as lanreotide and the recently developed pan-somatostatin agonist pasireotide, are also commercially available and display antitumor activity.21,22 However, in this review, we focus on octreotide, due to the fact that this synthetic somatostatin analogue exhibits the most extensively studied direct antineoplastic effects on malignant cells, both in vitro and in vivo, as discussed in the following paragraphs. In vitro and in vivo preclinical evidence and clinical data regarding the antitumorigenic function(s) of chloroquine and octreotide (Figure S1) strengthen the notion that even agents that are used for the management of rare medical conditions may also be used for the treatment of more frequently occurring pathological entities in oncology, due to their pleiotropic modes of action.
Overview of current clinical use of octreotide and chloroquine and their mechanisms of action
The cyclic octapeptide octreotide is a synthetic analogue of the hypothalamic hormone somatostatin. Besides its usage in the field of oncology, octreotide is used for the management of acromegaly. This is a rare pathological entity, with overall prevalence of 2.8–13.7 cases/100,000 individuals. It is mostly attributed to pituitary adenomas overproducing growth hormone (GH), and is associated with GH-mediated elevated hepatic synthesis of insulin-like growth factor 1 (IGF1).23,24 Five human somatostatin receptor subtypes, SSTR1–5, have been cloned. Octreotide selectively binds the receptor subtypes SSTR2 and SSTR5 and also binds at SSTR3 with lower affinity.25 SSTR binding by octreotide activates the mitogen-activated protein kinase (MAPK) cascade and affects serum levels of GH and IGF1 in patients.26 Octreotide has also been therapeutically used for the management of HIV-associated diarrhea,27 severe diabetic diarrhea,28 chylothorax,29 and congenital hyperinsulinemia (a rare disorder affecting 1/50,000 newborn babies).30,31
Given the well-established role of IGF132 and the role of the somatostatin-signaling axis33,34 in promoting and impeding tumorigenesis, respectively, the anticancer activity of octreotide is not a surprising finding. Indeed, radiolabeled octreotide currently has a diagnostic application in oncology, allowing the detection of SSTR-positive tumors via scintigraphy (octreoscan). At the therapeutic level, unradiolabeled octreotide has an application as an antisecretory agent to palliate the symptoms of bowel obstruction in patients with advanced gastrointestinal (GI), gynecological, or pelvic cancer35,36 and for the amelioration of carcinoid symptoms.37 It is also commonly used to alleviate symptoms associated with hypersecretion in patients with secretory neuroendocrine tumors (NETs). Additionally, it is used for the management of portal hypertension in subjects with cirrhosis.38 Furthermore, radiolabeled octreotide has an application as a radiopharmaceutical in so-called peptide receptor radionuclide therapy for tumors expressing SSTRs. However, experimental data have demonstrated that unradiolabeled octreotide also directly restrains tumor growth.39–46 The molecular mechanism(s) underlying the inhibitory effects of unradiolabeled octreotide on the growth of NET cells are being unveiled.47 Also, preclinical and clinical data indicate that the therapeutic exploitability of unradiolabeled octreotide could be expanded far beyond NET therapy to the treatment of non-NETs as well, through a plethora of molecular pathways. The multilevel functions of unradiolabeled octreotide against tumorigenesis are presented in the subsequent paragraphs.
In the pituitary cell lines GH4C1 and AtT20, prolonged exposure to somatostatin evokes desensitization. At the signaling level, this means that somatostatin cannot prevent the intracellular formation of cAMP, whereas cAMP formation in response to corticotrophin-releasing hormone, the adenylyl cyclase activator forskolin, or the unselective β-adrenergic agonist isoproterenol is enhanced. The phenomenon of desensitization to somatostatin takes place after receptor internalization, and in vitro experimentation (at least in AtT20 cells) has indicated that it is SSTR2 rather than SSTR5 that controls somatostatin desensitization.48 However, the extrapolation of these in vitro data to human pituitary receptor signaling or the functional and clinical relevance of desensitization to cancer treatment (ie, desensitization to radiolabeled octreotide and its possible association with the effects of peptide receptor radionuclide therapy) remains to be elucidated.
Chloroquine, a lysosomotropic autophagy inhibitor, is a 4-aminoquinoline that was introduced in clinical practice in the 1950s for the treatment and prophylaxis of malaria. The drug kills the hematophagous Plasmodium falciparum by entering parasites’ digestive vacuoles in infected erythrocytes. In this acidic compartment, chloroquine inhibits the biocrystallization of hemozoin, thereby leading to the accumulation of either toxic heme monomers or toxic chloroquine-bound heme complexes.49,50 As an antiparasitic agent, it also has application in the treatment of amoebic liver abscess.9 Additionally, it exhibits immunomodulatory effects and has application in the management of autoimmune disorders.8,10
Autophagy inhibition is an emerging anticancer strategy.16,51,52 The autophagic response is evoked by malignant cells in response to noxious agents, in order to damage and eliminate them, and this is the rationale of anticancer strategies targeting autophagy.20,53–55 Importantly, evidence presented herein suggests that chloroquine is a versatile pharmaceutical, affecting tumor cell biology, even in an autophagy-independent fashion.
Preclinical data: toward expanding the use of octreotide in the treatment of GI and liver tumors
In SGC-7901 human gastric adenocarcinoma cells, inhibition of proliferation by octreotide has been linked to drug-induced suppression of the activity of both telomerase and Akt kinase.56 Also, octreotide reduced the invasive potential of SGC-7901 cells and their ability to give rise to metastases in nude rats.57 According to more recent evidence, in SGC-7901 cells, the antigrowth effects of octreotide are mechanistically related to a decrease in phospho-Akt levels and a consequent increase in levels of a zinc-finger protein termed Zac.58 This protein is a well-known modulator of the histone acetyltransferase activity of the transcriptional coactivator p300, and it exhibits an antiproliferative function.59 The interplay between Zac and its partner p300 resulted in a decrease in the mitosis marker phosphohistone H3 (Ser10) and an elevation in acetylhistone H3 (Lys14).58 These data indicate that in SGC-7901 cells, octreotide engages an Akt-dependent pathway to impinge on histone posttranslational modifications and chromatin structure in a manner that inhibits cell proliferation.
A high-throughput analysis of the transcriptome in response to long-term (up to 16 months) treatment with 1 μM octreotide showed that the transcripts of six molecules were upregulated in small-intestine NET CNDT2.5 cells, which are known to express low levels of SSTRs. These six molecules included AnxA1, the RhoGAP family member ARHGAP18, TGFβR2, and the TNF-ligand family member TNFSF15. The expression of these molecules in response to octreotide was increased at both the mRNA and protein levels. Whether these molecules participate in parallel signaling pathways or are components of cross-talking pathways remains to be elucidated.60 In any case, the aforementioned study60 demonstrated that octreotide can restrain tumor cell growth in a manner that is reversible upon octreotide withdrawal. Most importantly, no drug resistance developed after long-term treatment of CNDT2.5 cells with octreotide.
Octreotide has been found to compromise the viability of SW480 colon cancer cells (non-NET cells) by negatively regulating the Wnt–β-catenin pathway.61 These data indicate the potential for a wider usage of octreotide, not only in the management of NETs arising from the small intestine but also for the treatment of human colon cancer. According to a previous study, in SW480 cells, octreotide inhibited growth as a result of both its cancer cell killing and cytostatic effects (G1 cell-cycle arrest), as shown by MTT assays and flow cytometry. Remarkably, the cancer-associated Wnt–β-catenin signaling pathway was targeted by octreotide at multiple levels in these cells. Microarray analysis revealed that the expression levels of 30 genes coding for Wnt–β-catenin pathway components were either negatively (17 genes) or positively (13 genes) affected in response to this somatostatin analogue, while β-catenin protein levels were negatively affected.62 This is a good example of a signaling axis which encompasses multiple points that can be targeted by antitumor pharmaceutical therapy (at least in vitro), and it may reflect the pathogenic importance of Wnt–β-catenin throughout colorectal carcinogenesis. In other colon cancer cell lines (HT29 and Caco2), the antiproliferative effects of octreotide were mediated by protein-tyrosine phosphatases, while in serum-free cultured Caco2 cells, the cytostatic effects of octreotide may also be associated with interference with telomerase signaling.63 These in vitro data indicate that different pathways are engaged by octreotide to suppress the growth of NETs or non-NETs from distinct regions of the GI tract.
Remarkably, octreotide selectively triggered apoptosis in human hepatocellular carcinoma (HCC) cell lines (SMMC-7721 and HepG2), but not in human fetal L-02 hepatocytes. The expression of SSTR2 and SSTR4 was a common molecular feature of both malignant HCC and L-02 cells, while SSTR3 was expressed only in HCC cells. As such, it could not be excluded that SSTR3-dependent signaling was involved in the differential responses of the malignant and nonmalignant liver cells to octreotide.64 Consistently with these data, SMMC-7721 cells have been found to undergo apoptosis in response to octreotide in a dose- and time-dependent fashion, and upregulation of the expression of Fas death receptor and its ligand, FasL, has been suggested as an underlying mechanism.65 In vivo data from rodent models of liver cancer metastasis, where researchers intraportally injected rat tumor cells, demonstrated the antimetastatic properties of octreotide, at least in this nonclinical experimental setup.66 In human HCC xenografts, octreotide halted tumor angiogenesis by reducing the level of VEGF and possibly by interfering with other pathways. In addition, octreotide hindered the incidence of primary cancer and distant (lung) metastases.67
Preclinical data: toward using octreotide in the treatment of tumors that arise in non-gastroenterological/non-hepatic tissue
In clinical specimens of prostate carcinoma, the area of the cancer nests in lymph nodes exhibits selective positivity for SSTR2A. In vitro experimentation in SSTR2A-positive human prostate cancer cell lines (PC3 and DU145 cells) demonstrated that octreotide hindered invasiveness and metastatic potential in a ROCK-inhibition-reversible manner. This was accompanied by cellular morphological alterations, suggesting actin cytoskeletal changes as a possible underlying mechanism of octreotide function in advanced prostate cancer.68 In fact, the SSTR2A-signaling axis has been proposed to be of therapeutic importance in prostate cancer.69
Remarkably, not only was the long-term usage of octreotide not associated with the development of resistance to its own effects60 but could also reverse resistance to cisplatin of SSTR2-positive SKOV3/DDP ovarian cancer cells by downregulating EGFR and the MRP2 transporter.70 Moreover, octreotide has been found to reverse the drug resistance of the A2780/Taxol ovarian cancer cell line, as it negatively modulated levels of SSTR2, VEGF, and MDR1.71
One study reported that there was no antiproliferative synergism between rapamycin (a stimulator of Akt that has been linked to drug resistance) and octreotide (which is an antagonist of Akt signaling) after treating the human bronchial or pancreatic NET cells NCI-H727 and BON-1, respectively, with these two agents. Further, no significant antitumor effects were observed in vitro when these cells were exposed to octreotide alone.72 Octreotide restrains the growth of follicular thyroid cancer cells73 and, when combined with the DNA topoisomerase I inhibitor topotecan, displays synergistic antileukemic effects in vitro and in animals implanted with murine P388 leukemia cells.74
Preclinical data suggesting a potential for repurposing chloroquine
In vitro and in vivo evidence
In DLD1 colon cancer cells, combined exposure to 10 μΜ 5-fluorouracil (a conventional chemotherapeutic agent) and 100 μΜ chloroquine resulted in cytostatic synergy by influencing different cell-cycle-inhibitory molecules exemplified by p53 and p27Kip1 (positive regulation) and cyclin D1 and CDK2 (negative regulation), suggesting a role for chloroquine as an adjunct in cancer therapeutics.75 In murine CT26 colon cancer cells, chloroquine triggered apoptosis, while in mice with CT26 tumors, chloroquine caused tumor shrinkage and prolonged survival.76 Therefore, these data indicate an antitumor function of chloroquine in colon cancer, irrespective of the human or murine origin of the malignant cells.
Chloroquine has been demonstrated to exert antigrowth effects in human ductal pancreatic adenocarcinoma PaTu-II cells.77 Additionally, in the pancreatic cancer cells Panc1 and MiaPaCa2, chloroquine exhibited cytostatic effects (induction of G2/M phase arrest), while it decreased the levels of antiapoptotic proteins and enhanced the growth-suppressive properties of TRAIL in xenografts.78 Evidence for the antitumor function of chloroquine in both metastatic S2VP10 and unaggressive MiaPaCa2 pancreatic cancer cells under hypoxic conditions indicated that chloroquine interfered with autophagy and substantially reduced the viability of malignant cells.79 The rationale for treating cancer cells with an autophagy inhibitor (such as chloroquine) in hypoxic conditions is that hypoxia evokes an autophagic response, which favors tumor-cell growth even under adverse conditions and endows cells with resistance to chemotherapeutic drugs.80
In the vast majority of HCC patients who undergo transcatheter arterial chemoembolization (TACE; a minimally invasive locoregional treatment approach to block the blood and nutrient supply to cancer cells and thus to damage them), autophagy occurs in response. Fortunately, experiments in the rabbit VX2 tumor model of human HCC suggest that the proapoptotic effects of TACE are enhanced by using chloroquine as an adjunct.81 In addition, autophagy was evoked in response to chemotherapeutic agents to enable escape from tumor dormancy, and pharmacological blockage of this response with chloroquine resulted in the prevention of tumor relapse in vitro, as evidenced by experiments in Neu-overexpressing MMC cells during treatment with adriamycin.82 In vitro and in vivo evidence corroborates the notion regarding the exploitability of chloroquine as either monotherapy or an adjunct in liver cancer therapeutics. In fact, exposure of Huh7 and HepG2 human HCC cells to chloroquine induced G0/G1 cell-cycle arrest and stimulated the mitochondrial pathway of apoptosis. Moreover, it has been reported that chloroquine induced DNA double-strand breaks and suppressed tumor growth in an orthotopic mouse model of human liver cancer.83
In human PC3 and DU145 prostate cancer cells, concomitant treatment with the Akt inhibitor AZD5363 and chloroquine unleashed the apoptosis-triggering effects of AZD5363. Significantly, chloroquine and AZD5363 synergistically promoted tumor shrinkage in xenografts involving the aforementioned prostate cancer cells.84 Also, chloroquine has been found to potentiate the proapoptotic effects of the Src-kinase inhibitor saracatinib in human PC3 prostate cancer cells and the antitumor efficacy of saracatinib in murine xenografts.85
A major factor that limits the cancer cell-killing effects (involving apoptosis) of the polyphenol resveratrol is that this naturally occurring compound evokes a growth-promoting autophagic response in cancer cells. Significantly, the concomitant exposure of Ishikawa endometrial cancer cells to resveratrol and chloroquine leads to a greater apoptotic response compared with resveratrol alone. The apoptosis-enhancing properties of chloroquine have been attributed to its inhibition of the autophagy pathway, as the synergistic effects of chloroquine on apoptosis can be phenocopied by silencing the expression of autophagy-related genes.86 As such, although chloroquine alone has been reported to suppress proliferation and stimulate apoptosis in parental Ishikawa and cisplatin-resistant Ishikawa cells,87 a combinational resveratrol/chloroquine regimen may also be a valuable treatment for endometrial cancer.86
The induction of apoptosis or necrosis has been found to mediate the cancer-cell-killing effects of chloroquine in A549 human non-small-cell lung cancer cells.88 In these cells, chloroquine has been reported to be an effective proapoptotic agent that decreased the Bcl2:Bax ratio, thereby strengthening the notion of its application in lung cancer therapeutics, even as a monotherapeutic agent.89 In murine xenografts bearing NETs, chloroquine potentiated the apoptosis-stimulating properties of the mTOR inhibitor everolimus.90 Further, cotreatment of pancreatic BON-1 NET cells with chloroquine and the mTOR inhibitor NVP-BEZ235 or Torin 1 has been shown to have synergistic effects on the induction of cancer-cell apoptosis.91 These findings contradict the previously mentioned findings that chloroquine displays no synergistic antitumor function when used in conjunction with the mTOR inhibitor rapamycin in BON-1 cells.72 The potential synergy between chloroquine and mTOR inhibitors may hold true for non-NET cells, and merits further investigation.
Cell lines of neuroblastomas (a common type of neonatal autonomic nervous system tumor) display a differential sensitivity to entrectinib. Entrectinib inhibits a neural RTK (ALK), which is etiopathologically linked to neuroblastoma and mutated in 8% of cases of sporadic neuroblastoma. In fact, NB1amp cells that harbor an amplified ALK gene are sensitive to entrectinib-induced apoptosis. In contrast, neuroblastoma SHSY5YF1174L cells positive for the ALKF1174L mutation are resistant to apoptosis triggered by entrectinib. Intriguingly, a Beclin 1-independent autophagic response has been identified as being involved in drug resistance in these cells, and treatment of SHSY5YF1174L with chloroquine may restore their sensitivity to entrectinib. Therefore, if entrectinib is approved as an antineuroblastoma agent and launched in the market, chloroquine could be repositioned in oncology for use in combination with entrectinib for the treatment of neuroblastomas that carry mutations associated with entrectinib resistance (as identified by molecular screening). Another important issue is that (at least in neuroblastoma cells) the autophagy-inhibition effect of chloroquine is effective as a cancer cell-elimination strategy in the presence of an anti-ALK agent, even when autophagy is Beclin 1-independent.92
Interestingly, experimental data indicate that chloroquine can be repositioned not only for the treatment of neuroblastoma but also for the treatment of glioblastoma (another tumor derived from nerve tissue and the most common brain tumor, characterized by high aggression). Specifically, evidence indicates that in U-373 and U-87 human glioblastoma cells, chloroquine reduces invasiveness, exhibits radiosensitizing effects, and prevents the production of TGF-β, a cytokine positively involved in the pathogenesis of glioblastoma.93 Chloroquine, the mTOR inhibitor rapamycin, and the alkylating agent temozolomide display synergistic anti-glioblastoma multiforme (GBM) activity as bioactive agents in a triple-combination regimen. The underlying mechanism involves cholesterol depletion, ROS formation, and lysosomal membrane destabilization, which eventually leads to the apoptotic death of various GBM cell lines (GBM8401, M059K, Hs683, and U87MG cells) and the suppression of GBM-xenograft growth. Therefore, this regimen seems to be effective in GBM and merits clinical investigation regarding its oncological use.94
Chloroquine exhibited cytotoxic effects both under conditions of normoxia and hypoxia independent of wild-type or mutant BRAF expression in various melanoma cell lines. However, under hypoxic conditions, when tumor cell growth was sustained by HIF1α, the exposure of cells to the HIF1α inhibitor echinomycin enhanced the cytotoxicity of chloroquine.95 The pharmacological inhibition of autophagy via exposure to chloroquine or via targeting autophagy-associated molecules (eg, Beclin 1 and ATG5) has more recently been found to stimulate the expression of the chemokine CCL5 via the PP2A–JNK–c-Jun pathway. This eventually led to the increased infiltration of melanoma tumors by natural killer (NK) cells and B16-F10 melanoma-tumor regression. Significantly, a correlation between elevated CCL5 expression and improved survival in melanoma patients has been reported. These data underline the possible exploitability of chloroquine or other autophagy-blocking methodologies as a novel antimelanoma strategy that is mechanistically linked to improvement in NK-cell-based cancer immunotherapy.96 Conceivably, chloroquine could be used as a pharmacological tool that switches the immunosuppressive nature of a tumor milieu to an immunosupportive one, at least via stimulation of the recruitment of NK cells into the tumor bed.
In human Bcap37 (ER-, p53-mutated) breast cancer cells, chloroquine halted cell-cycle progression and triggered apoptosis.97 In ER-positive breast cancer cells, chloroquine was recently reported to enhance the apoptosis triggered by the PI3K inhibitor GDC0941 in a Bim/PUMA-dependent fashion. In xenograft models of antiestrogen resistance, this pharmacological combination (chloroquine and the PI3K inhibitor GDC-0941) resulted in synergism regarding tumor regression.98
In triple-negative breast cancer (TNBC) cells, chloroquine exhibited anti-cancer stem cell (CSC) properties, as it effectively eliminated the CD44+/CD24−/low cellular subpopulation associated with relapse and drug resistance, as evidenced by in vitro and in vivo experiments.99,100 Bioinformatic modeling indicated the possible value of chloroquine as an anti-CSC agent in TNBC. Chloroquine alone displayed a marked ability to eliminate the CSC subpopulation among TNBC cells (decrease by 50 times), while it potentiated the tumor antigrowth effects of paclitaxel in orthotopic tumor models. At the molecular level, according to a methyl-CpG-binding domain protein-enriched DNA sequencing (MBDCap-Seq) analysis, chloroquine affected the methylome of MDA-MB231 cells, specifically regarding the methylation status of numerous proximal promoter regions of genes associated with diverse functions, such as binding of ATP, nucleotides, β-catenin, or metal ions. One pathway that may specifically account for at least part of the observed inhibitory synergy between chloroquine and paclitaxel regarding the CSC subpopulation is the DNMT1–SOCS–Jak2–STAT3 pathway. In this pathway, the chloroquine/paclitaxel regimen downregulated DNMT1. This caused a decrease in the expression of SOCS family member(s). In turn, this impaired Jak2/Stat3-dependent signaling, which is known to be critical for the maintenance and growth of breast CSCs.99 In 4T1 and 67NR mouse mammary tumor cells, chloroquine increased sensitivity to the mTOR inhibitor rapamycin and to the PI3K inhibitor LY294002, even in the absence of Beclin 1 and ATG12. These data suggest that chloroquine suppresses tumor-cell growth of mammalian cells by engaging diverge signaling pathways other than autophagy pathways.101
Pretreatment of human TNBC cells with chloroquine significantly counteracted their ability to give rise to lung metastasis in mice. Electron microscopy showed that chloroquine disrupted the structural integrity of mitochondria. This led to mitochondrial dysfunction, ROS generation, and oxidative DNA lesions in both CSCs and non-CSC subpopulations. Importantly, chloroquine combined with carboplatin inhibited the homologous recombination-mediated repair of DNA double-strand breaks by selectively decreasing Rad50 and Rad51 levels in the CSC subpopulation. In wild-type BRCA1 orthotopic TNBC xenografts, a chloroquine–carboplatin regimen effectively retarded tumor growth. This is of major importance, as in vivo preclinical data indicate that both chloroquine alone and carboplatin alone exhibit negligible antitumor growth effects in a wild-type BRCA1 molecular background. Altogether, these data indicate the possible clinical exploitability of chloroquine combined with carboplatin in the treatment of TNBC, which would harness the anti-CSC properties of chloroquine and the ability of chloroquine in conjunction with carboplatin to prevent the homologous recombination-mediated repair of DNA double-strand breaks. The prevention of DNA repair could force cancer cells to undergo apoptotic death.100 Table S1 summarizes the in vitro data that suggest the potential for repurposing chloroquine.
Chloroquine as a Par-4 secretagogue that stimulates apoptosis in p53-deficient cancer cells
The antitumor effects of chloroquine were identified in a series of in vitro experiments in p53+/+ mouse embryonic fibroblasts cocultured with p53-deficient lung cancer cells (eg, H1299), prostate cancer cells (PC3), or p53-expressing cancer cells (H460), as well as in models of metastasis. These antitumor effects can be harnessed for apoptosis stimulation, even in tumors that do not express functional p53, as a result of paracrine apoptosis. Indeed, chloroquine has been found to serve as a secretagogue of Par-4 (a tumor-suppressor protein): it activates p53 and stimulates the p53-dependent secretion of Par-4 by suppressing the expression of its binding partner UACA, thereby abolishing UACA-mediated sequestration of Par-4. Also, it stimulates the transcription of Rab8b, a protein that is involved in the vesicular traffic and secretion of Par-4, as the Rab8b promoter contains a p53-binding site. Both these events (inhibition of UACA expression and upregulation of Rab8b) occurred in a p53-dependent fashion in normal cells in response to chloroquine and led to paracrine apoptosis in nearby p53-deficient cancer cells in vitro. In addition, chloroquine prevented the spread of EO771 mouse mammary tumor cells to the lungs in Par-4+/+ mice, but not in Par-4-deficient mice.102
Of note, Arylquin 1, which shares a common quinoline ring with chloroquine, also acts as a Par-4 secretagogue. However, studies have focused on the p53-dependent stimulation by chloroquine of paracrine apoptosis in p53-deficient cells, because this antimalarial drug has already been approved by the US Food and Drug Administration (although it is not approved as an antineoplastic agent) and is relatively safe.102
Antigrowth and antiangiogenic properties of chloroquine may be harnessed for treatment of malignant pleural effusion
The interference of chloroquine with angiogenesis has been evaluated at multiple levels using various experimental settings. Specifically, chloroquine displayed antiangiogenic properties in an in vitro human umbilical vein endothelial cell (HUVEC) tube-formation assay, in which chloroquine suppressed the ability of HUVECs to give rise to tube-like structures. Also, chloroquine negated the proliferative and migratory potency of HUVECs. The antiangiogenic effects of chloroquine on HUVECs may be attributed to the downregulation of Ang2, phospho-Akt, and the Notch ligand JAG1. The fact that chloroquine counteracts angiogenesis has further been validated using the in vivo angiogenesis-screening chicken chorioallantoic membrane model, and chloroquine also decreased microvascular density in malignant pleural effusion (MPE) xenograft models. Additionally, chloroquine decreased pleural tumor foci and pleural efflux in MPE animals. These data indicate that chloroquine could possibly be used in the treatment of MPE.103 This is of major importance, because current options for the management of MPE are primarily palliative. Figure 1 collectively represents most of the well-described autophagy-independent signaling pathways that have been proposed to mediate the tumor-suppressive properties of chloroquine so far. It visually underlines the fact that the anticancer functions of chloroquine go far beyond autophagy blockage.
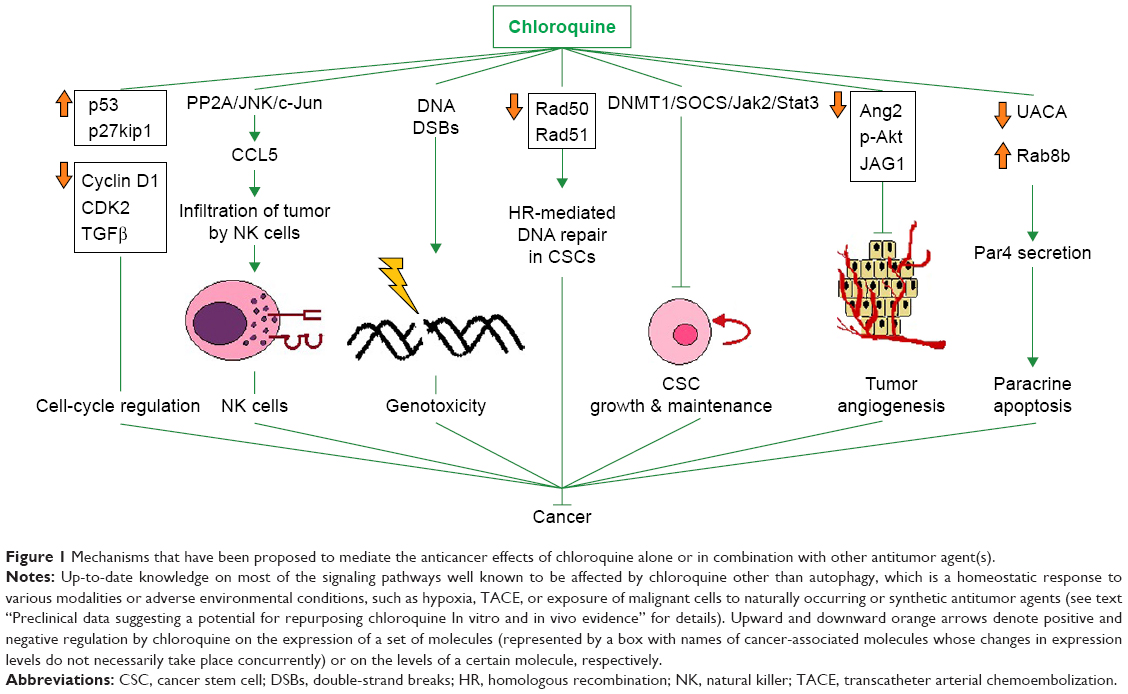 | Figure 1 Mechanisms that have been proposed to mediate the anticancer effects of chloroquine alone or in combination with other antitumor agent(s). |
Octreotide may interact directly with components of the Wnt pathway
As previously mentioned, an array-based study that was carried out a couple of years ago62 reported that octreotide influenced the expression of 30 Wnt-associated genes. These findings provided us with a reasonable impetus to explore whether octreotide also regulates the Wnt pathway at the molecular level. Of the 30 molecules, 12 (APC2, Axin1, GSK3β, β-catenin, SENP2, LRP5, Kremen1, DKK1, and the Frizzled family members FZD4, FZD7, FZD8, and FZD10) were randomly chosen, and possible protein–drug interactions were investigated using the AutoDock Vina program.
Octreotide possibly interacts with components of the β-catenin destruction complex
The predicted models according to AutoDock Vina regarding the binding of octreotide to Axin1 and β-catenin as well as the models of the APC2–Axin1 and β-catenin–Axin1 complexes are shown in Figures 2 and 3. According to AutoDock Vina, octreotide may bind to APC2 at two positions with affinity ΔG=−9.1 kcal/mol and ΔG=−7.2 kcal/mol, respectively. However, these positions do not correlate with APC2-binding sites for factors associated with the Wnt pathway. Nevertheless, it cannot be excluded that the suggested binding allosterically interferes with the binding of other factors. Remarkably, AutoDock Vina predicts that certain residues (yellow shaded residues in Table S2) of regulators of the G-protein signaling homologous domain of Axin1 mediate both APC2 binding (1EMU.pdb) and docking of octreotide.
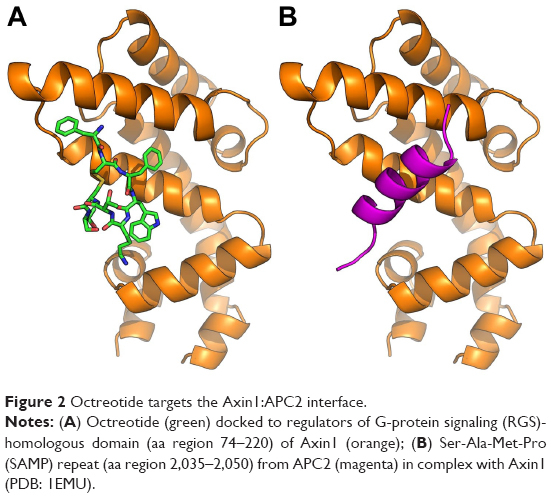 | Figure 2 Octreotide targets the Axin1:APC2 interface. |
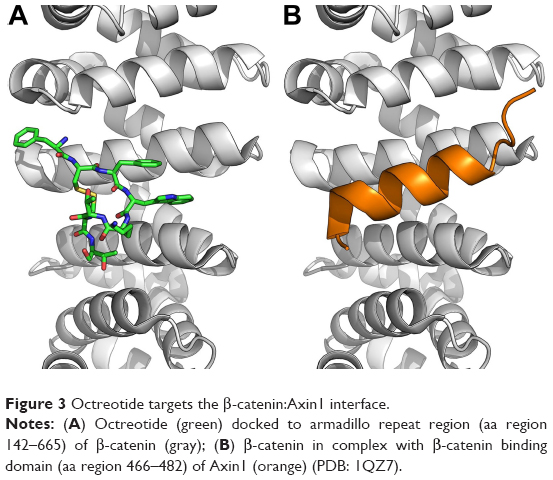 | Figure 3 Octreotide targets the β-catenin:Axin1 interface. |
Through a certain set of amino acid (aa) residues, β-catenin interacts with both Axin1 and octreotide (yellow shaded residues in Table S2). AutoDock Vina suggests a less stable binding (ΔG=−4.9 kcal/mol) of Axin1 to β-catenin (1QZ7.pdb) than the binding (ΔG=−7.3 kcal/mol) of octreotide to β-catenin. This indicates that octreotide probably antagonizes the binding of Axin1 to β-catenin. The model of the interaction between octreotide and SENP2 (a negative regulator of β-catenin) and the interaction between SENP2 and SMT3C (SUMO1; 1TGZ.pdb), according to AutoDock Vina, is depicted in Figure 4. Interestingly, SENP2 contains residues that mediate its interaction with both octreotide and SMT3C (yellow shaded residues in Table S2).
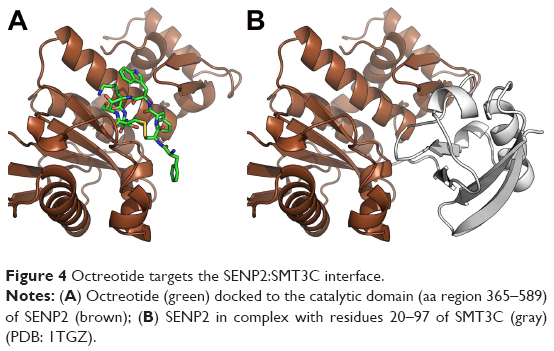 | Figure 4 Octreotide targets the SENP2:SMT3C interface. |
The C-terminal domain of Axin1 contains a DIX domain and two other distinct domains (ie, D and I domains) that are known to allow the homodimerization of Axin1. The formation of Axin1 dimers is thought to be essential for shutting off TCF–LEF1 transcription, while impaired homodimerization differentially affects Axin1-dependent pathways.104,105 In fact, 11 of 25 aa residues of chain A of Axin1 that make up the interface in the Axin1 homodimer model belong to the group of residues that potentially interact with octreotide (Table S2). Therefore, octreotide may prevent Axin1 homodimerization and impinge on Axin 1-dependent pathways.
According to LALIGN alignment software (https://www.ebi.ac.uk/tools/psa/lalign), Axin1 and Axin2 show 42.6% identity and 66.1% similarity in 886 aa overlap. Therefore, it is reasonable to assume that these molecules share common binding sites for octreotide. This merits further investigation. Axin2 residues that are predicted to interact with octreotide are shown in Table S2. Axin1 and octreotide do not share a common binding site on GSK-3β. Interestingly, as shown in Table S2, the binding site for octreotide on GSK-3β includes Y216 (1O9U.pdb). Given that autophosphorylation at Y216 is required for the formation of the kinase-active molecule p-GSK-3β-Υ216,106 the predicted interaction between octreotide and GSK-3β may affect the activity of GSK-3β.
Octreotide is predicted to interact with components of the DKK1–LRP5/6–Kremen1/2 ternary complex
According to AutoDock Vina, models of the binding of octreotide to DKK1 and Kremen1, as well as those of DKK1 with the LRP5 ectodomain and DKK1–Kremen1, are depicted in Figures 5 and 6. The DKK1 crystal structure 3S2K.pdb indicates two binding interfaces involving LRP5-ectodomain repeats 3 and 4 (the LRP5/6 ectodomain contains four repeating units that adopt a β-propeller structure). Using a homology model of LRP5 based on 3S2K.pdb (chains A and B), the docking procedure suggests two octreotide-binding sites, both at the interface between DKK1 and LRP5 (common residues through which DKK1 interacts both with octreotide and with LRP5 are shown in yellow in Table S2). The crystal structure 5FWW.pdb suggests a binding interface between DKK1 (chain C) and Kremen1 (chain B). Certain residues (shown in yellow in Table S2) of Kremen1 mediate its binding with both octreotide and DKK1. Therefore, octreotide may interfere with DKK1–Kremen1 complex formation.
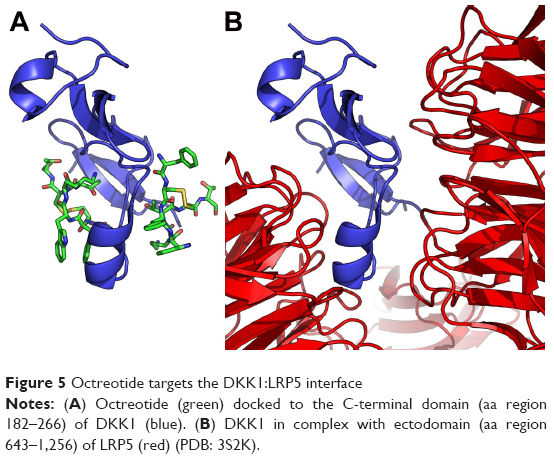 | Figure 5 Octreotide targets the DKK1:LRP5 interface |
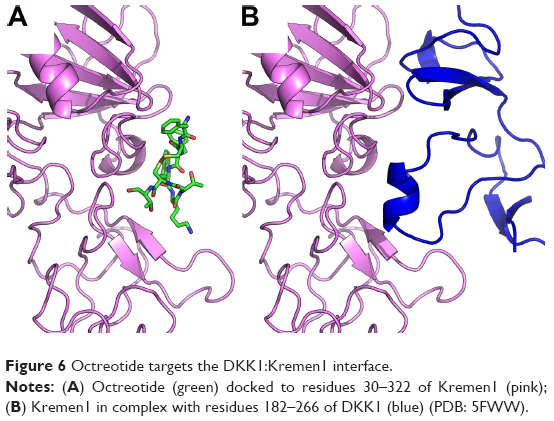 | Figure 6 Octreotide targets the DKK1:Kremen1 interface. |
Overall, in silico data suggest that octreotide interacts with proteins that form two ternary complexes of the Wnt pathway. The first ternary complex is the “β-catenin destruction complex”, which involves the tumor suppressor APC, the scaffolding protein Axin (either Axin1 or Axin2), and GSK-3β (which phosphorylates β-catenin and marks it for proteasomal degradation). This ternary complex binds with β-catenin, a positive effector of the Wnt pathway that is stabilized by Wnt and in turn drives the expression of Wnt targets.107–109 Second, octreotide interferes with components of the DKK1–LRP5/6–Kremen1/2 ternary complex encompassing the secreted Wnt inhibitor DKK1 and the DKK1-binding receptors LRP5/6 and Kremen1/2.110–112 Importantly, AutoDock Vina predicts that some of the aa residues that mediate the binding of octreotide to Axin1, β-catenin, SENP2, DKK1, and Kremen1, respectively. also mediate the interaction of the aforementioned proteins to APC2, Axin1, SMT3C, LRP5, and DKK1. Octreotide is predicted to interact with the de-SUMOylase SENP2, a protein that is known to stimulate the degradation of β-catenin in a manner that depends upon its SUMO-deconjugating activity.70,113 This adds another level of complexity to the mechanism by which octreotide possibly impinges on the Wnt pathway by interacting with multiple components of the Wnt-signaling axis. Therefore, it is increasingly clear that the Wnt pathway is highly wired not only with regard to the signaling complexity itself but also from a structural biology point of view. The predicted interference of octreotide with components of the aforementioned multiprotein complexes would result in the formation of numerous protein–protein and protein–drug interfaces.
Clinical data
Octreotide
Early clinical data on the use of octreotide as an antitumor agent in the treatment of advanced pancreatic cancer indicated clinical benefits (survival prolongation and disease stabilization) without compromising quality of life, even when octreotide was administered at high doses (2 mg three times/day).114 Patients with inoperable or resected pancreatic adenocarcinoma treated with a combinational regimen of octreotide (100 μg three times/day) and tamoxifen (10 mg twice/day) experienced a survival benefit compared with untreated patients.115 According to a prospective trial in recurrent pancreatic carcinoma patients, an octreotide–tamoxifen regimen (100 μg octreotide three times/day plus 20 mg tamoxifen once/day) increased life span and improved quality of life postoperatively without severe adverse effects, whereas these beneficial effects were not observed in best supportive care-treated patients.116 However, a Phase III study indicated that octreotide (200 μg three times/day) did not confer any clinical benefit to patients with advanced pancreatic cancer compared with those who were treated with conventional anticancer agents (5-fluorouracil with or without leucovorin).117 Nevertheless, data from a prospective Phase IV study indicated that the administration of 20 mg octreotide long-acting release (LAR) resulted in disease stabilization in a considerable proportion (38%) of patients with advanced pancreatic endocrine carcinoma, irrespective of intratumoral SSTR2- and SSTR5-transcript levels during a median follow-up of 49.5 months.118 In a prospective study, researchers recruited patients with progressive metastatic pancreatic endocrine tumor (gastrinoma). The initial round of treatment involved 200 μg octreotide twice a day followed by maintenance therapy involving 20–30 mg octreotide LAR monthly. Remarkably, there was a significant survival benefit among responders (patients who primarily experienced tumor stabilization and [less commonly] tumor shrinkage) compared with nonresponders, without any severe adverse effects.119
A randomized trial suggested that octreotide (200 μg three times a day, 5 of 7 days/week) given to patients with advanced GI cancer refractory to chemotherapeutic agents stabilized the disease in comparison with supportive care only.120 Survival prolongation in patients with advanced HCC who were treated with octreotide has also been reported in another randomized controlled trial.121 A Phase III clinical study involving patients with advanced NETs who were treated with octreotide LAR (20 mg every 3 weeks) combined either with IFNα2b (5 million units three times a week) or bevacizumab (15 mg/kg every 3 weeks) did not report any significant difference in progression-free survival (PFS) between these two arms (PFS of 16.6 vs 15.4 months in the bevacizumab and IFN arms, respectively).122 Data from the PROMID study group, which conducted a Phase IIIB trial, indicated that octreotide LAR (30 mg monthly) significantly delayed tumor progression in patients with metastatic midgut NETs compared to placebo.123 Hepatic tumor burden, rather than administration of octreotide LAR, seemed to affect overall survival in volunteers.124
Two Phase III clinical trials – MA.14 and B-29, addressed the possible clinical benefits of a combination octreotide–tamoxifen regimen compared to tamoxifen alone in early breast cancer patients. Unfortunately, for both trials, the sample of participants who were eventually successfully recruited was smaller than initially planned. In addition, in both trials, adverse effects associated with gallbladder physiology resulted in failure of the initially planned 5-year octreotide-treatment regimen. Therefore, it has been suggested that the aforementioned clinical studies underline the importance of interfering with insulin and IGF signaling in breast tumorigenesis by exploiting the anticancer properties of pharmacological agents other than octreotide, such as metformin or small molecules targeting the insulin–IGF axis.125 A Phase III study reported that octreotide LAR plus everolimus (10 mg/day per os) increased PFS in comparison to octreotide LAR plus placebo in patients with advanced NETs.126 Further clinical trials on the safety and antitumor efficacy of octreotide in patients with tumors (originating from the neuroendocrine system or not) are necessary to extend the usage of octreotide in the field of oncology.
Chloroquine
According to a Phase III clinical trial, a clinically achievable dosing regimen of chloroquine (150 mg/day per os for 1 year) in combination with conventional chemotherapy and radiotherapy conferred a substantial survival benefit in GBM patients.127 An ongoing Phase II clinical trial (NCT01446016) corroborated the notion that taxanes combined with chloroquine are clinically exploitable: in some women with treatment-refractory advanced or metastatic breast cancer, chloroquine plus paclitaxel resulted in a decrease in the CSC subpopulation of tumors up to sixfold.99 Interestingly, the number of autophagy vesicles/cells among peripheral blood mononuclear cells of study participants (based on electron microscopy) was a valuable biomarker of autophagy inhibition.128 Additional clinical studies are needed to assess the safety and anticancer potency of chloroquine. Table 1 summarizes selected completed or ongoing clinical trials (with various recruitment status) testing chloroquine in cancer patients. The publication of the results of these studies is eagerly anticipated.
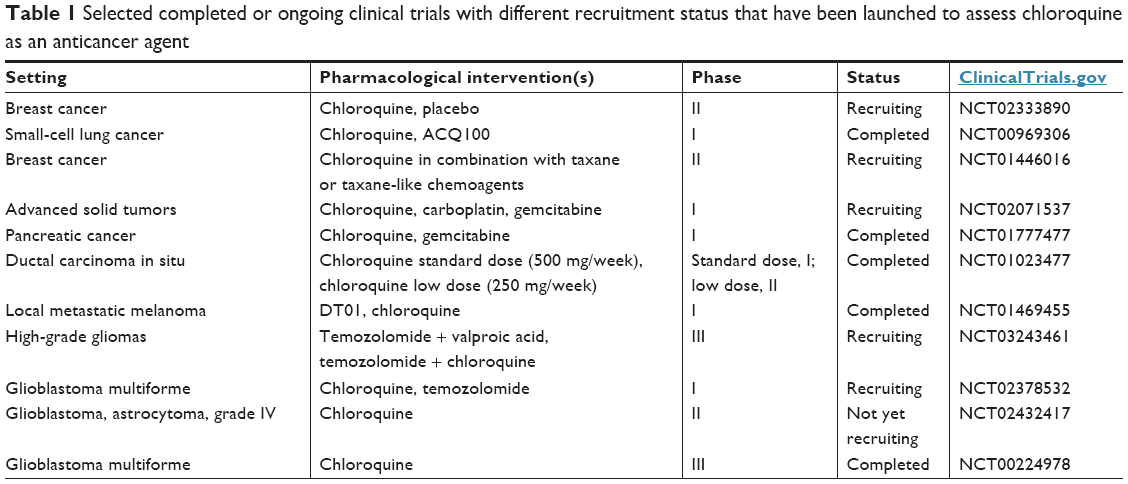 | Table 1 Selected completed or ongoing clinical trials with different recruitment status that have been launched to assess chloroquine as an anticancer agent |
Considerations for expanding the use of octreotide and for repurposing chloroquine
Clinical evidence suggests that octreotide is safe even when administered at high doses to patients with various diseases. Indeed, in acromegalic patients, a high-dose regimen of 60 mg every 4 weeks administered intramuscularly has been shown to be safe, with only minor side effects in some volunteers and no dose–response relationship regarding GI side effects.129 In patients with GI NETs who were treated with octreotide LAR at doses >30 mg/month, no severe toxic effects were reported, possibly indicating a favorable safety profile involving a scarcity of severe side effects.130 A patient who had received an esophagectomy developed chylothorax that was satisfactorily managed with high-dose octreotide (0.25 mg three times a day for 2 weeks), this noninvasive choice was proven safe.29
These data underline the notion that blocking autophagy with chloroquine is a promising anticancer strategy. Importantly, there are several decades of clinical experience regarding the use of this drug, and it could be used as an adjunct at clinically attainable doses (10 mg/kg/day).20 However, a series of safety issues arise regarding the use of chloroquine in oncology, due to the fact that it interferes with the homeostatic process of autophagy, which is pivotal for certain tissue, such as the myocardium and neuronal tissue. Indeed, there is in vivo preclinical evidence that cellular physiology is compromised in the proximal tubules of autophagy-deficient animals and that the sensitivity of kidneys to noxious stimuli (including cisplatin) is increased. As such, clinical data validating the safe use of chloroquine are necessary before its possible exploitation in the field of oncology. These data could resolve any doubt about the possible exacerbation of the myocardial toxicity of anthracyclines when they are administered with chloroquine, or even the possible tumor-promoting effects of chloroquine in different types of tissues, given that autophagy is known to be involved in the suppression of spontaneous tumorigenesis.33 Additionally, the ocular toxicity,131 neurotoxicity (drug-induced recurrent psychosis),132 and skin reactions133,134 that have been linked to the usage of chloroquine should be carefully considered before using chloroquine as an anticancer agent.
Conclusion
Exploiting the repositionability of distinct commercially available pharmaceuticals and/or the complete therapeutic potential of agents that are currently clinically underrated may be important strategies for adding “novel” anticancer drugs to the arsenal of oncologists. In comparison to newly designed/discovered pharmaceuticals, repositionable and/or underexploited agents are plausibly advantageous for many reasons, and are detailed in Figure 7.
 | Figure 7 Advantages of fully exploiting a marketed drug’s therapeutic potential and advantageous features of drug repositioning in oncology. |
Thus far, the therapeutic usage of unradiolabeled octreotide has an application in the management of NETs that are primarily located in the GI system. In addition, octreotide has long been believed to exhibit palliative value, rather than direct antitumor effects. However, both preclinical and clinical data indicate the direct antitumorigenic effects of octreotide, which could be harnessed for the management of tumors other than rare cases of GI NETs. Consistently with this, remarkable preclinical anticancer properties of octreotide have been reported in various non-NET cancer cell lines. This notion is also corroborated by clinical data, as octreotide has been found to be of therapeutic value not only in the management of advanced NETs but also in the treatment of advanced HCC and advanced GI cancer. At the mechanistic level, the anticancer function of octreotide has been linked to various molecular events, including interference with signaling mediated by multiple Wnt/β-catenin components, telomerase, protein-tyrosine phosphatases, VEGF, Fas/FaL, EGFR, MRP2, and MDR1 transporters or even actin cytoskeleton rearrangements (which affect cancer cell viability, cell cycle progression, metastatic potency, and angiogenesis in various experimental settings). Of note, in vitro data indicate that (at least in certain cancer cells) long-term exposure to octreotide does not result in the acquisition of drug resistance. More significantly, it reverses resistance to cisplatin and paclitaxel in ovarian cancer cell lines.
A major challenge for the future is to address whether the reported in vitro octreotide-induced reversal of drug resistance in ovarian cancer cells also occurs at the clinical level and is physiologically relevant for tumors other than ovarian cancer. In addition, another interesting topic for future investigation is the assessment of the therapeutic value of octreotide in the treatment of advanced pancreatic cancer, given that previous clinical research on this type of malignancy has yielded contradictory results. More preclinical and clinical experimentation is needed to enable the full therapeutic exploitation of octreotide in oncology.
The results of the in silico docking study presented here indicate that octreotide regulates Wnt not only at the level of gene expression but also at the level of molecular interactions with at least 12 Wnt-associated proteins (Table S2). These proteins are either predicted to be components of two ternary complexes, namely the β-catenin destruction complex (APC2, GSK-3β, and Axin1 or Axin2, along with β-catenin) and the DKK1–LRP5/6–Kremen1/2 complex (DKK1 or Kremen1), or they are predicted to be Frizzled protein family members (FZD4, FZD7, FZD8, and FZD10) or involved in β-catenin stability regulation (SENP2).
Whether the binding of octreotide to all these Wnt-associated proteins really takes place and the physiological relevance of these protein–drug interactions remain to be experimentally addressed via screening strategies or even through crystallography. The functional impact of the binding of octreotide to any of the aforementioned targets is not known. Another issue that warrants investigation is whether octreotide eventually precludes or enables the formation of Wnt-associated multiprotein complexes. All these topics merit future investigation. This may enable an expansion of the use of octreotide in oncology, given that deregulated Wnt signaling has been linked to the development of numerous types of human cancer.135–138 The docking predictions presented herein strengthen the notion that the anticancer value of octreotide is currently rather underestimated.
Questions remain regarding how the finding that octreotide binds to DKK1 (and probably interferes with the binding of its partners) could be therapeutically translated and the importance of this in silico prediction. In fact, functional assays should be performed in future to delineate whether the interaction of octreotide with DKK1 results in the activation or suppression of DKK1 activity and how this would affect tumorigenesis.
Significantly, data stemming from Phase IB clinical trials regarding the targeting of DKK1 by monoclonal antibodies, namely BHQ880 and DKN-01, in multiple myeloma and advanced tumors, respectively, are encouraging. In addition, interfering with DKK1-dependent signaling may not lead to widespread toxicity, owing to the expression pattern of DKK1 in adult tissues.139 Therefore, targeting DKK1 with octreotide, which is already being safely used in the management of pathologies other than cancer, merits further investigation.
The signaling pathway of autophagy encompasses several druggable components and can be targeted at multiple levels. Blocking autophagy with chloroquine would be advantageous, based on the multidecade experience regarding the use of this pharmacological agent, which exhibits profound anticancer activity in various types of human cancer cells and animal models of human malignancy. Experimentation has shown that chloroquine substantially overrides the drug-resistance and tumor growth-promoting effects of the autophagic response triggered in tumor cells by hypoxia (a common condition in solid tumors) and by therapeutic modalities, such as TACE, in HCC.
It is noteworthy that in MPE, for which only palliative treatment options exist, chloroquine exhibits profound antiangiogenic effects, while it may also be exploited in p53-deficient tumors (which account for ~50% of human malignancies) to suppress tumor growth by functioning as a Par-4 secretagogue. Owing to clinical experience regarding its use, chloroquine (either as a Par-4 secretagogue or as a DNA repair inhibitor) would be advantageous over Arylquin-1 or DT01 (which promote the secretion of Par-4 and inhibit DNA repair, respectively). The usefulness of chloroquine in difficult-to-treat medical conditions (TNBC or MPE) and in p53-deficient tumors (as identified by molecular screening) remains to be addressed. This is another major task for the future. In addition, the completion of several ongoing clinical studies addressing the exploitability of chloroquine in oncology (eg, NCT01446016 and NCT02333890) and the publication of their results is much awaited. Finally, another future challenge is to conduct clinical studies investigating the use of chloroquine to improve NK cell-based cancer immunotherapy, given the recently reported chloroquine-induced redirection of NK cells into melanomas.
Disclosure
The authors report no conflicts of interest in this work.
References
Perwitasari O, Bakre A, Tompkins SM, Tripp RA. siRNA genome screening approaches to therapeutic drug repositioning. Pharmaceuticals. 2013;6(2):124–160. | ||
Corbett A, Williams G, Ballard C. Drug repositioning: an opportunity to develop novel treatments for Alzheimer’s disease. Pharmaceuticals. 2013;6(10):1304–1321. | ||
Batchu RB, Gruzdyn OV, Bryant CS, et al. Ritonavir-mediated induction of apoptosis in pancreatic cancer occurs via the RB/E2F-1 and AKT pathways. Pharmaceuticals. 2014;7(1):46–57. | ||
Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci. 2014;10(7):654–663. | ||
Shameer K, Readhead B, Dudley JT. Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr Top Med Chem. 2015;15(1):5–20. | ||
Wick W, Kessler T. Drug repositioning meets precision in glioblastoma. Clin Cancer Res. 2018;24(2):256–258. | ||
Kim CH, Leblanc P, Kim KS. 4-amino-7-chloroquinoline derivatives for treating Parkinson’s disease: implications for drug discovery. Expert Opin Drug Discov. 2016;11(4):337–341. | ||
Freedman A, Steinberg VL. Chloroquine in rheumatoid arthritis; a double blindfold trial of treatment for one year. Ann Rheum Dis. 1960;19(3):243–250. | ||
Patel JC. Chloroquine in the treatment of amoebic liver abscess. Br Med J. 1953;1(4814):811–813. | ||
Lee SJ, Silverman E, Bargman JM. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat Rev Nephrol. 2011;7(12):718–729. | ||
Papanagnou P, Baltopoulos P, Tsironi M. Marketed nonsteroidal anti-inflammatory agents, antihypertensives, and human immunodeficiency virus protease inhibitors: as-yet-unused weapons of the oncologists’ arsenal. Ther Clin Risk Manag. 2015;11:807–819. | ||
Papanagnou P, Stivarou T, Tsironi M. Unexploited antineoplastic effects of commercially available anti-diabetic drugs. Pharmaceuticals. 2016;9(2):24. | ||
Papanagnou P, Stivarou T, Papageorgiou I, Papadopoulos G, Pappas A. Marketed drugs used for the management of hypercholesterolemia as anticancer armament. OncoTargets and Therapy. 2017;10:4393–4411. | ||
Tang C, Lei H, Zhang J, et al. Montelukast inhibits hypoxia inducible factor-1alpha translation in prostate cancer cells. Cancer Biology & Therapy. 2018:1–7. | ||
Shi TT, Yu XX, Yan LJ, Xiao HT. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol. 2017;79(2):287–294. | ||
Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci. 2017;18(6):E1279. | ||
Hall EA, Ramsey JE, Peng Z, et al. Novel organometallic chloroquine derivative inhibits tumor growth. J Cell Biochem. 2018;119(7):5921–5933. | ||
Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625(1–3):220–233. | ||
Zhang Y, Liao Z, Zhang LJ, Xiao HT. The utility of chloroquine in cancer therapy. Curr Med Res Opin. 2015;31(5):1009–1013. | ||
Pascolo S. Time to use a dose of Chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol. 2016;771:139–144. | ||
Borbath I, Leclercq IA, Sempoux C, Abarca-Quinones J, Desaeger C, Horsmans Y. Efficacy of lanreotide in preventing the occurrence of chemically induced hepatocellular carcinoma in rats. Chem Biol Interact. 2010;183(1):238–248. | ||
Graillon T, Romano D, Defilles C, et al. Pasireotide is more effective than octreotide, alone or combined with everolimus on human meningioma in vitro. Oncotarget. 2017;8(33):55361–55373. | ||
Khandelwal D, Khadgawat R, Mukund A, Suri A. Acromegaly with no pituitary adenoma and no evidence of ectopic source. Indian J Endocrinol Metab. 2011;15(Suppl 3):S250–S252. | ||
Lavrentaki A, Paluzzi A, Wass JA, Karavitaki N. Epidemiology of acromegaly: review of population studies. Pituitary. 2017;20(1):4–9. | ||
Hofland LJ, Lamberts SW. Somatostatin receptors and disease: role of receptor subtypes. Baillieres Clin Endocrinol Metab. 1996;10(1):163–176. | ||
Fougner SL, Bollerslev J, Latif F, et al. Low levels of raf kinase inhibitory protein in growth hormone-secreting pituitary adenomas correlate with poor response to octreotide treatment. J Clin Endocrinol Metab. 2008;93(4):1211–1216. | ||
Neild PJ, Evans DF, Castillo FD, et al. Effect of octreotide on small intestinal motility in HIV-infected patients with chronic refractory diarrhea. Dig Dis Sci. 2001;46(12):2636–2642. | ||
Corbould A, Campbell J. Efficacy of octreotide but not long-acting somatostatin analogue for severe refractory diabetic diarrhoea. Diabet Med. 2009;26(8):828–829. | ||
Bello SO, Rahamim J. High-dose intravenous octreotide is safe and may be superior to surgery in managing severe postesophagectomy chylothorax in high-risk patients. Ann Thorac Surg. 2015;100(1):297–299. | ||
Arnoux JB, Verkarre V, Saint-Martin C, et al. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis. 2011;6(1):63. | ||
Hosokawa Y, Kawakita R, Yokoya S, et al. Efficacy and safety of octreotide for the treatment of congenital hyperinsulinism: a prospective, open-label clinical trial and an observational study in Japan using a nationwide registry. Endocr J. 2017;64(9):867–880. | ||
Jung SY, Rohan T, Strickler H, et al. Genetic variants and traits related to insulin-like growth factor-I and insulin resistance and their interaction with lifestyles on postmenopausal colorectal cancer risk. PLoS One. 2017;12(10):e0186296. | ||
Barbieri F, Bajetto A, Pattarozzi A, et al. Peptide receptor targeting in cancer: the somatostatin paradigm. Int J Pept. 2013;2013(2):1–20. | ||
Peverelli E, Giardino E, Treppiedi D, et al. A novel pathway activated by somatostatin receptor type 2 (SST2): Inhibition of pituitary tumor cell migration and invasion through cytoskeleton protein recruitment. Int J Cancer. 2018;142(9):1842–1852. | ||
Davis MP, Nouneh C. Modern management of cancer-related intestinal obstruction. Curr Oncol Rep. 2000;2(4):343–350. | ||
Ripamonti C, Panzeri C, Groff L, Galeazzi G, Boffi R. The role of somatostatin and octreotide in bowel obstruction: pre-clinical and clinical results. Tumori. 2001;87(1):1–9. | ||
Pokuri VK, Fong MK, Iyer R. Octreotide and lanreotide in gastroenteropancreatic neuroendocrine tumors. Curr Oncol Rep. 2016;18(1):7. | ||
Li Y, Duan Z, Tian Y, Liu Z, Wang Q. A novel perspective and approach to intestinal octreotide absorption: sinomenine-mediated reversible tight junction opening and its molecular mechanism. Int J Mol Sci. 2013;14(6):12873–12892. | ||
Grimberg A. Somatostatin and cancer: applying endocrinology to oncology. Cancer Biol Ther. 2004;3(8):731–733. | ||
Muscarella LA, D’Alessandro V, La Torre A, et al. Gene expression of somatostatin receptor subtypes SSTR2a, SSTR3 and SSTR5 in peripheral blood of neuroendocrine lung cancer affected patients. Cell Oncol. 2011;34(5):435–441. | ||
Sidéris L, Dubé P, Rinke A. Antitumor effects of somatostatin analogs in neuroendocrine tumors. Oncologist. 2012;17(6):747–755. | ||
Mehta S, de Reuver PR, Gill P, et al. Somatostatin receptor SSTR-2a expression is a stronger predictor for survival than Ki-67 in pancreatic neuroendocrine tumors. Medicine. 2015;94(40):e1281. | ||
Phan AT, Halperin DM, Chan JA, et al. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumours: a multicentre, single-group, phase 2 study. Lancet Oncol. 2015;16(6):695–703. | ||
Maxwell JE, Howe JR. Imaging in neuroendocrine tumors: an update for the clinician. Int J Endocr Oncol. 2015;2(2):159–168. | ||
Hamiditabar M, Ali M, Roys J, et al. Peptide receptor radionuclide therapy with 177Lu-octreotate in patients with somatostatin receptor expressing neuroendocrine tumors: six years’ assessment. Clin Nucl Med. 2017;42(6):436–443. | ||
Yau H, Kinaan M, Quinn SL, Moraitis AG. Octreotide long-acting repeatable in the treatment of neuroendocrine tumors: patient selection and perspectives. Biologics. 2017;11:115–122. | ||
Aoki T, Motoi F, Sakata N, et al. Somatostatin analog inhibits the growth of insulinoma cells by p27-mediated G1 cell cycle arrest. Pancreas. 2014;43(5):720–729. | ||
Ben-Shlomo A, Melmed S. Pituitary somatostatin receptor signaling. Trends Endocrinol Metab. 2010;21(3):123–133. | ||
Lehane AM, Hayward R, Saliba KJ, Kirk K. A verapamil-sensitive chloroquine-associated H+ leak from the digestive vacuole in chloroquine-resistant malaria parasites. J Cell Sci. 2008;121(Pt 10):1624–1632. | ||
Coronado LM, Nadovich CT, Spadafora C. Malarial hemozoin: from target to tool. Biochim Biophys Acta. 2014;1840(6):2032–2041. | ||
Al-Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. 2015;70(6):1608–1621. | ||
Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016;35(1):1–11. | ||
Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9(12):2423–2433. | ||
McAfee Q, Zhang Z, Samanta A, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109(21):8253–8258. | ||
Yang A, Kimmelman AC. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy. 2014;10(9):1683–1684. | ||
Gao S, Yu BP, Li Y, Dong WG, Luo HS. Antiproliferative effect of octreotide on gastric cancer cells mediated by inhibition of Akt/PKB and telomerase. World J Gastroenterol. 2003;9(10):2362–2365. | ||
Wang C, Tang C. Inhibition of human gastric cancer metastasis by ocreotide in vitro and in vivo. Zhonghua Yi Xue Za Zhi. 2002;82(1):19–22. | ||
Wang L, Huang X, Chai Y, Zou L, Chedrawe M, Ding Y. Octreotide inhibits the proliferation of gastric cancer cells through P300-HAT activity and the interaction of ZAC and P300. Oncol Rep. 2017;37(4):2041–2048. | ||
Hoffmann A, Barz T, Spengler D. Multitasking C2H2 zinc fingers link Zac DNA binding to coordinated regulation of p300-histone acetyltransferase activity. Mol Cell Biol. 2006;26(14):5544–5557. | ||
Li SC, Martijn C, Cui T, et al. The somatostatin analogue octreotide inhibits growth of small intestine neuroendocrine tumour cells. PLoS One. 2012;7(10):e48411. | ||
Wang S, Bao Z, Liang QM, et al. Octreotide stimulates somatostatin receptor-induced apoptosis of SW480 colon cancer cells by activation of glycogen synthase kinase-3β, A Wnt/β-catenin pathway modulator. Hepatogastroenterology. 2013;60(127):1639–1646. | ||
Chen JS, Liang QM, Li HS, Yang J, Wang S, Long JW. Octreotide inhibits growth of colonic cancer SW480 cells by modulating the Wnt/P-catenin pathway. Pharmazie. 2009;64(2):126–131. | ||
Ayiomamitis GD, Notas G, Zaravinos A, et al. Effects of octreotide and insulin on colon cancer cellular proliferation and correlation with hTERT activity. Oncoscience. 2014;1(6):457–467. | ||
Liu HL, Huo L, Wang L. Octreotide inhibits proliferation and induces apoptosis of hepatocellular carcinoma cells. Acta Pharmacol Sin. 2004;25(10):1380–1386. | ||
Ma Q, Meng LQ, Liu JC, et al. Octreotide induces apoptosis of human hepatoma cells by the mechanism of facilitating the Fas/FasL gene expression therein. Zhonghua Yi Xue Za Zhi. 2008;88(10):716–718. | ||
Davies N, Kynaston H, Yates J, et al. Octreotide inhibits the growth and development of three types of experimental liver metastases. Br J Surg. 1995;82(6):840–843. | ||
Jia WD, Xu GL, Wang W, et al. A somatostatin analogue, octreotide, inhibits the occurrence of second primary tumors and lung metastasis after resection of hepatocellular carcinoma in mice. Tohoku J Exp Med. 2009;218(2):155–160. | ||
Tang X, Takekoshi S, Itoh J, et al. Somatostatin analogue inhibits the mobility of prostate carcinoma cells: a new therapeutic method for advanced prostate carcinoma. Int J Oncol. 2010;37(5):1077–1083. | ||
Hennigs JK, Müller J, Adam M, et al. Loss of somatostatin receptor subtype 2 in prostate cancer is linked to an aggressive cancer phenotype, high tumor cell proliferation and predicts early metastatic and biochemical relapse. PLoS One. 2014;9(7):e100469. | ||
Shen Y, Ren M, Shi Y, Zhang Y, Cai Y. Octreotide enhances the sensitivity of the SKOV3/DDP ovarian cancer cell line to cisplatin chemotherapy in vitro. Exp Ther Med. 2011;2(6):1171–1176. | ||
Shen Y, Zhang XY, Chen X, Ren ML, Cai YL. Octreotide reverses the resistance of A2780/Pacliaxel ovarian cancer cell line to paclitaxel chemotherapy in vitro. J Cancer Res Ther. 2016;12(2):657–662. | ||
Moreno A, Akcakanat A, Munsell MF, Soni A, Yao JC, Meric-Bernstam F. Antitumor activity of rapamycin and octreotide as single agents or in combination in neuroendocrine tumors. Endocr Relat Cancer. 2008;15(1):257–266. | ||
Hoelting T, Duh QY, Clark OH, Herfarth C. Somatostatin analog octreotide inhibits the growth of differentiated thyroid cancer cells in vitro, but not in vivo. J Clin Endocrinol Metab. 1996;81(7):2638–2641. | ||
Dalezis P, Geromichalos GD, Voyatzi S, et al. Combinational effect of topotecan and octreotide on murine leukemia cells in vivo and in vitro. J BUON. 2006;11(3):323–327. | ||
Choi JH, Yoon JS, Won YW, Park BB, Lee YY. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. APMIS. 2012;120(7):597–604. | ||
Zheng Y, Zhao YL, Deng X, et al. Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis. Cancer Invest. 2009;27(3):286–292. | ||
Zeilhofer HU, Mollenhauer J, Brune K. Selective growth inhibition of ductal pancreatic adenocarcinoma cells by the lysosomotropic agent chloroquine. Cancer Lett. 1989;44(1):61–66. | ||
Monma H, Iida Y, Moritani T, et al. Chloroquine augments TRAIL-induced apoptosis and induces G2/M phase arrest in human pancreatic cancer cells. PLoS One. 2018;13(3):e0193990. | ||
Frieboes HB, Huang JS, Yin WC, McNally LR. Chloroquine-mediated cell death in metastatic pancreatic adenocarcinoma through inhibition of autophagy. JOP. 2014;15(2):189–197. | ||
Song J, Qu Z, Guo X, et al. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy. 2009;5(8):1131–1144. | ||
Gao L, Song JR, Zhang JW, et al. Chloroquine promotes the anticancer effect of TACE in a rabbit VX2 liver tumor model. Int J Biol Sci. 2013;9(4):322–330. | ||
Aqbi HF, Tyutyunyk-Massey L, Keim RC, et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget. 2018;9(31):22113–22122. | ||
Hu T, Li P, Luo Z, et al. Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo. Oncol Rep. 2016;35(1):43–49. | ||
Lamoureux F, Zoubeidi A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy. 2013;9(7):1119–1120. | ||
Wu Z, Chang PC, Yang JC, et al. Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors. Genes Cancer. 2010;1(1):40–49. | ||
Fukuda T, Oda K, Wada-Hiraike O, et al. Autophagy inhibition augments resveratrol-induced apoptosis in Ishikawa endometrial cancer cells. Oncol Lett. 2016;12(4):2560–2566. | ||
Fukuda T, Oda K, Wada-Hiraike O, et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecol Oncol. 2015;137(3):538–545. | ||
Fan C, Wang W, Zhao B, Zhang S, Miao J. Chloroquine inhibits cell growth and induces cell death in A549 lung cancer cells. Bioorg Med Chem. 2006;14(9):3218–3222. | ||
Liu L, Han C, Yu H, et al. Chloroquine inhibits cell growth in human A549 lung cancer cells by blocking autophagy and inducing mitochondrialmediated apoptosis. Oncol Rep. 2018;39(6):2807–2816. | ||
Avniel-Polak S, Leibowitz G, Doviner V, Gross DJ, Grozinsky-Glasberg S. Combining chloroquine with RAD001 inhibits tumor growth in a NEN mouse model. Endocr Relat Cancer. 2018;25(6):677–686. | ||
Avniel-Polak S, Leibowitz G, Riahi Y, Glaser B, Gross DJ, Grozinsky-Glasberg S. Abrogation of autophagy by chloroquine alone or in combination with mTOR inhibitors induces apoptosis in neuroendocrine tumor cells. Neuroendocrinology. 2016;103(6):724–737. | ||
Aveic S, Pantile M, Seydel A, et al. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget. 2016;7(5):5646–5663. | ||
Roy LO, Poirier MB, Fortin D. Chloroquine inhibits the malignant phenotype of glioblastoma partially by suppressing TGF-beta. Invest New Drugs. 2015;33(5):1020–1031. | ||
Spc H, Kuo JS, Chiang HC, et al. Temozolomide, sirolimus and chloroquine is a new therapeutic combination that synergizes to disrupt lysosomal function and cholesterol homeostasis in GBM cells. Oncotarget. 2018;9(6):6883–6896. | ||
Egger ME, Huang JS, Yin W, McMasters KM, McNally LR. Inhibition of autophagy with chloroquine is effective in melanoma. J Surg Res. 2013;184(1):274–281. | ||
Mgrditchian T, Arakelian T, Paggetti J, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A. 2017;114(44):E9271–E9279. | ||
Jiang PD, Zhao YL, Shi W, et al. Cell growth inhibition, G2/M cell cycle arrest, and apoptosis induced by chloroquine in human breast cancer cell line Bcap-37. Cell Physiol Biochem. 2008;22(5–6):431–440. | ||
Yang W, Hosford SR, Traphagen NA, et al. Autophagy promotes escape from phosphatidylinositol 3-kinase inhibition in estrogen receptor-positive breast cancer. Faseb J. 2018;32(3):1222–1235. | ||
Choi DS, Blanco E, Kim YS, et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells. 2014;32(9):2309–2323. | ||
Liang DH, Choi DS, Ensor JE, Kaipparettu BA, Bass BL, Chang JC. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016;376(2):249–258. | ||
Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8(2):200–212. | ||
Burikhanov R, Hebbar N, Noothi SK, et al. Chloroquine-inducible Par-4 secretion is essential for tumor cell apoptosis and inhibition of metastasis. Cell Rep. 2017;18(2):508–519. | ||
Li Q, Yuan DM, Ma LH, et al. Chloroquine inhibits tumor growth and angiogenesis in malignant pleural effusion. Tumour Biol. 2016:16249–16258. | ||
Dahmen RP, Koch A, Denkhaus D, et al. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res. 2001;61(19):7039–7043. | ||
Luo W, Zou H, Jin L, et al. Axin contains three separable domains that confer intramolecular, homodimeric, and heterodimeric interactions involved in distinct functions. J Biol Chem. 2005;280(6):5054–5060. | ||
Credle JJ, George JL, Wills J, et al. GSK-3β dysregulation contributes to parkinson’s-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and α-synuclein. Cell Death Differ. 2015;22(5):838–851. | ||
Spink KE, Polakis P, Weis WI. Structural basis of the Axin-adenomatous polyposis coli interaction. Embo J. 2000;19(10):2270–2279. | ||
Wu ZQ, Brabletz T, Fearon E, et al. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc Natl Acad Sci U S A. 2012;109(28):11312–11317. | ||
Stamos JL, Weis WI. The β-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5(1):a007898. | ||
Dun X, Jiang H, Zou J, et al. Differential expression of DKK-1 binding receptors on stromal cells and myeloma cells results in their distinct response to secreted DKK-1 in myeloma. Mol Cancer. 2010;9:247. | ||
Ahn VE, Chu ML, Choi HJ, Tran D, Abo A, Weis WI. Structural basis of Wnt signaling inhibition by Dickkopf binding to LRP5/6. Dev Cell. 2011;21(5):862–873. | ||
Joiner DM, Ke J, Zhong Z, Xu HE, Williams BO. LRP5 and LRP6 in development and disease. Trends Endocrinol Metab. 2013;24(1):31–39. | ||
Jiang QF, Tian YW, Shen Q, Xue HZ, Li K. SENP2 regulated the stability of β-catenin through WWOX in hepatocellular carcinoma cell. Tumour Biol. 2014;35(10):9677–9682. | ||
Ebert M, Friess H, Beger HG, Büchler MW. Role of octreotide in the treatment of pancreatic cancer. Digestion. 1994;55(Suppl 1):48–51. | ||
Rosenberg L, Barkun AN, Denis MH, Pollak M. Low dose octreotide and tamoxifen in the treatment of adenocarcinoma of the pancreas. Cancer. 1995;75(1):23–28. | ||
Wenger FA, Jacobi CA, Siderow A, Zieren J, Zieren HU, Muller JM. Hormone therapy of postoperative recurrent pancreatic carcinoma with octreotide and tamoxifen. Der Chirurg; Zeitschrift fur alle Gebiete der operativen Medizen. 1999;70(6):694–699. | ||
Burch PA, Block M, Schroeder G, et al. Phase III evaluation of octreotide versus chemotherapy with 5-fluorouracil or 5-fluorouracil plus leucovorin in advanced exocrine pancreatic cancer: a North Central Cancer Treatment Group study. Clin Cancer Res. 2000;6(9):3486–3492. | ||
Butturini G, Bettini R, Missiaglia E, et al. Predictive factors of efficacy of the somatostatin analogue octreotide as first line therapy for advanced pancreatic endocrine carcinoma. Endocr Relat Cancer. 2006;13(4):1213–1221. | ||
Shojamanesh H, Gibril F, Louie A, et al. Prospective study of the antitumor efficacy of long-term octreotide treatment in patients with progressive metastatic gastrinoma. Cancer. 2002;94(2):331–343. | ||
Cascinu S, Del Ferro E, Catalano G. A randomised trial of octreotide vs best supportive care only in advanced gastrointestinal cancer patients refractory to chemotherapy. Br J Cancer. 1995;71(1):97–101. | ||
Kouroumalis EA. Octreotide for cancer of the liver and biliary tree. Chemotherapy. 2001;47(2):150–161. | ||
Yao JC, Guthrie KA, Moran C, et al. Phase III prospective randomized comparison trial of depot octreotide plus interferon Alfa-2b versus depot octreotide plus bevacizumab in patients with advanced carcinoid tumors: SWOG S0518. J Clin Oncol. 2017;35(15):1695–1703. | ||
Rinke A, Müller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663. | ||
Rinke A, Wittenberg M, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors (PROMID): results of long-term survival. Neuroendocrinology. 2017;104(1):26–32. | ||
Chapman JA, Costantino JP, Dong B, et al. Octreotide LAR and tamoxifen versus tamoxifen in phase III randomize early breast cancer trials: NCIC CTG MA.14 and NSABP B-29. Breast Cancer Res Treat. 2015;153(2):353–360. | ||
Pavel ME, Hainsworth JD, Baudin E, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378(9808):2005–2012. | ||
Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144(5):337–343. | ||
Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1391–1402. | ||
Giustina A, Bonadonna S, Bugari G, et al. High-dose intramuscular octreotide in patients with acromegaly inadequately controlled on conventional somatostatin analogue therapy: a randomised controlled trial. Eur J Endocrinol. 2009;161(2):331–338. | ||
Broder MS, Beenhouwer D, Strosberg JR, Neary MP, Cherepanov D. Gastrointestinal neuroendocrine tumors treated with high dose octreotide-LAR: a systematic literature review. World J Gastroenterol. 2015;21(6):1945–1955. | ||
Tzekov R. Ocular toxicity due to chloroquine and hydroxychloroquine: electrophysiological and visual function correlates. Doc Ophthalmol. 2005;110(1):111–120. | ||
Sahoo S, Kumar M, Sinha VK. Chloroquine-induced recurrent psychosis. American Journal of Therapeutics. 2007;14(4):406–407. | ||
Adebayo RA, Sofowora GG, Onayemi O, Udoh SJ, Ajayi AA. Chloroquine-induced pruritus in malaria fever: contribution of malaria parasitaemia and the effects of prednisolone, niacin, and their combination, compared with antihistamine. Br J Clin Pharmacol. 1997;44(2):157–161. | ||
Rustogi A, Munshi A, Jalali R. Unexpected skin reaction induced by radiotherapy after chloroquine use. Lancet Oncol. 2006;7(7):608–609. | ||
Tai D, Wells K, Arcaroli J, et al. Targeting the WNT signaling pathway in cancer therapeutics. Oncologist. 2015;20(10):1189–1198. | ||
Pai SG, Carneiro BA, Mota JM, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10(1):101. | ||
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–1473. | ||
Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50–60. | ||
Kagey MH, He X. Rationale for targeting the Wnt signalling modulator Dickkopf-1 for oncology. Br J Pharmacol. 2017;174(24):4637–4650. | ||
Vokinger KN, Kesselheim AS, Avorn J, Sarpatwari A. Strategies that delay market entry of generic drugs. JAMA Intern Med. 2017;177(11):1665–1669. | ||
Huang M, Shen A, Ding J, Geng M. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci. 2014;35(1):41–50. | ||
Printz C. Failure rate: Why many cancer drugs don’t receive FDA approval, and what can be done about it. Cancer. 2015;121(10):1529–1530. |
Supplementary materials
Supplementary materials and methods
In silico study
A docking study has been conducted of octreotide against 12 protein targets implicated in the wnt pathway. Protein-ligang docking presented herein focuses on components of two ternary compexes of the Wnt pathway, the β-catenin destruction complex and the DKK1-LRP5/6-Kremen1/2 ternary complex.
Necessary structures
For all docking trials we used a pdb file for octreotide generated from the CCDC file 1302496.cif obtained from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/).
The necessary protein target structures have been obtained as it follows:
For AXIN1 the RGS-homologous domain of AXIN (74–220) of 1DK8.pdb has been used, while for DKK1 (182–266) chain C of 3S2K.pdb. Similarly, for KREMEN1 (30–322) and SENP2 (366–589) chain A of 5FWS.pdb and 1TGZ.pdb has been used respectively. For β-CATENIN (142–665) we used chain A of 1QZ7.pdb. For GSK-3β we used chain A of 1O9U.pdb which includes a phosphorylated Y216. Structures for APC2, AXIN2, FZD4, FZD7, FZD8, FZD10, LRP5 have been modeled using the protein modeling server SWISS-MODEL (https://swissmodel.expasy.org/).25,26
Protein-ligand docking
Before docking all hetero atoms have been removed from the original pdb files. All docking trials of octreotide to the above protein targets have been performed using the program AutoDock Vina24 allowing only ligand flexibility. Binding poses with ΔG > −6.5 kcal/mol have been rejected as well as those located in the lipid-protein interface of the transmembrane proteins. Alternative binding pockets have been accepted for APC2, AXIN1, DKK1, FZD10, KREMEN1 and LRP5 after visual inspection.
 | Figure S1 Chemical structure of octreotide and chloroquine. |
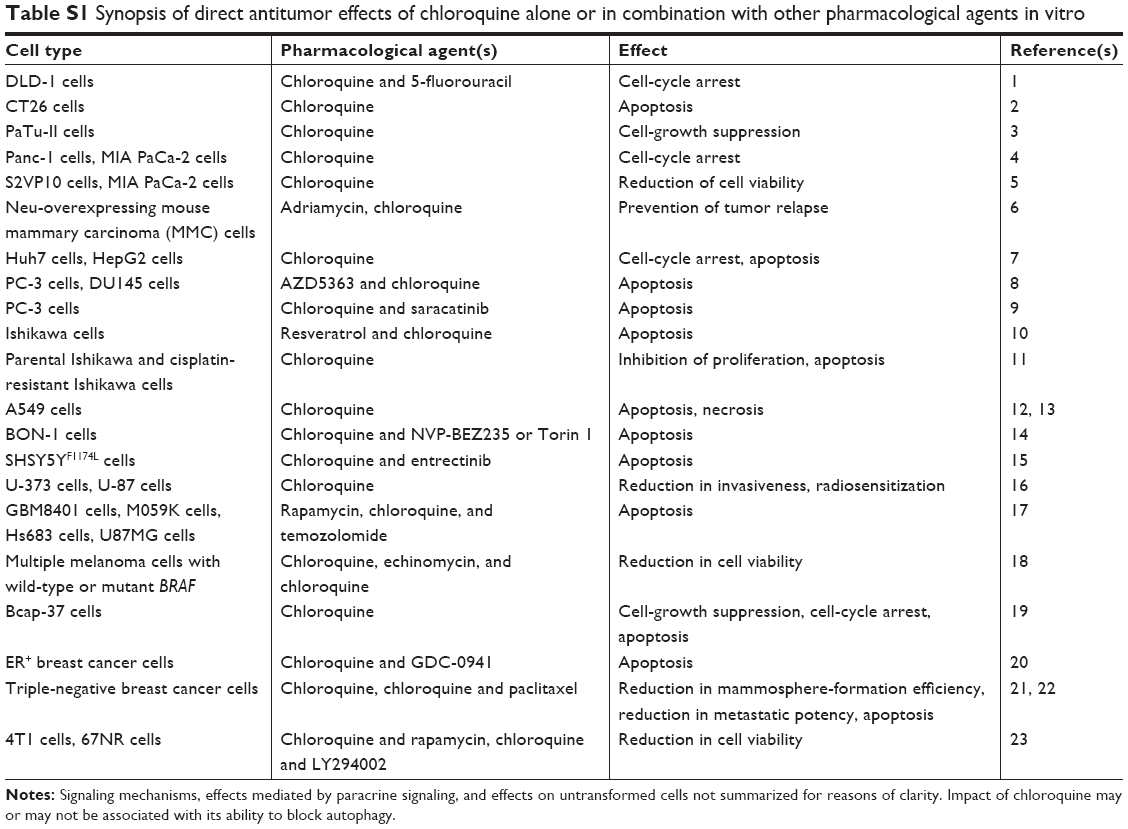 | Table S1 Synopsis of direct antitumor effects of chloroquine alone or in combination with other pharmacological agents in vitro |
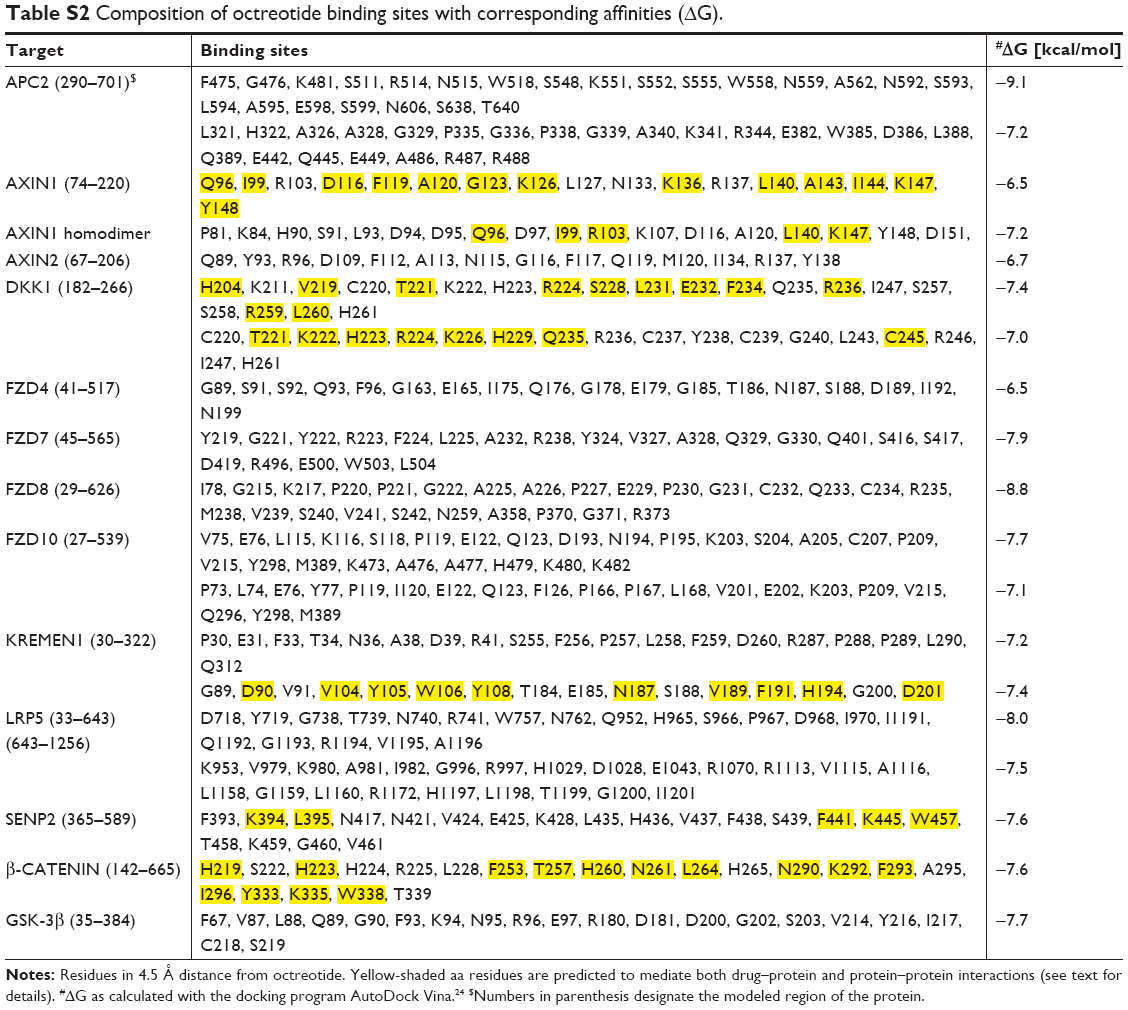 | Table S2 Composition of octreotide binding sites with corresponding affinities (ΔG). |
References
Choi JH, Yoon JS, Won YW, Park BB, Lee YY. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. APMIS. 2012;120(7):597–604. | ||
Zheng Y, Zhao YL, Deng X, et al. Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis. Cancer Invest. 2009;27(3):286–292. | ||
Zeilhofer HU, Mollenhauer J, Brune K. Selective growth inhibition of ductal pancreatic adenocarcinoma cells by the lysosomotropic agent chloroquine. Cancer Lett. 1989;44(1):61–66. | ||
Monma H, Iida Y, Moritani T, et al. Chloroquine augments TRAIL-induced apoptosis and induces G2/M phase arrest in human pancreatic cancer cells. PLoS One. 2018;13(3):e0193990. | ||
Frieboes HB, Huang JS, Yin WC, McNally LR. Chloroquine-mediated cell death in metastatic pancreatic adenocarcinoma through inhibition of autophagy. JOP. 2014;15(2):189–197. | ||
Aqbi HF, Tyutyunyk-Massey L, Keim RC, et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget. 2018;9(31):22113–22122. | ||
Hu T, Li P, Luo Z, et al. Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo. Oncol Rep. 2016;35(1):43–49. | ||
Lamoureux F, Zoubeidi A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy. 2013;9(7):1119–1120. | ||
Wu Z, Chang PC, Yang JC, et al. Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors. Genes Cancer. 2010;1(1):40–49. | ||
Fukuda T, Oda K, Wada-Hiraike O, et al. Autophagy inhibition augments resveratrol-induced apoptosis in Ishikawa endometrial cancer cells. Oncol Lett. 2016;12(4):2560–2566. | ||
Fukuda T, Oda K, Wada-Hiraike O, et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecol Oncol. 2015;137(3):538–545. | ||
Fan C, Wang W, Zhao B, Zhang S, Miao J. Chloroquine inhibits cell growth and induces cell death in A549 lung cancer cells. Bioorg Med Chem. 2006;14(9):3218–3222. | ||
Liu L, Han C, Yu H, et al. Chloroquine inhibits cell growth in human A549 lung cancer cells by blocking autophagy and inducing mitochondrialmediated apoptosis. Oncol Rep. 2018;39(6):2807–2816. | ||
Avniel-Polak S, Leibowitz G, Riahi Y, Glaser B, Gross DJ, Grozinsky-Glasberg S. Abrogation of autophagy by chloroquine alone or in combination with mTOR inhibitors induces apoptosis in neuroendocrine tumor cells. Neuroendocrinology. 2016;103(6):724–737. | ||
Aveic S, Pantile M, Seydel A, et al. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget. 2016;7(5):5646–5663. OncoTargets and Therapy 2019:12 | ||
Roy LO, Poirier MB, Fortin D. Chloroquine inhibits the malignant phenotype of glioblastoma partially by suppressing TGF-beta. Invest New Drugs. 2015;33(5):1020–1031. | ||
Spc H, Kuo JS, Chiang HC, et al. Temozolomide, sirolimus and chloroquine is a new therapeutic combination that synergizes to disrupt lysosomal function and cholesterol homeostasis in GBM cells. Oncotarget. 2018;9(6):6883–6896. | ||
Egger ME, Huang JS, Yin W, McMasters KM, McNally LR. Inhibition of autophagy with chloroquine is effective in melanoma. J Surg Res. 2013;184(1):274–281. | ||
Jiang PD, Zhao YL, Shi W, et al. Cell growth inhibition, G2/M cell cycle arrest, and apoptosis induced by chloroquine in human breast cancer cell line Bcap-37. Cell Physiol Biochem. 2008;22(5–6):431–440. | ||
Yang W, Hosford SR, Traphagen NA, et al. Autophagy promotes escape from phosphatidylinositol 3-kinase inhibition in estrogen receptor-positive breast cancer. Faseb J. 2018;32(3):1222–1235. | ||
Choi DS, Blanco E, Kim YS, et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells. 2014;32(9):2309–2323. | ||
Liang DH, Choi DS, Ensor JE, Kaipparettu BA, Bass BL, Chang JC. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016;376(2):249–258. | ||
Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8(2):200–212. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(Web Server issue):W252–W258. | ||
Bienert S, Waterhouse A, de Beer TA, et al. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017;45(D1):D313–D319. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.