Back to Journals » Clinical Ophthalmology » Volume 12
Topical ocular 0.1% cyclosporine A cationic emulsion in dry eye disease patients with severe keratitis: experience through the French early-access program
Authors Pisella P, Labetoulle M, Doan S, Cochener-Lamard B, Amrane M, Ismail D, Creuzot-Garcher C, Baudouin C ![]()
Received 6 September 2017
Accepted for publication 25 October 2017
Published 5 February 2018 Volume 2018:12 Pages 289—299
DOI https://doi.org/10.2147/OPTH.S150957
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Pierre-Jean Pisella,1 Marc Labetoulle,2 Serge Doan,3 Beatrice Cochener-Lamard,4 Mourad Amrane,5 Dahlia Ismail,5 Catherine Creuzot-Garcher,6,7 Christophe Baudouin8–10
1Department of Ophthalmology, Tours University Hospital, University François Rabelais, Bretonneau Hospital, Tours, 2Department of Ophthalmology, Bicêtre Hospital, APHP, Paris-Sud University, Le Kremlin-Bicêtre, 3Department of Ophthalmology, Bichat Hospital and Fondation A de Rothschild, Paris, 4Brest University Medical School, Morvan Hospital, Brest, 5Santen SAS, Evry, 6Department of Ophthalmology, University Hospital, 7Department of Ophthalmology III, Eye and Nutrition Research Group, Burgundy, Dijon, 8Research Team S12, Quinze-Vingts National Ophthalmology Hospital, 9Department of Ophthalmology, Ambroise-Paré Hospital, APHP, UPMC University, Paris 6, Vision Institute, INSERM UMRS968, CNRS UMR7210, Paris, 10University of Versailles Saint-Quentin en Yvelines, Versailles, France
Purpose: The objective of this study was to report the evaluation of efficacy and safety of cyclosporine A cationic emulsion (CsA CE) 0.1% for the treatment of severe keratitis in adults with dry eye disease (DED) in a French early-access program.
Methods: Patients with DED and severe keratitis (corneal fluorescein staining [CFS] score of 3–5 on the Oxford scale and/or the presence of corneal lesions [filaments or ulcers]) were enrolled in a compassionate use program (Authorization for Temporary Use [ATU]) for once-daily CsA CE, which was approved by French health authorities prior to its registration. Efficacy and safety at 1, 3, 6, and 12-month follow-up visits were evaluated.
Results: The ATU cohort (n=1,212; mean age =60.5 years; 79.5% female; 98.1% with severe keratitis; 74.5% with corneal lesions) consisted of 601 CsA-naïve patients and 611 patients treated previously with other CsA formulations. The primary DED etiology was Sjögren’s syndrome (48.7%). Clinical benefit could be discerned among 548 evaluable patients from months 1 to 12: keratitis improvement, 44.8% at month 1 and 42.1% at month 12; keratitis stabilization, 47.2% and 45.7%, respectively; symptom improvement, 47.2% and 48.6%; and symptom stabilization, 44.8% and 45.0%. Corneal clearing (CFS score =0) increased from 4.8% (month 1) to 11.4% (month 12). No unexpected safety concerns were identified; instillation site pain (10.2%) and eye irritation (7.8%) were the most common adverse events.
Conclusion: The French ATU cohort provides supportive data on the clinical benefit of CsA CE in improving/stabilizing symptoms and corneal damage in DED patients with severe keratitis in real-world clinical practice.
Keywords: dry eye, keratoconjunctivitis sicca, cyclosporine A, Sjögren’s syndrome, inflammation, cationic emulsion, severe keratitis, Ikervis
Introduction
According to the newly released TFOS DEWS II™ Report, dry eye disease (DED) – also known as keratoconjunctivitis sicca – is a
multifactorial disease of the ocular surface characterized by a loss of homeostasis of the tear film, and accompanied by ocular symptoms, in which tear film instability and hyperosmolarity, ocular surface inflammation and damage, and neurosensory abnormalities play etiological roles.1
The prevalence of DED ranges from 5% to 50% worldwide, depending on the definition of the disease applied.2 The pathogenesis of DED stems from a vicious inflammatory cycle that results in reduced tear flow, tear film instability, and tear hyperosmolarity.3 Patients with DED may experience ocular symptoms such as irritation, pain, dryness, burning, stinging, foreign body sensation, and visual disturbance, which are not well correlated with objective clinical findings.4,5 In severe cases and in later stages of the disease, conjunctival scarring, filamentary keratitis, epithelial defects, and corneal ulceration may occur.6 DED adversely affects health-related quality of life, with the burden directly proportional to disease severity.7
Topical corticosteroids improve the signs and symptoms of DED, but side effects such as intraocular hypertension and cataracts limit the duration of therapy with these agents.8,9 Artificial tear formulations have also proven useful for the treatment of DED symptoms, but they provide relief only on a short-term basis.10 The anti-inflammatory agent cyclosporine A (CsA) has become an increasingly useful treatment option in the past few decades.11 Clinical trials with CsA have shown that it can provide effective relief of DED signs and symptoms, and can be administered on a long-term basis.11,12 Restasis® (Allergan, Irvine, CA, USA), an anionic emulsion of CsA 0.05%, was approved by the US Food and Drug Administration for treatment of DED in 2003.
A CsA cationic emulsion formulation (CsA CE 0.1% [1 mg/mL]; Ikervis®, Santen SAS, Evry, France) was registered in the European Economic Area in 2015 for the treatment of severe keratitis in adult patients with DED that has not improved despite the use of tear substitutes.13 The CE formulation increases the bioavailability of CsA in the tear film compared with previously available CsA formulations.14 In the phase 3 SANSIKA study, once-daily instillation of CsA CE significantly improved the condition of the cornea over time, as measured by corneal fluorescein staining (CFS), compared with vehicle in DED patients with severe keratitis.15 This agent also reduced ocular surface inflammation, as measured by human leukocyte antigen–antigen D-related (HLA-DR) expression at the conjunctival cell surface, and was generally well tolerated. Prior to its approval by the European Medicines Agency, CsA CE was made available to patients in France through a cohort Authorization for Temporary Use (ATU) program approved by the French National Agency for Medicines and Health Products Safety (ANSM). Herein, we present results from this French ATU program, which offered a unique opportunity to evaluate the efficacy and safety of CsA CE in DED patients with severe keratitis in a real-world clinical practice setting.
Methods
ATU program and patients
The French ANSM granted authorization for the compassionate use of CsA CE 0.1% (1 mg/mL) in October 2013. The protocol for therapeutic use was approved in December 2013 (version 1) and was amended in March 2014 (version 2) and March 2015 (version 3). This report describes patients enrolled in the ATU cohort from January 2014 to June 2015.
Male and female patients aged ≥18 years with severe DED-related keratitis were eligible to participate in the ATU program. Eligibility required an ocular examination to assess clinical keratoconjunctivitis sicca within a context of hyposecretion, which included objective signs of severe corneoconjunctival impairment using fluorescein staining (CFS score of 3–5 on the Oxford scale16 and/or the presence of corneal filaments or ulcer), clinical confirmation of ocular symptoms, and failure of prior treatment with tear substitutes. Patients with active or suspected ocular or periocular infection or hypersensitivity to CsA or any excipient in CsA CE were excluded. Patients receiving anionic CsA emulsion (Restasis [cyclosporine ophthalmic emulsion] 0.05%; Allergan) through a previous named-patient ATU discontinued its use and then switched to CsA CE per ANSM request. Patients were instructed to instill 1 drop of CsA CE (~30 μg of CsA) once daily at bedtime. Prior to ATU participation, the objectives of CsA CE treatment were explained and discussed with each patient; written informed consent was not required because treatment was provided under compassionate use authorized by the ANSM and not within a clinical trial framework.
Assessments
Patients underwent ocular examinations at screening, at the start of treatment (day 0), and at follow-up visits after 1, 3, 6, 12, and 18 months of treatment. The requirement for a visit at 1 month was removed by an amendment to version 2 of the protocol. Efficacy was assessed by the proportion of patients demonstrating changes from the start of CsA CE treatment in severity of signs (keratitis) and symptoms. At each visit, the clinician classified the ocular signs and symptoms as resolved, improved, no change, or exacerbated. Safety was assessed at each visit by monitoring of adverse events (AEs) and serious AEs (SAEs).
Data analysis
Efficacy and safety data were categorized according to CsA treatment history (CsA-treatment naïve, previously treated with an anionic CsA emulsion, or previously treated with a hospital-compounded CsA formulation), as well as by baseline etiology (Sjögren’s syndrome vs other etiologies), and then analyzed by using descriptive statistics.
Results
Patient disposition and characteristics
Treatment applications of 1,244 patients were accepted for participation in the ATU program; 26 patients did not start treatment, and 6 patients were excluded from the analysis (Figure 1). As a result, a total of 1,212 patients were included in the analysis of CsA CE under the ATU. The mean age of the ATU cohort was 60.5 years (SD =16.3; age range =18–100 years); 37.6% of the patients were aged 65–74 years, and 30.5% were aged ≥75 years. The majority were women (79.5%). Of the 1,212 patients, 601 (49.6%) were CsA-treatment naïve, 531 (43.8%) had been treated with anionic CsA emulsion under a named-patient ATU, and 80 (6.6%) had been treated with a hospital-compounded CsA formulation. Table 1 shows the baseline clinical characteristics of the ATU cohort. Consistent with eligibility criteria, almost all the patients had severe keratitis (98.1%) and reported having no improvement in symptoms despite treatment with tear substitutes (99.0%). Overall, the patients had DED for a mean of 5.0 years (median =3.1), with patients previously treated with anionic CsA emulsion having a longer disease duration than CsA-treatment naïve patients (median =4.7 vs 1.6 years). Nearly half of the ATU cohort had a diagnosis of Sjögren’s syndrome (n=590; 48.7%); the subset with Sjögren’s syndrome had a mean age of 62.7 years (SD =14.5) and included 534 women (91.1%). Other DED etiologies included graft-versus-host disease (n=108; 8.9%), rheumatoid arthritis (n=89; 7.3%), and ocular rosacea (n=71; 5.9%).
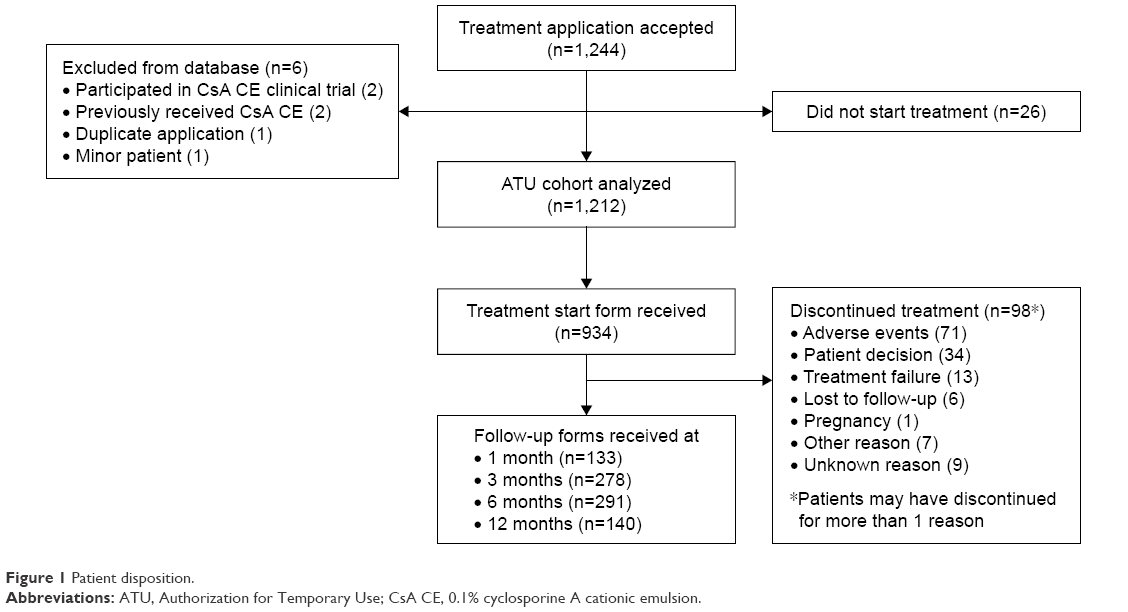 | Figure 1 Patient disposition. |
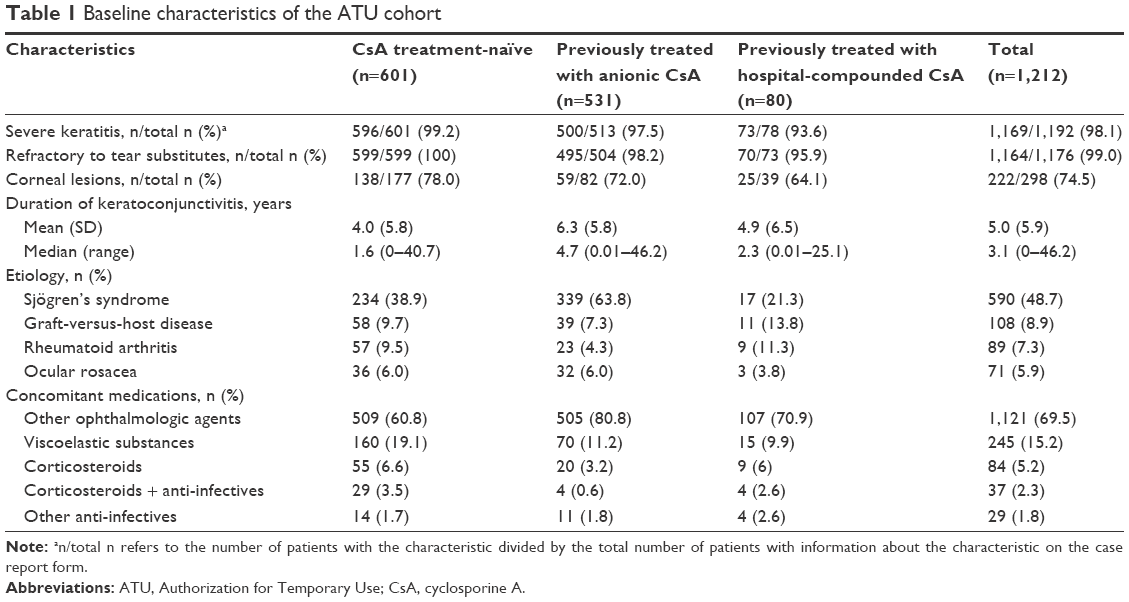 | Table 1 Baseline characteristics of the ATU cohort |
Following initiation of the ATU, a total of 1,782 report forms were analyzed for 939 patients (Figure 1). A treatment start form was received at baseline for 934 patients, and follow-up forms at 1, 3, 6, and 12 months were available for 133, 278, 291, and 140 patients, respectively; follow-up forms were available for only 6 patients at 18 months, and these data are not included in any of the analyses presented in this manuscript.
Among the patients who had ≥1 visit, mean treatment duration for CsA CE was available for 530 patients and was 27.7 weeks (SD =16.5; range =0.1–82.1). Treatment with CsA CE was stopped temporarily in 34 of 496 patients (6.9%) with available data, including 17 of 217 CsA-naïve patients (7.8%) and 17 of 252 patients (6.7%) previously treated with anionic CsA emulsion. A total of 98 of 548 patients (17.9%) with available data discontinued CsA CE permanently, most commonly due to AEs and patient decision (Figure 1). Discontinuation rates were independent of CsA treatment history.
Efficacy
The signs (keratitis) and symptoms of DED were assessed as improved or unchanged in most patients at every time point assessed, regardless of CsA treatment history. Improvements in signs and symptoms were reported in 44.8% and 47.2% of patients, respectively, at month 1, 48.3% and 53.3% at month 3, 45.8% and 50.0% at month 6, and 42.1% and 48.6% at month 12 (Figure 2). Signs and symptoms were unchanged (stabilized) in 47.2% and 44.8% of patients, respectively, at month 1, 43.5% and 43.1% at month 3, 42.5% and 45.1% at month 6, and 45.7% and 45.0% at month 12. In the overall population, clinicians also reported that some patients experienced a complete resolution of signs (corneal clearing [CFS =0]; 6.7% at 3 months, increasing to 11.4% at 12 months; Figure 3) and symptoms (1.4% at 3 months, increasing to 6.4% at 12 months; Figure 2). Exacerbation of signs and symptoms was reported rarely and declined in incidence over time: for signs, 3%, 2%, 2%, and 0% of the total population at months 1, 3, 6, and 12, respectively, and for symptoms, 5%, 2%, 1%, and 0% (Figure 2).
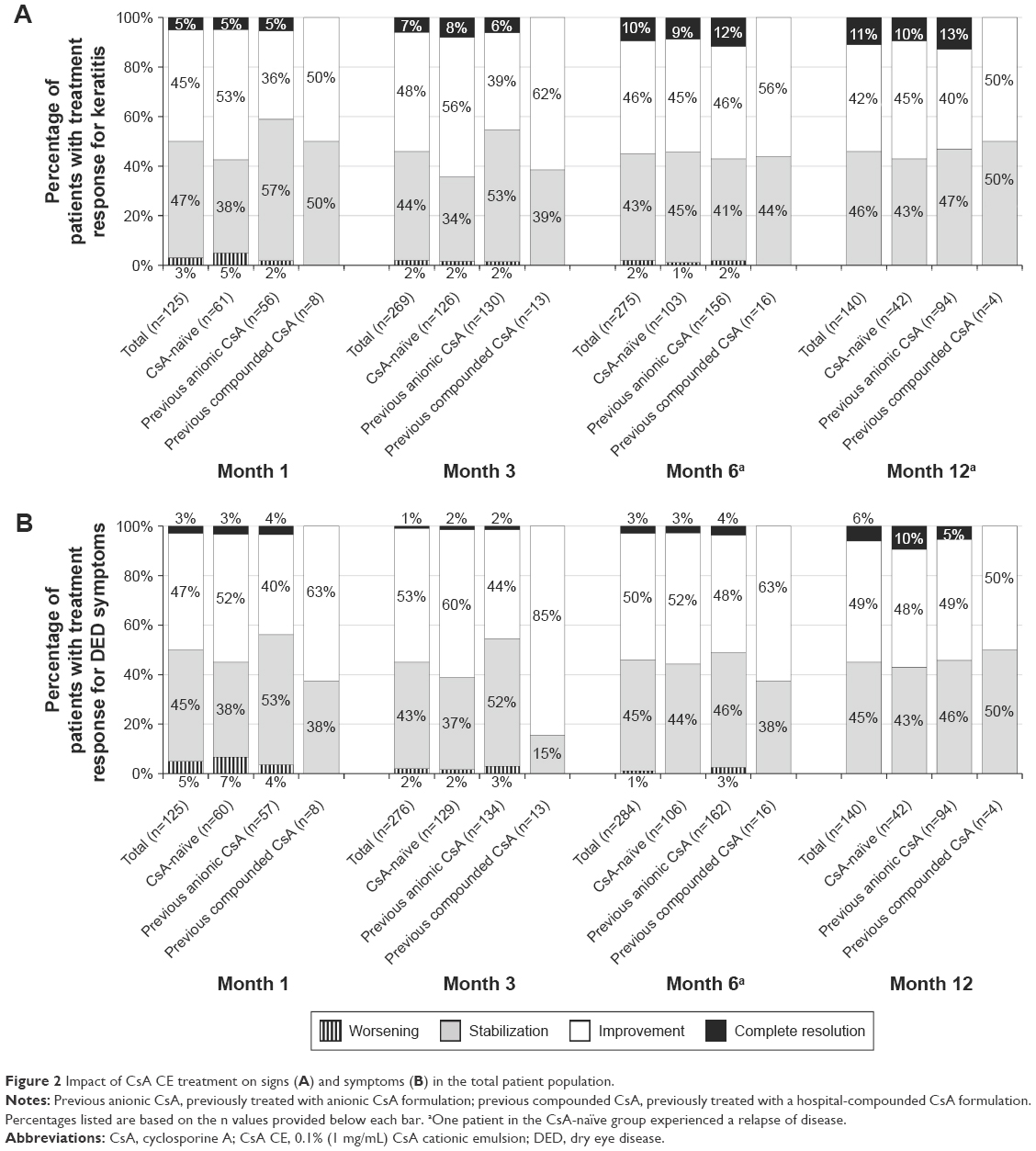 | Figure 2 Impact of CsA CE treatment on signs (A) and symptoms (B) in the total patient population. |
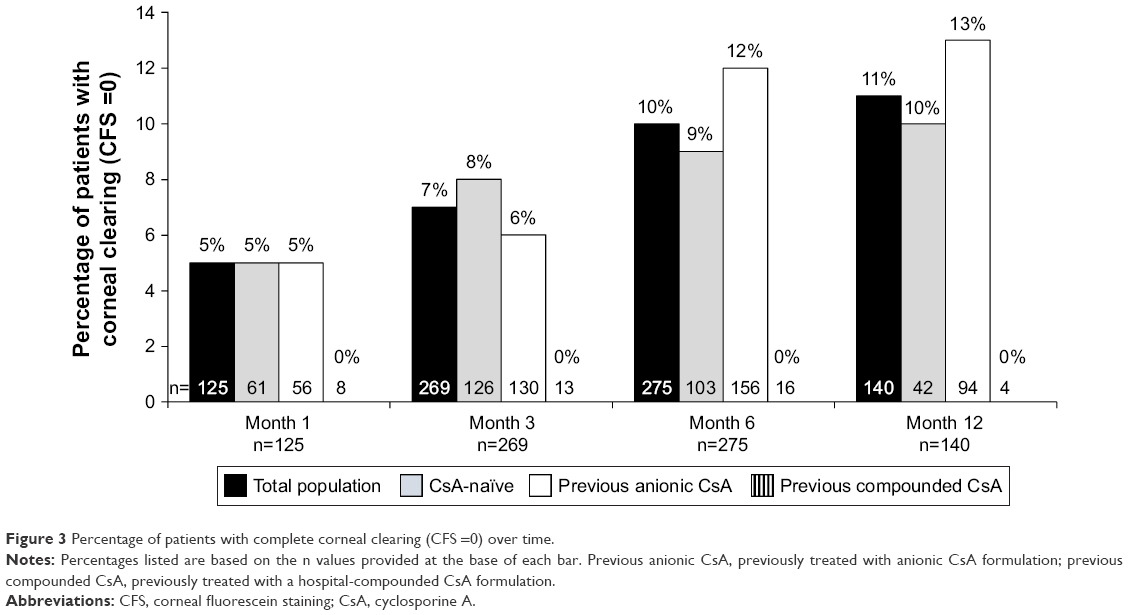 | Figure 3 Percentage of patients with complete corneal clearing (CFS =0) over time. |
Efficacy findings in the subset of patients with Sjögren’s syndrome were similar to those for the entire ATU cohort in that signs and symptoms were reported as improved or stable in most patients at each time point, regardless of CsA treatment history. As with the overall patient population, treatment effects were noted at month 1 when signs and symptoms were considered improved in 46% and 51% of patients, respectively, and unchanged (stabilized) in 48% and 44% of patients, respectively (Figure 4). At 3 months, signs and symptoms were considered improved in 49% and 51% of patients, respectively, and stabilized in 46% and 46% of patients, respectively. The corresponding proportions were similar at 6 and 12 months: improvements in signs and symptoms were reported in 48% and 52% of patients, respectively, at 6 months and in 38% and 49% of patients, respectively, at 12 months; stabilization was reported in 41% and 44% at 6 months and in 47% and 45% at 12 months. The resolution of signs (CFS =0) was reported in 5% of patients at 3 months, increasing to 14% at 12 months, and the resolution of symptoms was reported in 2% at 3 months, increasing to 6% at 12 months. As with the overall population, exacerbation of signs and symptoms was reported rarely in the Sjögren’s population and declined in incidence over time, with no reports of exacerbation at month 12. Similar response patterns for signs and symptoms were observed in the subset of patients without Sjögren’s syndrome.
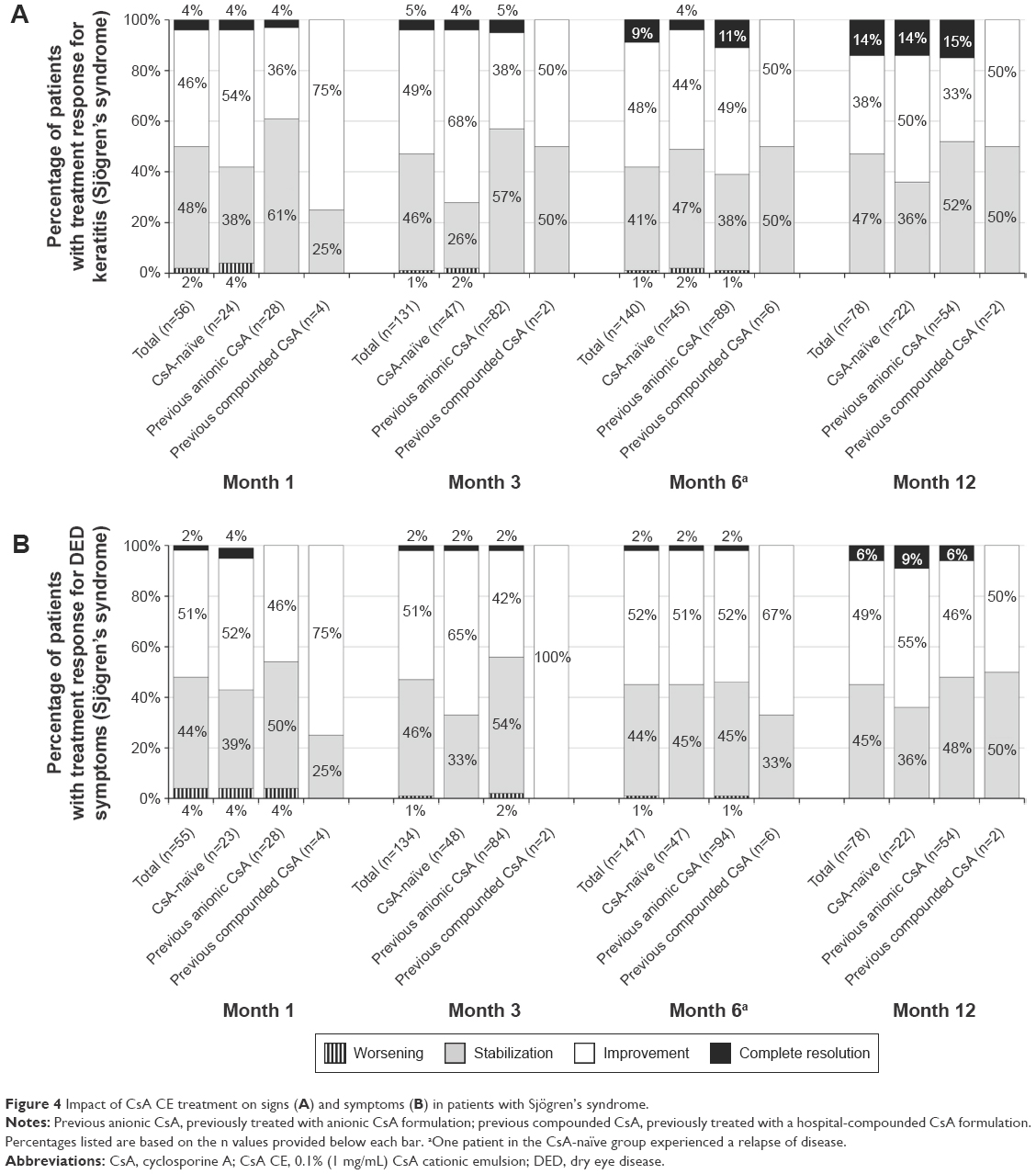 | Figure 4 Impact of CsA CE treatment on signs (A) and symptoms (B) in patients with Sjögren’s syndrome. |
In addition to the descriptive analysis performed at each time point, a longitudinal assessment of the evolution of patients’ signs and symptoms that categorized the totality of changes for individual patients (as opposed to focusing solely on a descriptive global assessment at each time point) was also performed. In this analysis, patients were pooled into clinical subgroups according to the following rules, irrespective of their number of follow-up visits:
- If the patient experienced a worsening at least once, even if he or she had an improvement/stabilization during the follow-up, the condition was considered as “worsened/exacerbated.”
- If the patient had a resolution without any worsening during the follow-up, the condition was considered as “resolved.”
- If the patient had improvement without any worsening during the follow-up, the condition was considered as “improved.”
- If the patient had stabilization compared with baseline without any worsening or improvement, the condition was considered as “stabilized.”
With respect to keratitis, in the CsA-naïve patients, the participating clinicians reported that 31.07% of patients had no change from baseline; however, 54.37% and 11.17% had improvement and complete resolution, respectively (Figure 5). In the group who received anionic CsA before starting the ATU, 36.59% of patients had no change after the switch to CsA CE, whereas 48.78% and 12.6% showed improvement and complete resolution of keratitis, respectively. In the group of patients who received hospital-compounded CsA before starting the ATU, 46.15% of patients had no change after the switch to CsA CE, whereas 53.85% of patients showed improvement and 0% had complete resolution. In the total population, only 2 of 478 patients (0.42%) showed worsening of keratitis after improvement/stabilization.
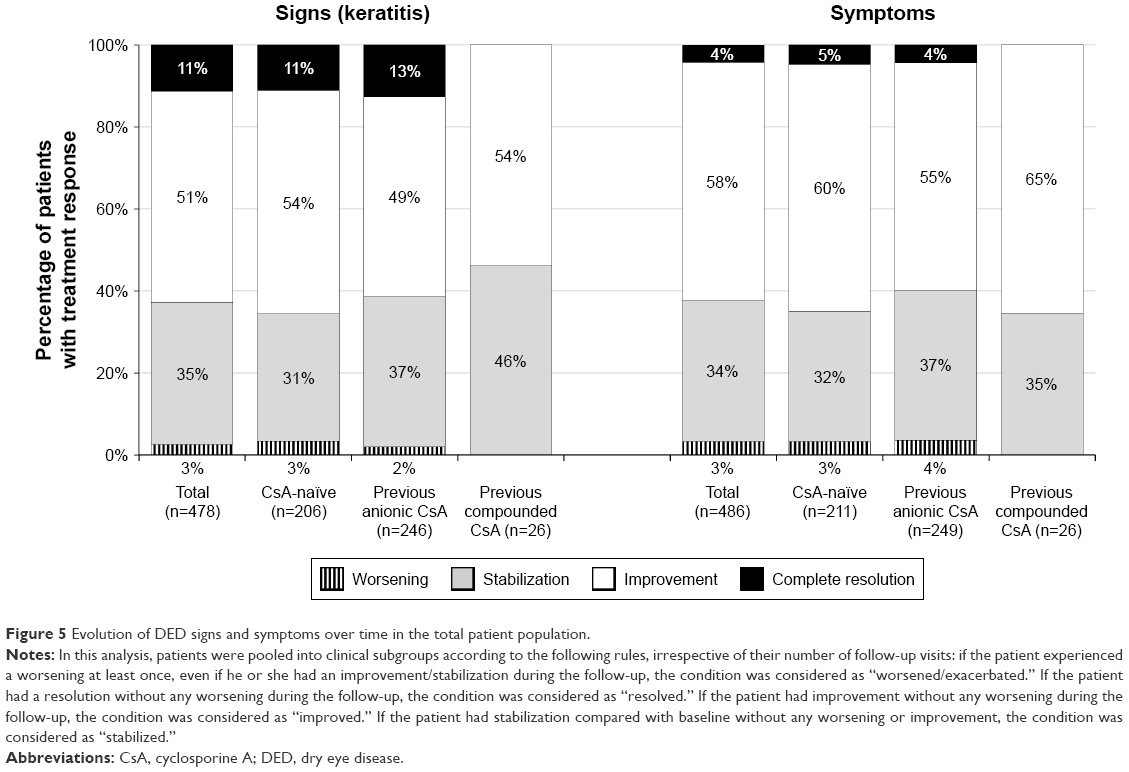 | Figure 5 Evolution of DED signs and symptoms over time in the total patient population. |
With respect to symptoms, the clinicians reported that 31.75% of CsA-naïve patients had no change from baseline; however, 60.19% and 4.74% of patients had improvement and complete resolution, respectively (Figure 5). In the group of patients who received anionic CsA before starting the ATU, 36.55% had no change after the switch to CsA CE, whereas 55.42% and 4.42% showed improvement and complete resolution, respectively. In the group of patients who received hospital-compounded CsA before starting the ATU, 34.62% of patients had no change after the switch to CsA CE, whereas 65.38% showed an improvement in their symptoms and 0% had complete resolution. In the total population, only 4 of 486 patients (0.82%) showed worsening of symptoms after improvement/stabilization.
Similar patterns with respect to the evolution of signs and symptoms were seen in the subgroup of patients with Sjögren’s syndrome.
Safety
AEs were reported with the use of CsA CE in 197 of 548 evaluable patients (35.9%; Table 2). Most AEs were ocular in nature and expected with the use of CsA.11 The most common AEs were instillation site pain (10.8%) and eye irritation (8.2%). Nine patients (1.6%) had a total of 20 SAEs, including eye irritation (3 patients); eye pain (3 patients); ulcerative keratitis/corneal ulceration (2 patients); disease exacerbation/condition aggravated (2 patients); and reduced visual acuity, ocular hyperemia, conjunctivitis, instillation site pain, skin rash, skin ulcer, jaw pain, rhinorrhea, sinus headaches, and death (1 patient each). All of the SAEs were considered to be treatment-related by the investigators, with the exception of the death, which occurred in a patient with a history of acute myeloid leukemia who died due to hematologic disease. All the patients with SAEs discontinued CsA CE, with the exception of 1 patient with skin ulcer. Similar discontinuation rates were observed across patient subgroups, regardless of prior treatment history. Sixty-eight patients (12.4%) permanently discontinued due to nonserious AEs (mainly instillation site pain).
 | Table 2 Summary of overall AEs and SAEs in ATU cohort |
Discussion
The results of this compassionate use/cohort ATU program support the efficacy and safety of CsA CE in DED patients with severe keratitis in real-world clinical practice. For most patients, treatment with CsA CE improved or stabilized keratitis and symptoms associated with DED, with 42.1% and 48.6% of patients experiencing improvement in signs and symptoms, respectively, at month 12 and 45.7% and 45.0% of patients experiencing stabilization. In addition, some patients experienced a complete resolution of signs and/or symptoms (11.4% and 6.4%, respectively, at month 12). This treatment benefit was observed across all patient subpopulations, regardless of CsA treatment history (CsA-treatment naïve or previously treated with anionic CsA emulsion or with a hospital-compounded CsA formulation) or underlying etiology (Sjögren’s syndrome vs other causes), and was reported beginning at month 1. The benefit of CsA CE in patients with severe keratitis is noteworthy as this form of the disease can be difficult to treat17,18 and may result in corneal ulceration or perforation.6 In this French ATU program, there was no reported corneal perforation, and only 2 patients (0.4%) were reported with corneal ulcerations.
The CsA CE treatment benefit documented in this French ATU cohort reinforces previous findings regarding this formulation’s efficacy in treating DED patients with severe keratitis. In the multicenter, randomized, phase 3 SANSIKA study, once-daily treatment with CsA CE significantly improved the CFS score compared with vehicle over the course of the 6-month trial.15 Inflammation was assessed by the level of expression of class II antigens (HLA-DR)19 at the conjunctival cell surface and was also improved significantly with CsA CE compared with the vehicle. The symptom scores improved to similar extents with CsA CE and the vehicle, likely reflecting the ability of the vehicle (a cationic oil-in-water nanoemulsion) to increase the retention time of nanodroplets on the ocular surface. In the ATU cohort, similar degrees of improvement were reported for both keratitis and symptoms. This observation is noteworthy in that signs and symptoms are often poorly correlated in DED, particularly in severe disease where reduced corneal sensation may reduce discomfort.20 It is also worth noting that the majority of ATU patients who had previously received anionic CsA (Restasis) and hospital-compounded CsA formulations showed an improvement in their condition.
With regard to previously reported data with CsA formulations, our findings of improvements in CFS scores as early as month 1 with CsA CE are consistent with earlier findings for this formulation. In the multicenter, randomized, phase 3 SICCANOVE study, once-daily treatment with CsA CE significantly improved CFS score compared with the vehicle as early as month 1 in patients with moderate-to-severe keratitis.21 However, it should be noted that available data with other CsA formulations suggest that at least 2–3 months of treatment is necessary for CsA to obtain its optimal effect.22,23 It is also interesting to note that the incidence of reduced visual acuity reported in the overall population of our study (0.2%) was substantially lower than that reported in a group of 412 patients with moderate-to severe DED who received a CsA 0.1% anionic emulsion twice daily for up to 3 years (3.2%).12
The recently released TFOS DEWS II Report concludes that there is “strong level 1 evidence to support the use of cyclosporine in the treatment of DED,” citing numerous beneficial effects, including reduction of inflammatory markers and tear osmolarity, antiapoptotic effects relevant to the known reversal of the normal epithelial cell/leukocyte relationship in DED, and recovery of reduced conjunctival goblet cell density.18 The report also notes, however, that treatment may be required for extended periods of time to achieve the resolution of symptoms. In this regard, it is interesting to note that Straub et al24 found that a median treatment period of 23 months resulted in the resolution of symptoms of severe DED with the anionic emulsion (Restasis) in a series of 26 patients (22 of them with Sjögren’s syndrome). On a 10-year follow-up, DED signs (as assessed by Schirmer test, CFS score, lissamine green staining, and tear breakup time) were all significantly improved versus the study baseline (P≤0.03); symptoms (as measured by the Ocular Surface Disease Index©) were also improved, but not to a significant degree (P=0.15).
Overall, there were no appreciable differences in the proportions of patients with improvement and stabilization between the subset with and without Sjögren’s syndrome in the ATU cohort. This finding is consistent with the hypothesis that the self-perpetuating inflammatory cascade that is both the cause and consequence of DED essentially becomes disconnected from the underlying cause of the disease.3 For this reason, the efficacy of CsA CE, which hinges on its immune-modulating/anti-inflammatory effects, would be expected to be similar in severe DED caused by Sjögren’s syndrome or by alternative etiologies.
The safety and tolerability profile of CsA CE in the French ATU cohort was acceptable and consistent with the findings from the phase 3 clinical studies.15,21 Most AEs were categorized as eye disorders (most commonly eye irritation or eye pain) or as related to treatment administration (instillation site pain). These AEs are consistent with those observed previously with other ocular CsA formulations.11 Importantly, treatment with CsA CE over 12 months did not identify any new or unexpected safety concerns (although it should be noted that the month 12 population was small compared with the original baseline population). Overall, the safety profile combined with the treatment benefit indicates a favorable benefit/risk ratio for CsA CE in the treatment of DED patients with severe keratitis.
Several limitations should be noted. The French ATU program did not include a placebo control arm; however, this was not intended to be a clinical trial of CsA CE but was rather designed to give early access to CsA CE before marketing authorization and to generate data regarding its efficacy and safety in real-world clinical practice. Efficacy measures were not collected using standardized methodology (eg, the Ocular Surface Disease Index for DED symptoms) or standardized site-monitoring practices (eg, for CFS assessments for keratitis, as were employed in the aforementioned phase 3 SANSIKA trial,15 or for monitoring of treatment instillation with respect to proper technique and compliance), and data for individual patients (eg, for CFS scores, symptoms, and corneal ulcer size) were not available through the ATU protocol per ANSM request (as this was not a clinical trial), prohibiting further analyses of the overall patient population or intergroup comparisons. The classification of ocular signs and symptoms as resolved, improved, no change, or exacerbated required the physician to make comparisons with the patients’ clinical state at the previous visit and was thus subject to recall bias; however, it should be noted that the participating ophthalmologists were experts in DED. It should also be noted that our report focused primarily on descriptive comparisons of treatment effects at each time point versus baseline; however, an additional longitudinal analysis was performed in order to obtain a more comprehensive view of the evolution of DED signs and symptoms over time (ie, the totality of changes occurring between time points), and this analysis reinforced the findings of the primary analysis that the majority of patients in the total population and the prior treatment symptoms experienced improvement in signs and symptoms. Finally, the ATU cohort included 1,212 patients, but only 548 patients were evaluable for efficacy and safety, and for many patients in this subgroup, follow-up forms were available for analysis at only 1 or 2 time points, with only 16 patients having follow-up data for all 4 time points. It was not possible to determine why data were missing for any given time point, as this information was not available to us through the ATU protocol. Because this protocol, as compared with those used for traditional clinical trials, did not have a monitoring component to ensure the compliance of the participants, it is possible that clinicians did not return some of the follow-up forms; it is also possible that patients did not return to the hospital as required by the protocol. Nevertheless, despite all of the limitations listed above, the treatment benefits observed at each time point were consistent, suggesting that the effects of CsA CE are maintained for ≥12 months (bearing in mind that the study sample was substantially diminished by month 12). It should also be noted that many of the aforementioned limitations are consistent with the reality of clinical practice in terms of the assessments that clinicians typically make with their patients.
In summary, the trend for clinical benefit observed with CsA CE in this “real-life” French ATU program is consistent with the efficacy and safety profile reported in previous clinical studies, including the SANSIKA trial, and provides supportive evidence for the clinical effectiveness and tolerability of CsA CE in the treatment of DED patients with severe keratitis in clinical practice.
Acknowledgments
This compassionate use/cohort ATU program was supported by Santen SAS, Evry, France. The authors thank Chameleon Communications International Ltd., London, UK, for medical writing, and BioScience Communications, New York, NY, USA, for medical writing and copyediting, which was funded by Santen SAS. Neither honoraria nor payments were made for authorship.
Selected data in this manuscript have been presented in abstract/poster format at the Association for Research in Vision and Ophthalmology meeting, April 30–May 4, 2016, Seattle, WA, USA, and the Tear Film and Ocular Surface Society meeting, September 7–10, 2016, Montpellier, France.
Author contributions
All the authors contributed toward data interpretation, drafting, and critically revising the paper; gave final approval for the version to be published; and agreed to be accountable for all aspects of the work.
Disclosure
Pierre-Jean Pisella, Serge Doan, and Christophe Baudouin are consultants for or have received research grants from Alcon, Allergan, Santen, and Théa and have served as clinical investigators for Santen. Marc Labetoulle is a consultant for or has received a research grant from Alcon, Allergan, Bausch & Lomb, Dompè, Merck Sharpe & Dhome, Novartis, Santen, Shire, and Théa and has served as a clinical investigator for Santen. Beatrice Cochener has served as a consultant and clinical investigator for Alcon, Allergan, AMO Pharma, Cutting Edge, Dompè, Hoya, Santen, Théa, and Zeiss. Mourad Amrane and Dahlia Ismail are employees of Santen SAS. Catherine Creuzot-Garcher is a consultant for or has received a research grant from Alcon, Allergan, Bayer, Horus, Novartis, and Théa and has served as a clinical investigator for Santen. The authors report no other conflicts of interest in this work.
References
Craig JP, Nichols KK, Akpek EK, et al. TFOS DEWS II definition and classification report. Ocul Surf. 2017;15(3):276–283. | ||
Stapleton F, Alves M, Bunya VY, et al. TFOS DEWS II epidemiology report. Ocul Surf. 2017;15(3):334–365. | ||
Baudouin C, Aragona P, Messmer EM, et al. Role of hyperosmolarity in the pathogenesis and management of dry eye disease: proceedings of the OCEAN Group meeting. Ocul Surf. 2013;11(4):246–258. | ||
Bartlett JD, Keith MS, Sudharshan L, Snedecor SJ. Associations between signs and symptoms of dry eye disease: a systematic review. Clin Ophthalmol. 2015;9:1719–1730. | ||
Wolffsohn JS, Arita R, Chalmers R, et al. TFOS DEWS II diagnostic methodology report. Ocul Surf. 2017;15(3):539–574. | ||
Messmer EM. The pathophysiology, diagnosis, and treatment of dry eye disease. Dtsch Arztebl Int. 2015;112(5):71–82. | ||
McDonald M, Patel DA, Keith MS, Snedecor SJ. Economic and humanistic burden of dry eye disease in Europe, North America, and Asia: a systematic literature review. Ocul Surf. 2016;14(2):144–167. | ||
Marsh P, Pflugfelder SC. Topical nonpreserved methylprednisolone therapy for keratoconjunctivitis sicca in Sjogren syndrome. Ophthalmology. 1999;106(4):811–816. | ||
Pflugfelder SC. Antiinflammatory therapy for dry eye. Am J Ophthalmol. 2004;137(2):337–342. | ||
Gayton JL. Etiology, prevalence, and treatment of dry eye disease. Clin Ophthalmol. 2009;3:405–412. | ||
Schultz C. Safety and efficacy of cyclosporine in the treatment of chronic dry eye. Ophthalmol Eye Dis. 2014;6:37–42. | ||
Barber LD, Pflugfelder SC, Tauber J, Foulks GN. Phase III safety evaluation of cyclosporine 0.1% ophthalmic emulsion administered twice daily to dry eye disease patients for up to 3 years. Ophthalmology. 2005;112(10):1790–1794. | ||
Ikervis [Summary of Product Characteristics]. Evry, France: Santen SAS; 2016. | ||
Lallemand F, Daull P, Benita S, Buggage R, Garrigue JS. Successfully improving ocular drug delivery using the cationic nanoemulsion, novasorb. J Drug Dev. 2012;2012:6044204. | ||
Leonardi A, Van Setten G, Amrane M, et al. Efficacy and safety of 0.1% cyclosporine A cationic emulsion in the treatment of severe dry eye disease: a multicenter randomized trial. Eur J Ophthalmol. 2016;26(4):287–296. | ||
Bron AJ, Evans VE, Smith JA. Grading of corneal and conjunctival staining in the context of other dry eye tests. Cornea. 2003;22(7):640–650. | ||
Foulks GN. Treatment of dry eye disease by the non-ophthalmologist. Rheum Dis Clin North Am. 2008;34(4):987–1000. | ||
Jones L, Downie LE, Korb D, et al. TFOS DEWS II management and therapy report. Ocul Surf. 2017;15(3):575–628. | ||
Brignole-Baudouin F, Riancho L, Ismail D, Deniaud M, Amrane M, Baudouin C. Correlation between the inflammatory marker HLA-DR and signs and symptoms in moderate to severe dry eye disease. Invest Ophthalmol Vis Sci. 2017;58(4):2438–2448. | ||
Baudouin C, Aragona P, Van Setten G, et al. Diagnosing the severity of dry eye: a clear and practical algorithm. Br J Ophthalmol. 2014;98(9):1168–1176. | ||
Baudouin C, Figueiredo FC, Messmer EM, et al. A randomized study of the efficacy and safety of 0.1% cyclosporine A cationic emulsion in treatment of moderate to severe dry eye. Eur J Ophthalmol. 2017;27(5):520–530. | ||
Perry HD, Solomon R, Donnenfeld ED, et al. Evaluation of topical cyclosporine for the treatment of dry eye disease. Arch Ophthalmol. 2008;126(8):1046–1050. | ||
Aragona P. Topical cyclosporine: are all indications justified? Br J Ophthalmol. 2014;98(8):1001–1002. | ||
Straub M, Bron AM, Muselier-Mathieu A, Creuzot-Garcher C. Long-term outcome after topical ciclosporin in severe dry eye disease with a 10-year follow-up. Br J Ophthalmol. 2016;100(11):1547–1550. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.