Back to Journals » International Journal of Nanomedicine » Volume 11
Toll-like receptor 3-induced immune response by poly(D,L-lactide-co-glycolide) nanoparticles for dendritic cell-based cancer immunotherapy
Authors Han HD ![]() , Byeon YS, Kang TH, Jung ID, Lee JW
, Byeon YS, Kang TH, Jung ID, Lee JW ![]() , Shin BC, Lee YJ, Sood AK, Park YM
, Shin BC, Lee YJ, Sood AK, Park YM
Received 22 March 2016
Accepted for publication 24 September 2016
Published 2 November 2016 Volume 2016:11 Pages 5729—5742
DOI https://doi.org/10.2147/IJN.S109001
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Hee Dong Han,1,* Yeongseon Byeon,1,* Tae Heung Kang,1 In Duk Jung,1 Jeong-Won Lee,2 Byung Cheol Shin,3 Young Joo Lee,4 Anil K Sood,5–7 Yeong-Min Park1
1Department of Immunology, School of Medicine, Konkuk University, Chungwondaero, Chungju-Si, Chungcheongbuk-Do, 2Department of Obstetrics and Gynecology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, 3Bio/Drug Discovery Division, Korea Research Institute of Chemical Technology, Yuseong-gu, Daejeon, 4Department of Bioscience and Biotechnology, Sejong University, Kwang-Jin-Gu, Seoul, South Korea; 5Department of Gynecologic Oncology and Reproductive Medicine, 6Department of Cancer Biology, 7Center for RNA Interference and Non-coding RNA, The University of Texas MD Anderson Cancer Center, TX, USA
*These authors contributed equally to this work
Abstract: Dendritic cells (DCs) are potent professional antigen-presenting cells that are capable of initiating a primary immune response and activating T cells, and they play a pivotal role in the immune responses of the host to cancer. Prior to antigen presentation, efficient antigen and adjuvant uptake by DCs is necessary to induce their maturation and cytokine generation. Nanoparticles (NPs) are capable of intracellular delivery of both antigen and adjuvant to DCs. Here, we developed an advanced poly(D,L-lactide-co-glycolide) (PLGA)-NP encapsulating both ovalbumin (OVA) as a model antigen and polyinosinic-polycytidylic acid sodium salt (Toll-like receptor 3 ligand) as an adjuvant to increase intracellular delivery and promote DC maturation. The PLGA-NPs were taken up by DCs, and their uptake greatly facilitated major histocompatibility class I antigen presentation in vitro. Moreover, vaccination with PLGA-NP-treated DCs led to the generation of ovalbumin-specific CD8+ T cells, and the resulting antitumor efficacy was significantly increased in EG.7 and TC-1 tumor-bearing mice compared to control mice (P<0.01). Taken together, these findings demonstrated that the PLGA-NP platform may be an effective method for delivering tumor-specific antigens or adjuvants to DCs.
Keywords: cancer immunotherapy, PLGA nanoparticles, antigen delivery
Introduction
Multifunctional nanoparticles (NPs) have attracted interest in various biomedical fields for disease prevention and therapy.1,2 Several types of NPs have been used for the delivery of adjuvants and antigens to immune cells. These components can elicit strong immunotherapeutic responses to cancer and infectious diseases. In cancer immunotherapy, dendritic cell (DC)-based approaches hold great potential for antigen cross-presentation.3–5 DCs are the most effective antigen-presenting cells, which present antigens to CD8+ T cells and secrete proinflammatory cytokines, resulting in tumor antigen-specific activation of CD8+ T cells that contribute to tumor inhibition. After internalizing antigen and adjuvant into DCs, DCs can exhibit antigen cross-presentation to cytotoxic T cells through extracellular major histocompatibility complex (MHC)-I molecules.6 Therefore, effective maturation of DCs is the key first step in DC-based cancer immunotherapy. Because antigen or adjuvant show limited penetration into DCs, efficient delivery systems are highly suitable for both antigen and adjuvant delivery into DCs. Recent studies have shown that antigen-loaded NPs increase DC maturation, enhancing antigen cross-presentation in DCs and inducing cytotoxic T cell responses.7
As DCs require high antigen uptake, various nanomaterials have been developed with specific physicochemical properties and are currently under extensive investigation for their potential utilization as drug delivery systems for immunotherapy8,9 and antitumor chemotherapeutic drugs.10,11 Poly(D,L-lactide-co-glycolide) (PLGA)-NPs, which are particularly attractive for clinical and biological applications because of their low toxicity, low immunogenicity, biocompatibility, and biodegradability,12,13 have been widely utilized as drug carriers in nanomedicine.14,15 These studies motivated us to know whether PLGA-NPs can increase the uptake efficiency of adjuvant or antigen by DCs and lead to an increase in antigen-specific CD8+ T cell responses.
Polyinosinic-polycytidylic acid (poly I:C) as an adjuvant promotes maturation of DCs through its interaction with Toll-like receptor 3 (TLR3), and could therefore improve antigen-mediated cross-presentation to antigen-specific CD8+ T cells. The potency of poly I:C has been demonstrated by its immunomodulatory effects in mice.16–18 Poly I:C mimics viral double-stranded RNA, which is a promising immunostimulatory candidate for vaccines directed against intracellular pathogens. However, TLR3 is localized in the endosome of DCs. Therefore, to activate TLR3, poly I:C has to penetrate the DCs. To overcome this limitation, a carrier system is needed to enhance intracellular delivery of poly I:C. Activation of TLR3 in DCs leads to the expression of proinflammatory cytokines such as interleukin (IL)-12, IL-6, and tumor necrosis factor-alpha (TNF-α), resulting in enhanced innate and adaptive immune responses.
Here, we developed a PLGA-NP system encapsulating both ovalbumin (OVA) and poly I:C to increase the efficiency of their intracellular delivery into DCs and to promote DC maturation and antigen-specific cross-presentation (Figure 1). Taken together, our results demonstrated that PLGA-NPs were a highly efficient delivery system with therapeutic efficacy in EG.7 and TC-1 animal tumor models.
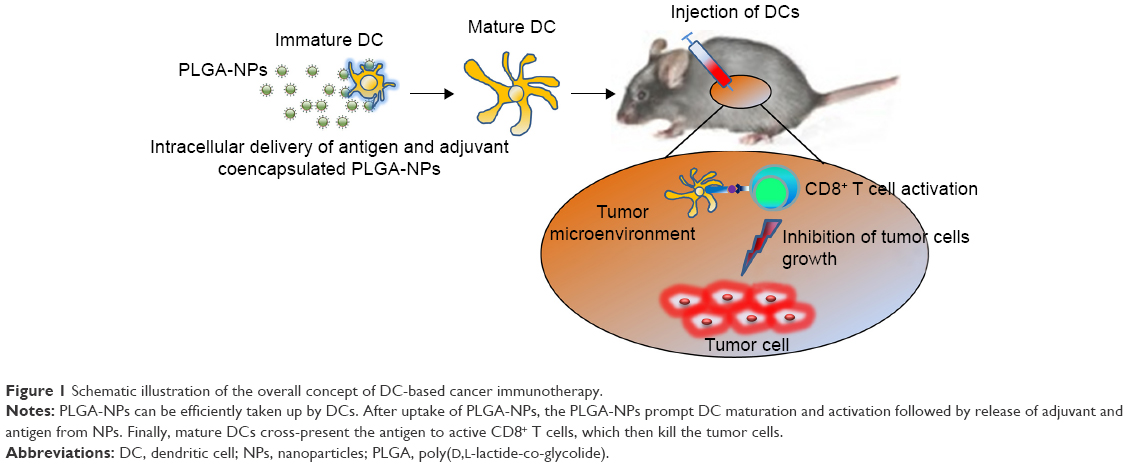 | Figure 1 Schematic illustration of the overall concept of DC-based cancer immunotherapy. |
Materials and methods
Materials
PLGA (Resomer RG502H, monomer ratio 50:50, molecular weight [MW] 10–12 kDa) was purchased from Boehringer Ingelheim (Ingelheim, Germany). Polyvinyl alcohol (80% hydrolyzed, MW 9–10 kDa), chicken egg OVA, and poly I:C sodium salt were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Fluorescein isothiocyanate (FITC)-conjugated antimouse interferon (IFN)-γ, R-phycoerythrin (PE)-conjugated antimouse CD40, OVA-specific (SIINFEKL/H-2Kb) antibody, and mouse TNF-α, IL-1β, IL-6, IL-12p70 enzyme-linked immunosorbent assay (ELISA) Ready-SET-Go kit were purchased from eBioscience (San Diego, CA, USA). FITC-conjugated antimouse CD11c antibody, PE-conjugated antimouse CD8a, CD80, CD86, MHC class I, MHC class II, and mouse IFN-β ELISA kit were purchased from Biolegend (San Diego, CA, USA). Roswell Park Memorial Institute Medium (RPMI) 1640 and fetal bovine serum (FBS) were purchased from Biowest (Nuaille, France). Granulocyte-macrophage colony-stimulating factor was purchased from JW CreaGene (Gyeonggi, South Korea). E7 peptide (MW 2.4 kDa, 42–64 amino acids, AGQAEPDRAHYNIVTFCCKCDS) was purchased from AnyGen Co. (Seoul, South Korea). Anti-CD8 antibody for CD8+ T cell depletion was purchased from BioXcell (West Lebanon, USA). All other materials were of analytical grade and used without further purification.
Preparation of PLGA-NPs
PLGA-NPs containing both OVA and poly I:C (PLGA [OVA + poly I:C]-NPs) were prepared by a water-in-oil-in-water (w/o/w) double emulsion solvent evaporation method.19,20 Briefly, 1 mg OVA and 2 mg poly I:C were dissolved in 0.2 mL deionized water and mixed with 2 mL chloroform containing 40 mg PLGA using a probe-type sonicator at 4°C for 1 min (20 pulses of 5 s with a 3 s gap). The primary emulsion was further emulsified with a secondary aqueous phase (10 mL of 2.0% w/v polyvinyl alcohol) at 4°C for 10 min to form a secondary emulsion. To remove chloroform completely, the emulsion was evaporated using a rotary evaporator at 30°C under vacuum. After evaporation, the suspension of PLGA-NPs was washed three times with deionized water at 4°C by centrifugation at 13,000 rpm for 20 min. To quantify encapsulated OVA or poly I:C, the PLGA-NPs were dissolved in 0.1 M NaOH and 0.1% sodium dodecyl sulfate, incubated at 27°C overnight, and then measured using a bicinchoinic acid protein assay kit (Pierce Biotechnology, Rockford, IL, USA) for OVA or a NanoDrop 2,000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) for poly I:C absorbance at 260 nm in ultraviolet light.21 The size and surface charge of the PLGA-NPs were analyzed by dynamic light scattering using an electrophoretic light scattering photometer (ELS-Z; Otsuka Electronics, Osaka, Japan). In addition, the morphology of PLGA-NPs was monitored using scanning electron microscopy (Mira 3 LMU FEG; Tescan, Brno, Czech Republic).
Mice and cell lines
Female C57BL/6 mice (5–6 weeks old, 20 g) were purchased from ORIENT (Gapyeong, South Korea) and maintained under a protocol approved by the Konkuk University Institutional Animal Care and Use Committee (Ref No: KU14157). All procedures were performed according to approved protocols and were in accordance with recommendations for the proper care and use of animals at the specific pathogen-free housing facility at Konkuk University. In this study, we used 168 mice for in vivo cytotoxic CD8+ T cell activation (30 mice), cytotoxic T lymphocyte (CTL) assays (30 mice), therapeutic efficacy tests for the EG.7 and TC-1 tumor model (72 mice), and therapeutic efficacy with CD8+ T cell depletion (36 mice). OVA expressing EG.7 cells (EL4 cell line transfected with the gene encoding OVA) and TC-1 cells expressing HPV16 and HPV-E7 proteins were cultured in RPMI 1640 medium supplemented with 0.1% gentamycin and 10% FBS.
Generation of DCs from mouse bone marrow
DCs were harvested from the bone marrow of C57BL/6 mice.22 Briefly, bone marrow was collected from the tibias and femurs. Red blood cells were depleted using 0.83 M NH4Cl buffer (Sigma-Aldrich Co.). The bone marrow cells (2×106 cells/well) were collected and cultured in a six-well culture plate containing 6 mL culture medium supplemented with 10% FBS, 0.1% gentamycin, and 20 ng/mL mouse recombinant granulocyte-macrophage colony-stimulating factor.
Intracellular uptake assay
Before testing for intracellular uptake of PLGA-NPs, we first conjugated tetramethylrhodamine with OVA or FITC with poly I:C for flow cytometric and confocal microscopic analyses, respectively. Briefly, the DCs were incubated with PLGA-NPs including OVA and poly I:C for 30 min at 37°C. After incubation, PLGA-NP uptake by the DCs was analyzed by flow cytometry (FACSCalibur with CELLQuest software; BD Biosciences, Franklin Lakes, NJ, USA) and confocal microscopy (DeltaVision; GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA).
Cell viability
The viability of cells treated with PLGA-NPs was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.23 DCs were transferred to 96-well plates at 5×104 cells per well and incubated in a 5% CO2 incubator overnight to allow the cells to attach. PLGA-NPs were added to the wells, and the cells were incubated for an additional 24 or 48 hr. Absorbance was read using a microplate reader (EL808; Bio-Tek, Winooski, VT, USA) at 590 nm. The means and standard deviations of triplicates were calculated.
DC maturation and cytokine generation
To confirm DC maturation, DCs were cultured in 6-well plates at a density of 2×106 cells per well and allowed to adhere overnight. DCs alone as a control, PLGA-NPs, PLGA (OVA)-NPs, PLGA (poly I:C)-NPs, or PLGA (OVA + poly I:C)-NPs containing OVA (100 μg/mL) and poly I:C (100 μg/mL) were added to the wells. After 48 hr of incubation, DC maturation was analyzed by flow cytometry. DCs were stained with FITC-conjugated anti-CD11c and PE-conjugated anti-CD40, anti-CD80, anti-CD86, anti-MHC class I, anti-MHC class II, and anti-OVA-specific (SIINFEKL/H-2Kb) MHC class I antibodies. In addition, proinflammatory cytokines (IL-1β, IL-6, IL-12p70, and TNF-α) secreted from DCs during maturation were analyzed using cytokine-specific ELISA kit (eBioscience).
In vivo CD8+ T cell activation
To determine CD8+ T cell activation, DCs treated with PLGA-NPs were injected subcutaneously (sc) into C57BL6 mice (five mice/group). Mice were monitored daily for adverse effects of vaccination and were sacrificed at 7 days after the last immunization (two times weekly). DCs treated with PLGA-NPs were administered in the right flank region with (1) DCs only as a control, (2) DCs treated with OVA solution, (3) DCs treated with PLGA-NPs, (4) DCs treated with PLGA (OVA)-NPs, (5) DCs treated with PLGA (poly I:C)-NPs, or (6) DCs treated with PLGA (OVA + poly I:C)-NPs. At 7 days after the last immunization, 1×107 splenocytes were harvested from the vaccinated mice, suspended in 1 mL of RPMI with 10% FBS, 0.1% gentamycin, and 0.5% 2-mercaptoethanol, and incubated for 16 hr with GolgiPlug (BD Biosciences) and OVA peptide (1 μg/mL).24,25 Cells were washed, stained with PE-conjugated anti-CD8a antibody, fixed, permeabilized, and stained with FITC-conjugated anti-IFN-γ antibody. Cells were analyzed by flow cytometry.
In vivo CTL assay
For CTL assays, DCs treated with PLGA-NPs (2×106 cells/mouse) were injected sc three times, once per week, into C57BL6 mice.26 Four groups (five mice/group) were immunized in the right flank region with (1) DCs only as a control, (2) DCs treated with OVA solution, (3) DCs treated with PLGA-NPs, (4) DCs treated with PLGA (OVA)-NPs, (5) DCs treated with PLGA (poly I:C)-NPs, or (6) DCs treated with PLGA (OVA + poly I:C)-NPs. At 7 days after the last immunization (two times weekly), the splenocytes were harvested and pulsed with or without OVA peptide (10 μg/mL) for 45 min. The OVA peptide-pulsed or unpulsed populations were then incubated with either 10 μM (high) or 1 μM (low) carboxyfluorescein succinimidyl ester (CFSE) at 37°C for 10 min. The two populations were then mixed at a 1:1 ratio. Finally, the mixed splenocytes were injected (107 cells/mouse) intravenously through the tail vein into the immunized C57BL6 mice. After 18 hr, the splenocytes were isolated and the numbers of CFSEhigh and CFSElow cells were measured by flow cytometry.
Antitumor efficacy of DCs treated with PLGA-NPs
To produce tumors, EG.7 lymphoma cells (1×106 cells per 0.1 mL Hank’s Balanced Salt Solution [HBSS]) or TC-1 cervical tumor cells (1×106 cells per 0.1 mL HBSS) were injected sc into mice. We selected EG.7 tumor cells, which express OVA peptide,27 and TC-1 tumor cells, which express HPV16 E7 protein.28 Therefore, EG.7 and TC-1 tumor models would exhibit antigen-specific immune responses against cytotoxic CD8+ T cells, resulting in antigen-specific immune responses to EG.7 (OVA specific) and TC-1 (E7 specific) cells. One week following injection, the mice were randomly allocated into the following groups (n=6 mice per group): (1) DCs only as a control, (2) DCs treated with OVA solution, (3) DCs treated with PLGA-NPs, (4) DCs treated with PLGA (OVA)-NPs, (5) DCs treated with PLGA (poly I:C)-NPs, and (6) DCs treated with PLGA (OVA + poly I:C)-NPs. DCs were administered three times (sc, 2×106 cells/mouse, 1 week intervals for 3 weeks) in the right flank. Tumor volume and survival were recorded. Tumors were measured using calipers, and the tumor volume was calculated using the following formula: tumor volume (mm3) = length × (width)2/2. In addition, we confirmed CD8+ T cell localization using anti-CD8 antibody and tumor cell apoptosis using terminal deoxynucleotidyl transferase dUTP nick-end labeling assays in the tumor tissue by immunohistochemical staining analysis, as described previously using formalin-fixed, paraffin-embedded EG.7 and TC-1 tumor tissue specimens.29 The analysis was recorded in five random fields for each slide and was performed by two investigators in a blinded fashion.
In addition, we performed in vivo antibody depletion assay to confirm CD8+ T cell-mediated antitumor therapeutic efficacy.27,30 We used monoclonal antibody 2.43 for CD8+ T cell depletion. One week after tumor cell injection, the mice were randomly allocated into the following groups (n=6 mice per group): (1) DCs only as a control, (2) DCs treated with PLGA (OVA + poly I:C)-NPs with CD8+ T cell depletion, and (3) DCs treated with PLGA (OVA)-NPs. DCs were administered three times (sc, 2×106 cells/mouse, 1 week intervals for 3 weeks) in the right flank. Depletions (100 μg of anti-CD8 antibody, once daily) were initiated 5 days after tumor cell injection and lasted till the end of the experimental period. The completeness of depletion was confirmed by flow cytometry analysis. More than 90%–95% depletion of the appropriate subset was achieved with normal levels of the other lymphocyte subsets. Tumor was measured using calipers, and the tumor volume was calculated with the above equation.
Statistical analysis
Differences in continuous variables were analyzed using Student’s t-test for comparing two groups. Nonparametric one-way or two-way analysis of variance was performed with Statistical Package for the Social Sciences (SPSS, Inc.), which was used for all statistical analyses. Results with P-value of <0.05 were considered statistically significant.
Results
Characteristics of PLGA-NPs
In this study, we selected PLGA as the polymer matrix because it is particularly attractive for clinical and biological applications due to its low toxicity, biocompatibility, biodegradability, and low immunogenicity.12 Here, we successfully prepared PLGA-NPs using a w/o/w double emulsion method to encapsulate both OVA and poly I:C. We first confirmed the physical properties of PLGA-NPs, PLGA (OVA)-NPs, PLGA (poly I:C)-NPs, and PLGA (OVA + poly I:C)-NPs (Figure 2). The mean particle size and zeta potential were around 200±3.69 nm and −30 mV, respectively (Figure 2A and B). Representative histograms of the particle size distribution are shown in Figure 2C. These data indicated that the encapsulation of either OVA or poly I:C did not affect the formation or physicochemical properties of these PLGA-NP formulations. Additionally, the morphology of these PLGA-NPs was confirmed by scanning electron microscopy. The NPs were spherical and the size was 200 nm (Figure 2C). The loading efficiency of OVA and poly I:C was 90% and 50%, respectively (Figure 2D). In addition, we also confirmed the release of OVA from PLGA (OVA + poly I:C)-NPs, indicating that OVA was easily released in an acidic environment at 37°C (Figure S1).
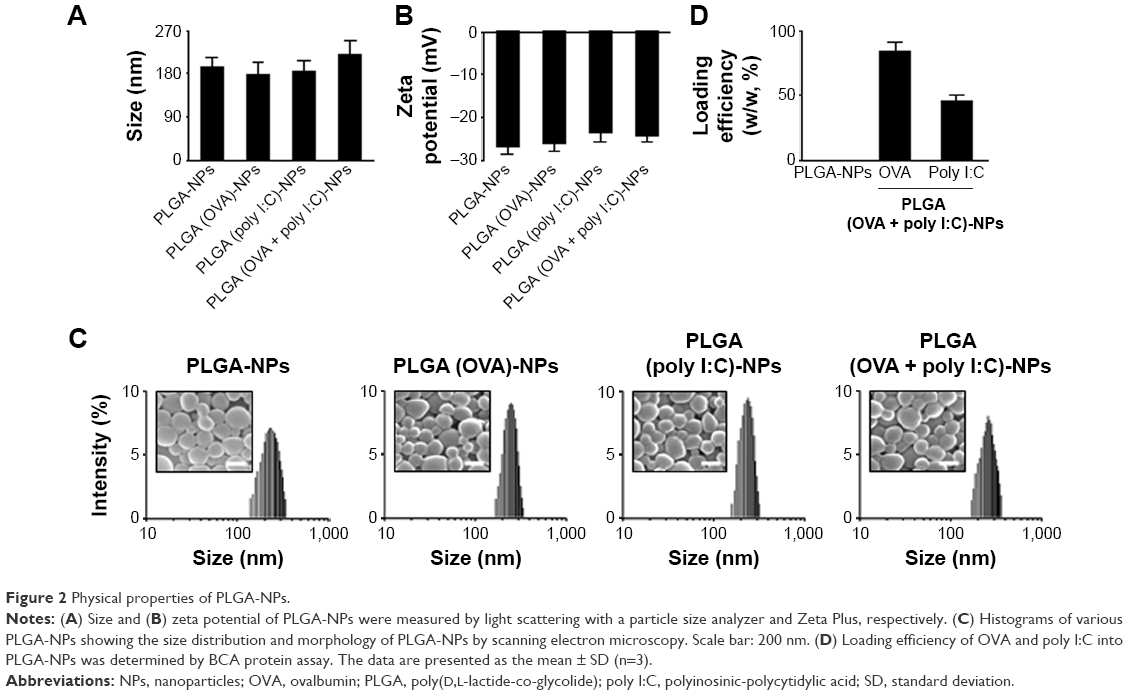 | Figure 2 Physical properties of PLGA-NPs. |
Intracellular uptake of PLGA-NPs in DCs
We next assessed the intracellular uptake of PLGA (OVA + poly I:C)-NPs by flow cytometry and confocal microscopy (Figures 3 and S2). Flow cytometric analysis indicated that the PLGA-NPs exhibited highly efficient and dose-dependent intracellular delivery compared with control and OVA solution (Figure 3A). In addition, confocal microscopic analysis (Figure 3B) showed that uptake of PLGA (OVA + poly I:C)-NPs by DCs was consistent with the flow cytometric data.
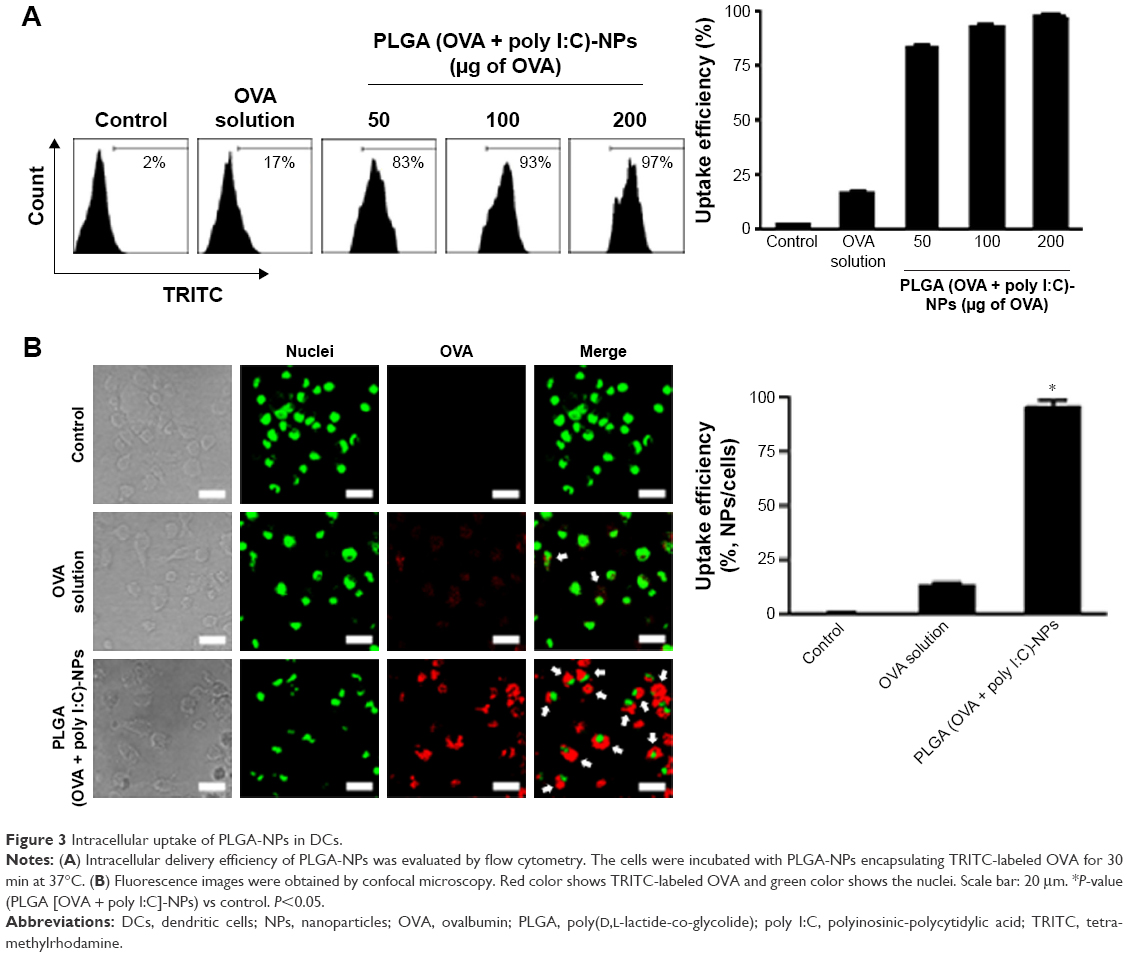 | Figure 3 Intracellular uptake of PLGA-NPs in DCs. |
Cell viability of PLGA-NPs
To assess the possible cytotoxic effect of PLGA-NP formulations on DCs, we tested the viability of DCs after uptake of PLGA-NPs with (Figure 4A) or without encapsulated OVA or poly I:C (Figure 4B). As shown in Figure 4A and B, cell viability was as high as 90%, even with increasing PLGA concentration, indicating that the base PLGA-NP composition was not appreciably toxic to the DCs.
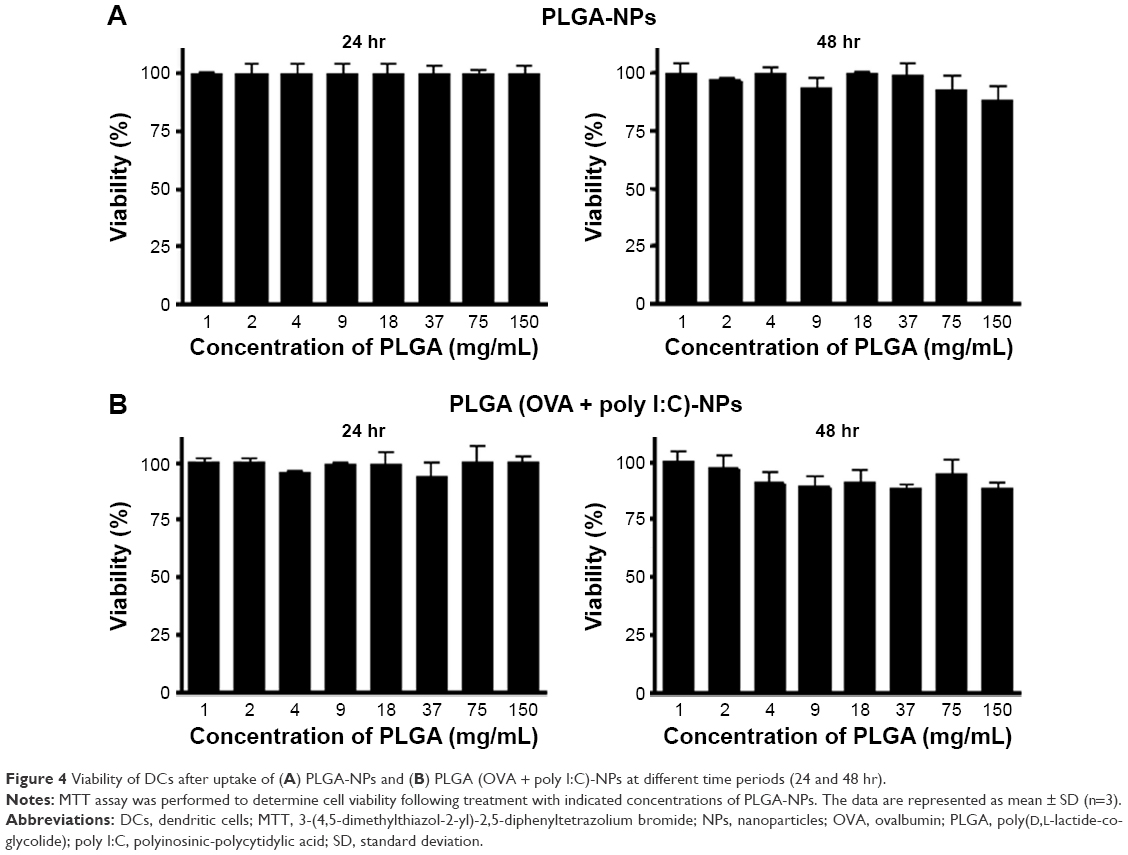 | Figure 4 Viability of DCs after uptake of (A) PLGA-NPs and (B) PLGA (OVA + poly I:C)-NPs at different time periods (24 and 48 hr). |
DC maturation and activation after uptake of PLGA-NPs
To assess maturation and activation, we characterized the presence of activation markers and proinflammatory cytokine expression in DCs. DCs were isolated from the bone marrow of mice and incubated with or without PLGA-NPs (100 μg poly I:C). Various maturation and activation markers were then assessed using flow cytometry and ELISA. DCs treated with PLGA (OVA + poly I:C)-NPs expressed significantly higher levels of the maturation surface markers CD40, CD80, CD86, MHC class I, and MHC class II compared to control DCs, which exhibited expression levels similar to those treated with empty PLGA-NPs as a negative control (Figure 5A). These results indicated that PLGA (OVA + poly I:C)-NPs activated and induced maturation of DCs. We also confirmed OVA-specific MHC I-mediated antigen cross-presentation. As shown in Figure 5A, OVA-dependent MHC I expression was significantly higher than that in control DCs (Figure 5A). In addition, incubation with PLGA (OVA + poly I:C)-NPs significantly increased the expression of proinflammatory cytokines IL-1β, IL-6, IL-12p70, and TNF-α during DC maturation relative to that in control DCs (Figure 5B). Collectively, these data demonstrated that PLGA (OVA + poly I:C)-NPs could elicit maturation, activation, and induce antigen-specific cross-presentation in DCs.
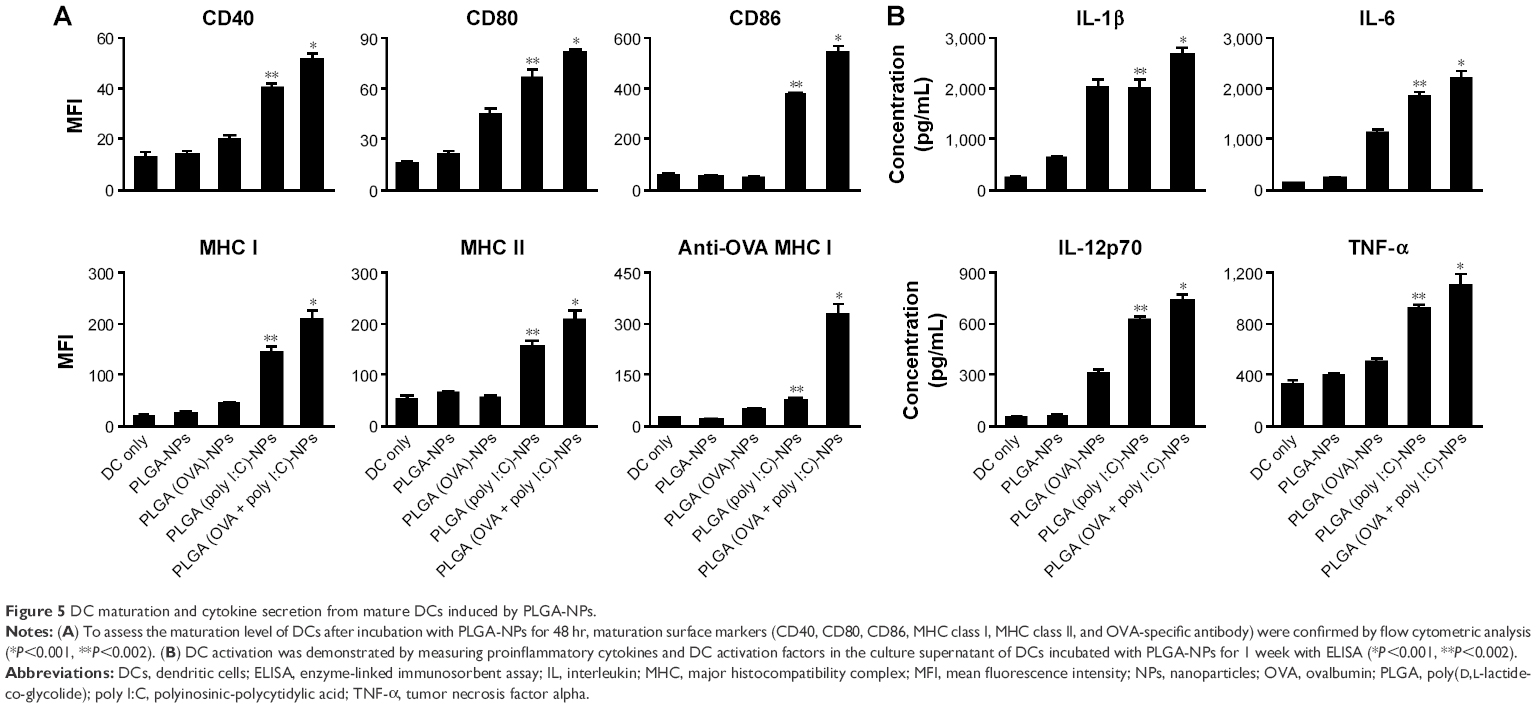 | Figure 5 DC maturation and cytokine secretion from mature DCs induced by PLGA-NPs. |
In vivo antigen-specific CD8+ T cell activation and CTL assay
We next evaluated the potential of PLGA (OVA + poly I:C)-NPs to generate antigen-specific adaptive cellular immune responses using PLGA (OVA + poly I:C)-NPs activated DCs as a DC-based vaccination. C57BL/6 mice (five per group) were immunized with DCs treated with PLGA-NPs (2×106) pulsed with or without OVA peptide (1 μg/mL), and splenocytes were harvested and analyzed by flow cytometry 7 days after the last immunization. Mice vaccinated with PLGA (OVA + poly I:C)-NP-treated DCs pulsed with OVA peptides generated significantly higher numbers of activated CD8+ T cells as measured by IFN-γ secretion (Figure 6A). This result indicated that PLGA (OVA + poly I:C)-NP-treated DCs could activate antigen-specific CD8+ T cells, which were capable of tumor protection.
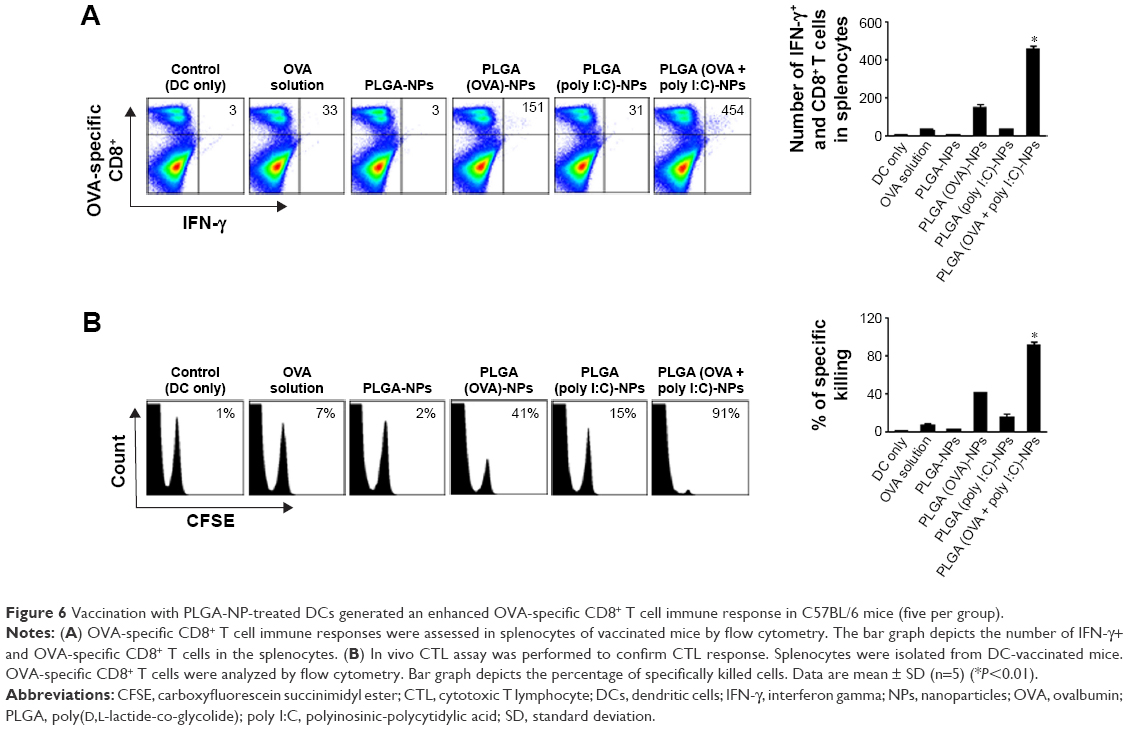 | Figure 6 Vaccination with PLGA-NP-treated DCs generated an enhanced OVA-specific CD8+ T cell immune response in C57BL/6 mice (five per group). |
In addition, we confirmed the ability of activated CD8+ T cells stimulated by DCs treated with PLGA (OVA + poly I:C)-NPs to kill tumor cells using an in vivo CTL activity assay. C57BL/6 mice (five per group) were immunized with DCs treated with PLGA-NPs (2×106) pulsed with or without OVA peptide (1 μg/mL), and splenocytes were harvested and analyzed by flow cytometry 7 days after the last immunization. We observed significantly higher target cell lysis in mice that received PLGA (OVA + poly I:C)-NP-treated DCs than that in mice immunized with DCs only, DCs treated with PLGA-NPs, or DCs treated with OVA solution (Figure 6B). These results indicated that PLGA (OVA + poly I:C)-NP-treated DCs activated cytotoxic CD8+ T cells, and thus served as a cancer vaccine against OVA-positive tumor cells by enhancing cytotoxic activity.
Therapeutic efficacy of vaccination with PLGA-NP-treated DCs
To determine the potential therapeutic efficacy of the PLGA-NPs, we selected EG.7-OVA cells, which are an attractive tumor model for OVA-based experimental models of anticancer vaccines. Seven days following the sc injection of tumor cells, mice were randomly allocated to the following groups (n=6 mice/group): (1) DCs only, (2) DCs treated with OVA solution, (3) DCs treated with PLGA-NPs, (4) DCs treated with PLGA (OVA)-NPs, (5) DCs treated with PLGA (poly I:C)-NPs, and (6) DCs treated with PLGA (OVA + poly I:C)-NPs. DCs were injected sc 7 days after tumor inoculation three times at 1 week intervals (Figure 7A). DCs treated with PLGA (OVA + poly I:C)-NPs showed significantly higher inhibition of tumor growth compared to DCs alone (control group, 93% reduction; P<0.005), OVA solution (90% reduction; P<0.007), and PLGA-NPs (93% reduction; P<0.004) (Figure 7B and D). Notably, 100% of mice vaccinated with PLGA (OVA + poly I:C)-NPs-treated DCs survived for up to 45 days, while mice in the other vaccinated groups died within 45 days after tumor inoculation (Figure 7C). The data suggested that vaccination with PLGA (OVA + poly I:C)-NP-treated DCs could enhance therapeutic antitumor efficacy and prolong survival. There were no differences in total body weight, feeding habits, or behavior between the groups, suggesting that there were no overt toxicities related to the therapy. In addition, we examined the tumors for markers of anti-CD8 antibody to determine CD8+ T cells in the tumor tissue. PLGA (OVA + poly I:C)-NP-treated DCs showed a significantly higher population of CD8+ T cells in the tumor tissue compared to control or other treatment groups, and apoptosis was increased (Figure 7E).
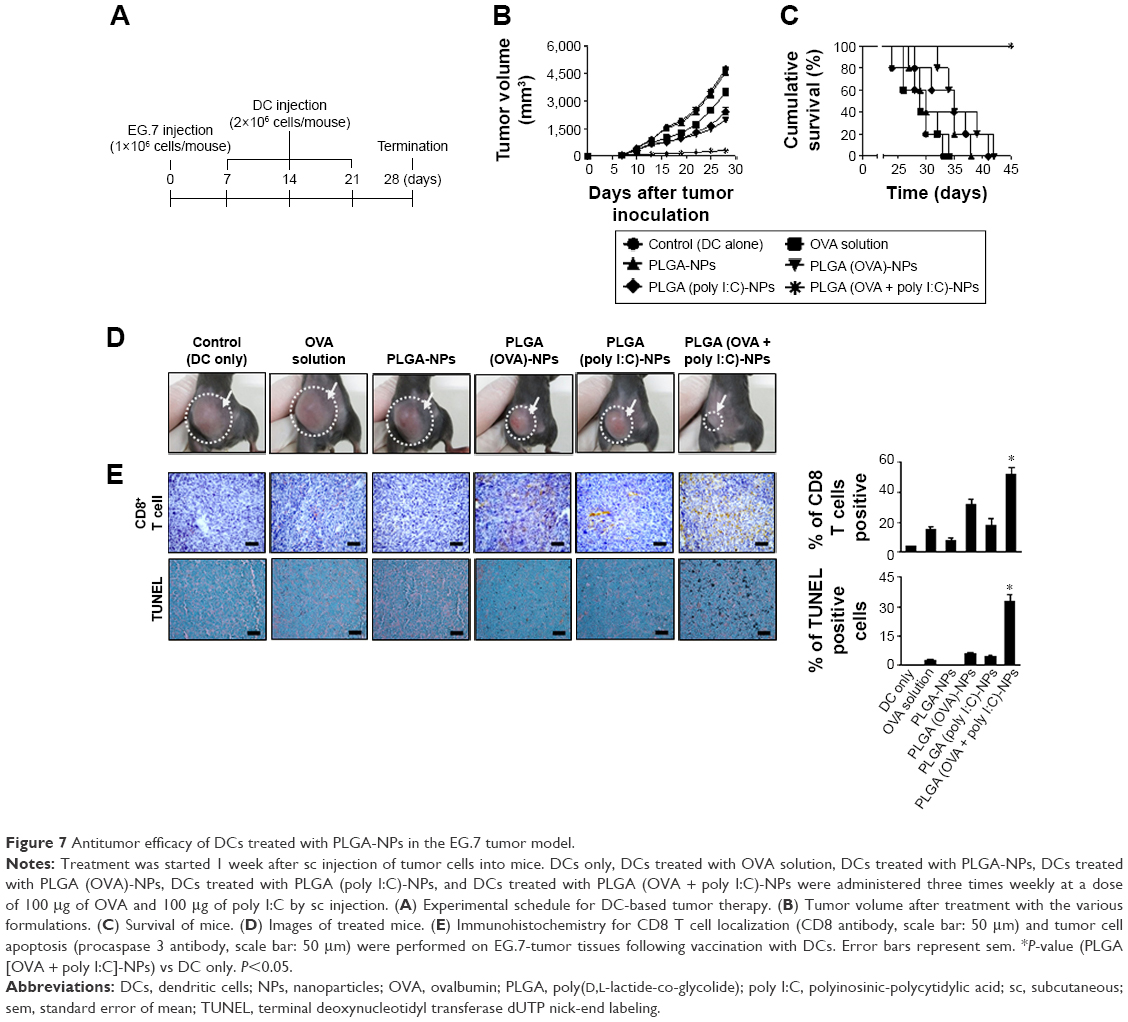 | Figure 7 Antitumor efficacy of DCs treated with PLGA-NPs in the EG.7 tumor model. |
To establish that the therapeutic effects of PLGA-NPs are not unique to just one target antigen, we also performed an in vivo experiment with additional target E7 antigens. We prepared E7 peptide (AGQAEPDRAHYNIVTFCCKCDS)-incorporated PLGA (E7 + poly I:C)-NPs against the TC-1 tumor model, which expresses HPV16 and HPV-E7 proteins. Mice were randomly allocated to the following groups (n=6 mice/group): (1) DCs only, (2) DCs treated with E7 solution (100 μg), (3) DCs treated with PLGA-NPs, (4) DCs treated with PLGA (E7)-NPs, (5) DCs treated with PLGA (poly I:C)-NPs, and (6) DCs treated with PLGA (E7 + poly I:C)-NPs. The experimental groups underwent three intraperitoneal injections at weekly intervals after 7 days of tumor cell injection (Figure 8A). Treatment with PLGA (E7 + poly I:C)-NPs resulted in significant inhibition of tumor growth as compared to DC alone (control group, 91% reduction; P<0.01), E7 solution (89% reduction; P<0.01), and PLGA-NPs (92% reduction; P<0.01) (Figure 8B and D). Notably, 100% of mice vaccinated with DCs treated with PLGA (E7 + poly I:C)-NPs survived over 40 days, while mice vaccinated with control DCs, E7 solution, and PLGA-NPs died within 35 days after tumor inoculation (Figure 8C). In addition, PLGA (E7 + poly I:C)-NP-treated DCs showed significantly higher populations of CD8+ T cells in the tumor tissue compared to the other treatment groups, and apoptosis was increased (Figure 8E).
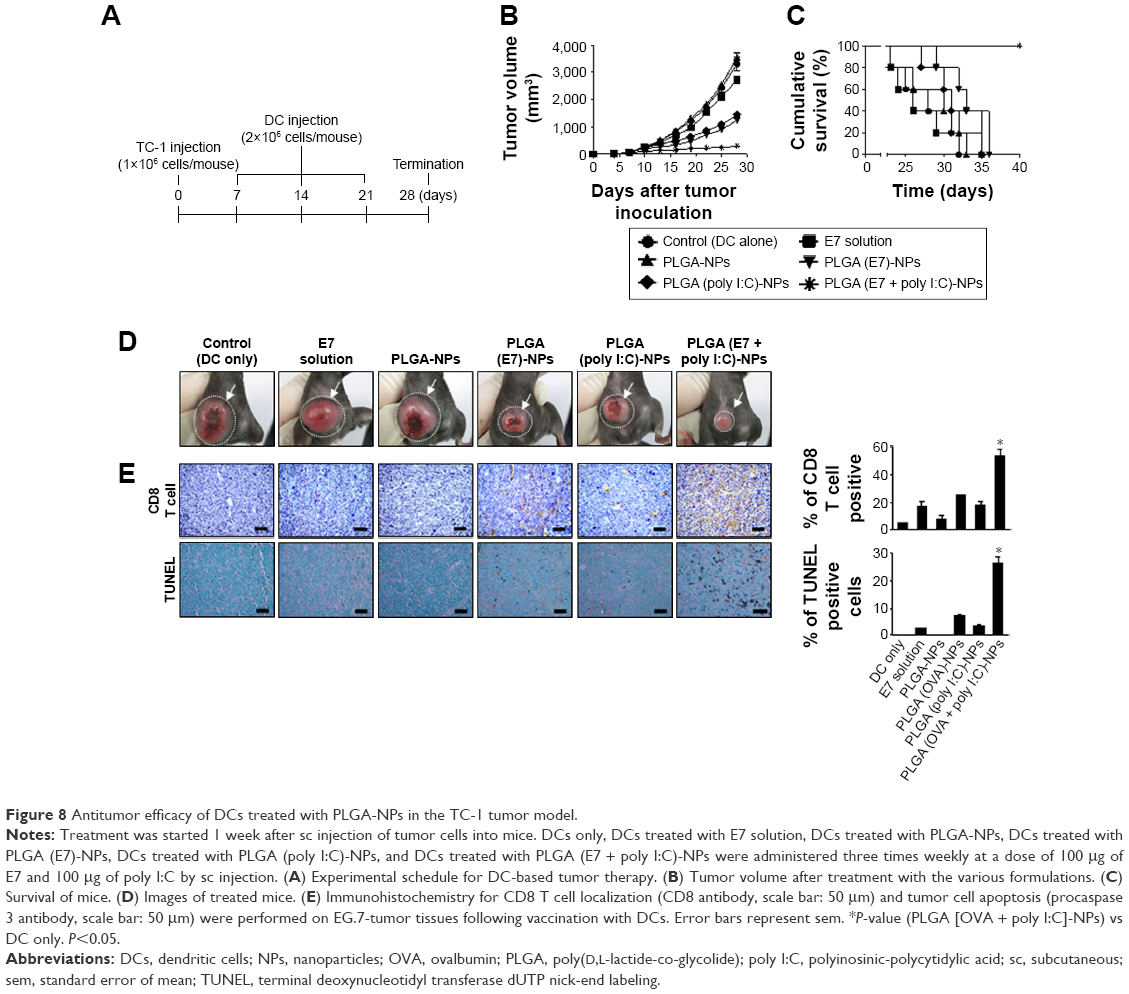 | Figure 8 Antitumor efficacy of DCs treated with PLGA-NPs in the TC-1 tumor model. |
To determine CD8+ T cell-mediated immune response, we performed tumor protection experiment with antibody depletion (Figure 9). Mice were randomly allocated to the following groups (n=6 mice/group): (1) DCs only, (2) DCs treated with PLGA (OVA + poly I:C)-NPs with CD8+ T cell depletion, and (3) DCs treated with PLGA (poly I:C)-NPs. The experimental schedule followed for mice treatment is presented in Figure 9A. Treatment with PLGA (OVA + poly I:C)-NPs with CD8+ T cell depletion showed nearly the same volume of tumor as compared to DC alone (control group; P<0.65) (Figure 9B), and the tumor mass is shown in Figure 9C.
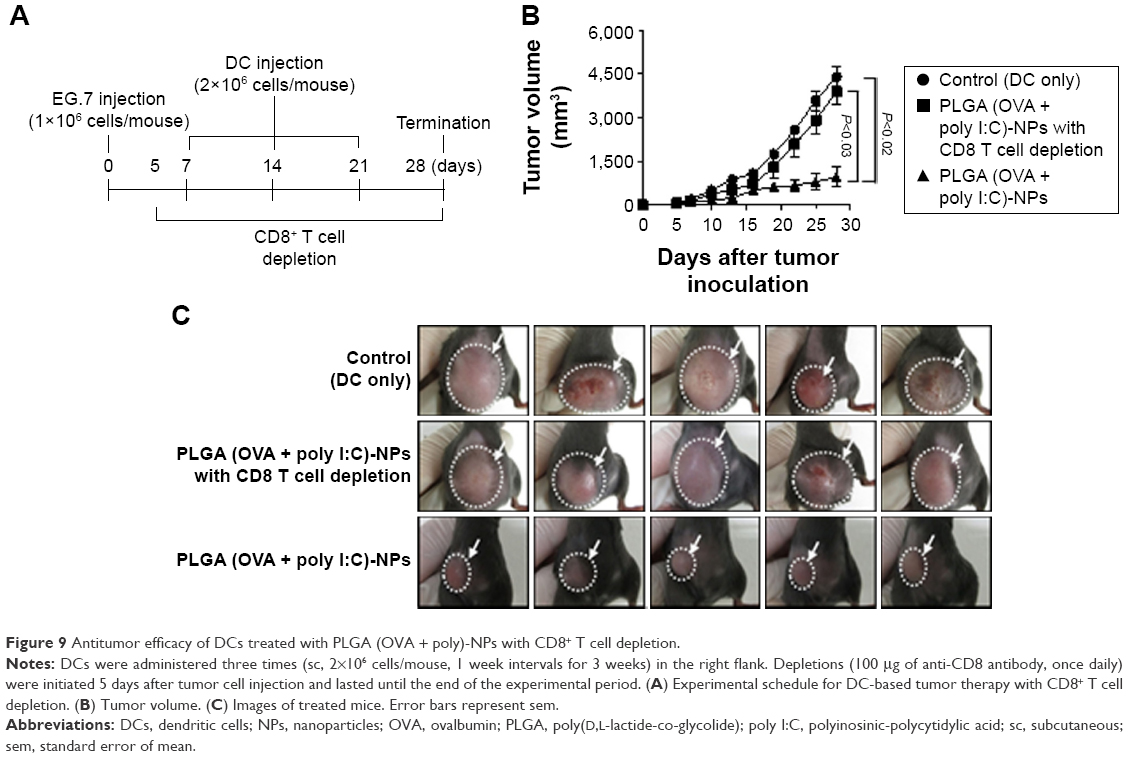 | Figure 9 Antitumor efficacy of DCs treated with PLGA (OVA + poly)-NPs with CD8+ T cell depletion. |
Discussion
We demonstrated here that an effective PLGA-NP-based delivery system loaded with adjuvant and antigen to increase the maturation of DCs leads to potent antigen-specific CD8+ T cell immunity in EG.7 and TC-1 tumor-bearing mice. This approach has broad utility for adjuvant delivery as well as tumor-specific antigen delivery. The PLGA-NPs were effective in delivering both adjuvant and antigen and in achieving high therapeutic efficacy.
Adjuvant and antigen delivery to DCs is a key parameter for medical and pharmaceutical applications of DC-based cancer immunotherapies to initiate the maturation of DCs. However, limited intracellular delivery of the target molecules must be overcome in order to realize the full potential of this therapeutic strategy in the clinical setting. Moreover, the delivery efficiency of free adjuvant or antigen without the use of NPs is quite low, and most of the adjuvant or antigen is rapidly degraded following injection.18 Therefore, to overcome this limitation, effective delivery systems are needed.
While a number of NP systems have been utilized for therapeutic applications, most of these systems result in wide distribution of NPs throughout the body and can lead to toxicity in normal tissues. In addition, single-adjuvant delivery may require higher doses for effective intracellular delivery to DCs. These systems include microparticles,31 nanofibers,32 metal-based particles,33 and emulsions.34 Although many types of compounds have potential utility as delivery agents, some are associated with concerns regarding their safety and evoked immune responses. The development of a DC-based vaccination for immunotherapy, therefore, requires clinically suitable, safe, and effective delivery systems. NPs are promising and highly desirable for their potential to overcome the limitations of conventional compound delivery. Moreover, therapeutic payloads packaged into NPs should be a clinically viable approach for the development of vaccine-related immunotherapy.
PLGA-NPs are an attractive platform for payload delivery because of their biocompatibility, biodegradability, low toxicity, and low immunogenicity, which are key parameters for medical and pharmaceutical applications.13 Moreover, payloads are frequently loaded within the particles, and their type and number may not affect the pharmacokinetics and biodistribution of the NPs. Recent work comparing adjuvant delivery has shown that the primary role is to increase DC maturation. Here, we loaded poly I:C into PLGA-NPs as an adjuvant, which can stimulate TLR3 receptor in the endosome within the DCs, leading to DC maturation. Poly I:C is a promising immunostimulant for cancer vaccines directed against intracellular pathogens and strongly drives cell-mediated immunity and type I IFN response.18,35 In addition, we demonstrated the therapeutic efficacy of DC-based immunotherapy using PLGA (OVA + poly I:C)-NPs in different tumor models and confirmed that antigen-specific CD8+ T cell activation contributed to inhibition of tumor growth.
NP-based systems have been used as effective antigen or adjuvant delivery carriers to increase penetration into DCs through ex vivo manipulation. Moreover, NP systems have also been utilized for delivery of cargo by intratumoral injection to promote immune responses in the tumor microenvironment. In this study, we established the optimum formulation to increase the coloading efficiency of antigen and adjuvant. Combinations of different antigens or, more importantly, combinations of antigens and adjuvants in the same particle can produce effective immune response. Antigens and adjuvants have to be codelivered to the same cell. PLGA (OVA + poly I:C)-NPs with high coloading efficiency and containing low doses of antigens and adjuvants were able to induce strong DC maturation and antigen-specific activation. The use of lower doses in our system would be advantageous not only for minimizing the potential side effects associated with the use of adjuvants, but also from an economical point of view. In addition, we demonstrated the therapeutic efficacy of DC-based cancer immunotherapy in different tumor models. PLGA (antigen + poly I:C) treatment of DCs in vitro resulted in antigen-specific DC maturation. After injection of mature DC into tumor-bearing mice, we confirmed antigen-specific CD8+ T cell activation leading to increased therapeutic efficacy.
DC-based cancer immunotherapy for adjuvant and/or antigen delivery leads to potent immune responses in mice. This study provides a rational approach to improving the safety and reducing the toxicity of adjuvants. In addition, the NP system allows for the codelivery of therapeutic payloads, such as an antigen or adjuvant, which could further enhance the antigen-specific immune response without increasing toxicity. This NP-based adjuvant delivery system may be attractive for many diverse biomedical applications. Although the PLGA-NP system could be useful for diseases associated with the immune system and for enhancing the immune responses, additional possibilities for immune modulation using the PLGA-NP platform may be explored and developed for research purposes.
Conclusion
We demonstrated the ability of the PLGA-NP platform to enhance the potency of DC-based vaccination in tumor-bearing mice. We showed that PLGA (OVA + poly I:C)-NPs induced DC activation and maturation. Furthermore, PLGA (OVA + poly I:C)-NP-treated DCs pulsed with OVA peptides generated higher numbers of OVA-specific CD8+ T cells, which led to potent antitumor efficacy in EG.7 and TC-1 tumor-bearing mice. In addition, the antigen-specific CD8+ T cell immune response elicited by the PLGA (OVA + poly I:C)-NP-treated DC vaccine led to more potent antitumor effects and prolonged survival in tumor-bearing mice. This PLGA-NP-based delivery system will also be useful for delivering many other target adjuvants or antigens to DCs. In addition, the PLGA-NP system could be expanded and developed to include additional therapeutic and experimental approaches. The PLGA-NP-based strategy presented here has broad potential as a delivery platform for increasing the immune responses to DC-based cancer immunotherapy and could be adapted for other immune-associated diseases.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (NRF-2015R1A2A1A13001713, NRF-2016R1A5A2012284, NRF-2015R1A2A2A04003620) (Y-MP and YJL). This work was supported by Basic Research Laboratory Program through the NRF funded by the Ministry of Science (NRF-2016R1A2B2007327), and ICT and Future Planning (No 2013R1A4A1069575) (HDH). This study was supported by a grant from the National R&D program for Cancer Control, Ministry for Health, Welfare and Family affairs, Republic of Korea (1520100) (J-WL, and HDH). The authors would like to express their thanks to Ga Hee Kim, Min Gi Kim, and Hyung Jun Ahn for data analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Heo MB, Cho MY, Lim YT. Polymer nanoparticles for enhanced immune response: combined delivery of tumor antigen and small interference RNA for immunosuppressive gene to dendritic cells. Acta Biomater. 2014;10(5):2169–2176. | ||
Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462(7272):449–460. | ||
Steinman RM. Dendritic cells in vivo: a key target for a new vaccine science. Immunity. 2008;29(3):319–324. | ||
Gelao L, Criscitiello C, Esposito A, et al. Dendritic cell-based vaccines: clinical applications in breast cancer. Immunotherapy. 2014;6(3):349–360. | ||
Chiang CL, Balint K, Coukos G, Kandalaft LE. Potential approaches for more successful dendritic cell-based immunotherapy. Expert Opin Biol Ther. 2015;15(4):569–582. | ||
Cruz LJ, Rosalia RA, Kleinovink JW, Rueda F, Lowik CW, Ossendorp F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8(+) T cell response: a comparative study. J Control Release. 2014;192:209–218. | ||
Reddy ST, Swartz MA, Hubbell JA. Targeting dendritic cells with biomaterials: developing the next generation of vaccines. Trends Immunol. 2006;27(12):573–579. | ||
Rosalia RA, Silva AL, Camps M, et al. Efficient ex vivo induction of T cells with potent anti-tumor activity by protein antigen encapsulated in nanoparticles. Cancer Immunol Immunother. 2013;62(7):1161–1173. | ||
Liu SY, Wei W, Yue H, et al. Nanoparticles-based multi-adjuvant whole cell tumor vaccine for cancer immunotherapy. Biomaterials. 2013;34(33):8291–8300. | ||
Felice B, Prabhakaran MP, Rodriguez AP, Ramakrishna S. Drug delivery vehicles on a nano-engineering perspective. Mater Sci Eng C Mater Biol Appl. 2014;41:178–195. | ||
van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, van Gaal EV. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65(10):1284–1298. | ||
Godsey ME, Suryaprakash S, Leong KW. Materials innovation for co-delivery of diverse therapeutic cargos. RSC Adv. 2013;3(47):24794–24811. | ||
Joshi VB, Geary SM, Salem AK. Biodegradable particles as vaccine antigen delivery systems for stimulating cellular immune responses. Hum Vaccin Immunother. 2013;9(12):2584–2590. | ||
Heo MB, Lim YT. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials. 2014;35(1):590–600. | ||
Zhang Z, Tongchusak S, Mizukami Y, et al. Induction of anti-tumor cytotoxic T cell responses through PLGA-nanoparticle mediated antigen delivery. Biomaterials. 2011;32(14):3666–3678. | ||
Perez-Giron JV, Belicha-Villanueva A, Hassan E, et al. Mucosal polyinosinic-polycytidylic acid improves protection elicited by replicating influenza vaccines via enhanced dendritic cell function and T cell immunity. J Immunol. 2014;193(3):1324–1332. | ||
Quinn KM, Yamamoto A, Costa A, et al. Coadministration of polyinosinic:polycytidylic acid and immunostimulatory complexes modifies antigen processing in dendritic cell subsets and enhances HIV gag-specific T cell immunity. J Immunol. 2013;191(10):5085–5096. | ||
Hafner AM, Corthesy B, Merkle HP. Particulate formulations for the delivery of poly (I:C) as vaccine adjuvant. Adv Drug Deliv Rev. 2013;65(10):1386–1399. | ||
Yao MH, Ma M, Chen Y, et al. Multifunctional Bi2S3/PLGA nanocapsule for combined HIFU/radiation therapy. Biomaterials. 2014;35(28):8197–8205. | ||
Zhang Y, Gou J, Sun F, et al. Impact of electrolytes on double emulsion systems (W/O/W) stabilized by an amphiphilic block copolymer. Colloids Surf B Biointerfaces. 2014;122:368–374. | ||
Dolen Y, Kreutz M, Gileadi U, et al. Co-delivery of PLGA encapsulated invariant NKT cell agonist with antigenic protein induce strong T cell-mediated antitumor immune responses. Oncoimmunology. 2015;5(1):e1068493. | ||
Zhou F, Ciric B, Zhang GX, Rostami A. Immunotherapy using lipopolysaccharide-stimulated bone marrow-derived dendritic cells to treat experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2014;178(3):447–458. | ||
Roh JW, Huang J, Hu W, et al. Biologic effects of platelet-derived growth factor receptor alpha blockade in uterine cancer. Clin Cancer Res. 2014;20(10):2740–2750. | ||
Kang TH, Kim YS, Kim S, et al. Pancreatic adenocarcinoma upregulated factor serves as adjuvant by activating dendritic cells through stimulation of TLR4. Oncotarget. 2015;6(29):27751–27762. | ||
Kang TH, Knoff J, Yeh WH, et al. Treatment of tumors with vitamin E suppresses myeloid derived suppressor cells and enhances CD8+ T cell-mediated antitumor effects. PLoS One. 2014;9(7):e103562. | ||
Jung ID, Shin SJ, Lee MG, et al. Enhancement of tumor-specific T cell-mediated immunity in dendritic cell-based vaccines by Mycobacterium tuberculosis heat shock protein X. J Immunol. 2014;193(3):1233–1245. | ||
Song CK, Han HD, Noh KH, et al. Chemotherapy enhances CD8(+) T cell-mediated antitumor immunity induced by vaccination with vaccinia virus. Mol Ther. 2007;15(8):1558–1563. | ||
Ramakrishnan R, Assudani D, Nagaraj S, et al. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest. 2010;120(4):1111–1124. | ||
Cho MS, Vasquez HG, Rupaimoole R, et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014;6(6):1085–1095. | ||
Carmi Y, Spitzer MH, Linde IL, et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature. 2015;521(7550):99–104. | ||
Singh M, Chakrapani A, O’Hagan D. Nanoparticles and microparticles as vaccine-delivery systems. Expert Rev Vaccines. 2007;6(5):797–808. | ||
Chen J, Pompano RR, Santiago FW, et al. The use of self-adjuvanting nanofiber vaccines to elicit high-affinity B cell responses to peptide antigens without inflammation. Biomaterials. 2013;34(34):8776–8785. | ||
Yu X, Feizpour A, Ramirez NG, et al. Glycosphingolipid-functionalized nanoparticles recapitulate CD169-dependent HIV-1 uptake and trafficking in dendritic cells. Nat Commun. 2014;5:4136. | ||
Zeng BJ, Chuan YP, O’Sullivan B, et al. Receptor-specific delivery of protein antigen to dendritic cells by a nanoemulsion formed using top-down non-covalent click self-assembly. Small. 2013;9(22):3736–3742. | ||
Ammi R, De Waele J, Willemen Y, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther. 2015;146:120–131. |
Supplementary materials
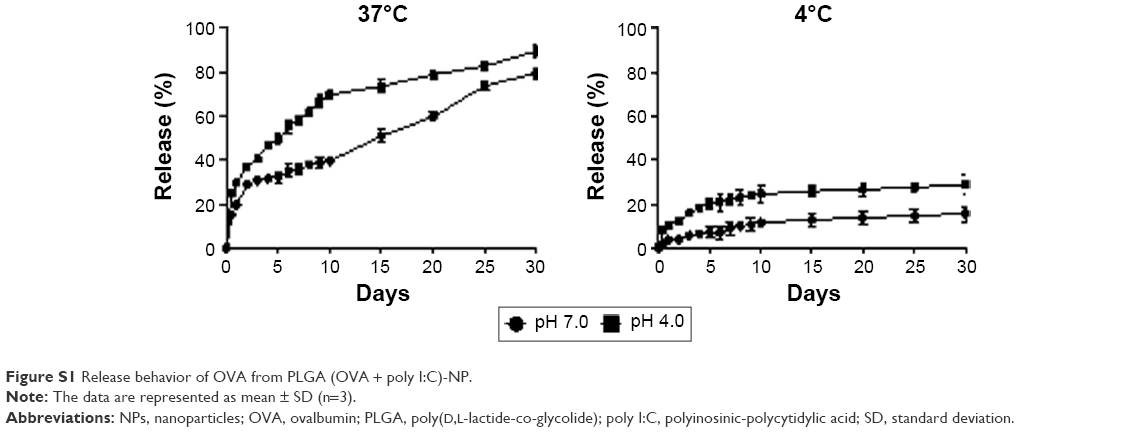 | Figure S1 Release behavior of OVA from PLGA (OVA + poly I:C)-NP. |
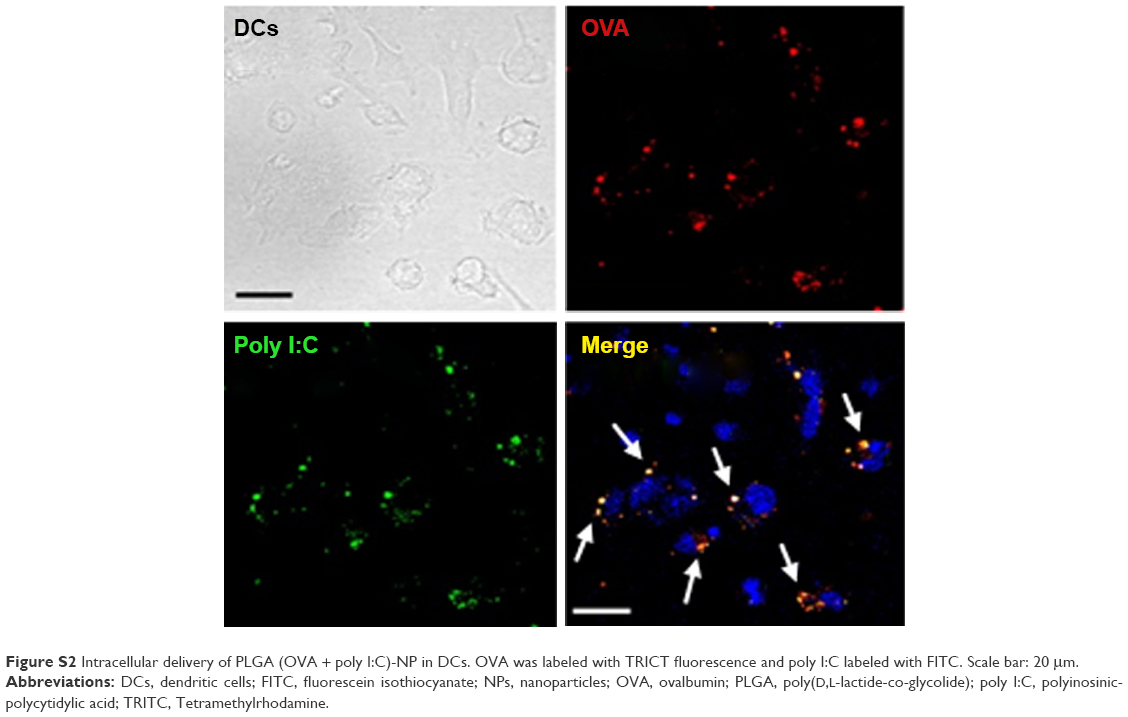 | Figure S2 Intracellular delivery of PLGA (OVA + poly I:C)-NP in DCs. OVA was labeled with TRICT fluorescence and poly I:C labeled with FITC. Scale bar: 20 μm. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.