Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
Thyroid cancer in a patient with Lynch syndrome – case report and literature review
Authors Fazekas-Lavu M, Parker A, Spigelman AD, Scott RJ, Epstein RJ, Jensen M ![]() , Samaras K
, Samaras K
Received 8 September 2016
Accepted for publication 10 March 2017
Published 21 July 2017 Volume 2017:13 Pages 915—918
DOI https://doi.org/10.2147/TCRM.S121812
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Video abstract presented by Monika Fazekas-Lavu
Views: 414
Monika Fazekas-Lavu,1 Andrew Parker,2 Allan D Spigelman,3,4 Rodney J Scott,5 Richard J Epstein,6 Michael Jensen,7 Katherine Samaras1,8
1Department of Endocrinology, 2Department of Pathology, St Vincent’s Hospital, Darlinghurst, NSW, Australia; 3Hereditary Cancer Clinic, St Vincent’s Cancer Genetics Service, Darlinghurst, NSW, Australia; 4University of NSW, St Vincent’s Clinical School, Darlinghurst, NSW, Australia; 5Division of Molecular Medicine, Pathology North, John Hunter Hospital and The Hunter Medical Research Institute, Newcastle, NSW, Australia; 6Department of Oncology, 7Department of Oncological Surgery/General Surgery, St Vincent’s Hospital, Darlinghurst, NSW, Australia; 8Diabetes and Metabolism Research Program, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia
Abstract: Lynch syndrome describes a familial cancer syndrome comprising germline mutations in one of four DNA mismatch repair genes, MLH1, MSH2, MSH6, and PMS2 and is characterized by colorectal, endometrial, and other epithelial malignancies. Thyroid cancer is not usually considered to be part of the constellation of Lynch syndrome cancers nor have Lynch syndrome tumor gene mutations been reported in thyroid malignancies. This study reports a woman with Lynch syndrome (colonic cancer and a DNA mismatch repair mutation in the MSH2 gene) with a synchronous papillary thyroid cancer. Six years later, she developed metachronous breast cancer. Metastatic bone disease developed after 3 years, and the disease burden was due to both breast and thyroid diseases. Despite multiple interventions for both metastatic breast and thyroid diseases, the patient’s metastatic burden progressed and she died of leptomeningeal metastatic disease. Two prior case reports suggested thyroid cancer may be an extraintestinal malignancy of the Lynch syndrome cancer group. Hence, this study examined the genetic relationship between the patient’s known Lynch syndrome and her thyroid cancer. The thyroid cancer tissue showed normal expression of MSH2, suggesting that the tumor was not due to the oncogenic mutation of Lynch syndrome, and molecular analysis confirmed BRAF V600E mutation. Although in this case the thyroid cancer was sporadic, it raises the importance of considering cancer genetics in familial cancer syndromes when other cancers do not fit the criteria of the syndrome. Careful documentation of other malignancies in patients with thyroid cancer and their families would assist in better understanding of any potential association. Appropriate genetic testing will clarify whether a common pathogenic mechanism links seemingly unrelated cancers.
Keywords: cancer genetics, familial cancer syndromes, thyroid and hereditary non-polyposis colorectal cancer
Introduction
Lynch syndrome (hereditary non-polyposis cancer) is an autosomal dominant disorder associated with germline mutations in one of four DNA mismatch repair genes. Approximately 90% of mutations are located in MLH1 or MSH2, the remainder in MSH6 and PMS2.1,2 Lynch syndrome carries increased risk of colorectal cancer, endometrial cancer and other epithelial malignancies.1
Lynch syndrome classifications do not include thyroid cancer. The Amsterdam criteria apply the “3-2-1 Rule”: three relatives with colorectal cancer, one a first degree relative of the other two; two successive generations affected; one diagnosed before the age of 50 years. The expanded Amsterdam II classification includes non-colorectal cancers: endometrial, small intestine, ureteric, or renal cancers.3
This study reports a female with Lynch syndrome-related colon cancer that developed synchronous thyroid cancer and 6 years later breast cancer with progression to metastatic disease due to thyroid and breast cancer burden. The aim of this study was to determine whether the thyroid cancer could be genetically linked to the same mismatch repair gene mutation as her Lynch syndrome colonic cancer.
Case report
Written informed consent was obtained from the patient’s next of kin for publication of this case report and the associated images. A 58-year-old female with widespread, progressive metastatic disease (breast, bowel, and thyroid cancer) was referred for advice on attribution of the metastatic burden to thyroid cancer and thyroid-specific intervention. Past history included metallic aortic valve replacement (at age 34 years) and aortic arch replacement (at age 52 years), and prophylactic total hysterectomy (at age 41 years). The family history included ovarian, endometrial, and colon cancer in several first-degree relatives.
Synchronous thyroid and colorectal malignancies were detected at 47 years. The cecal carcinoma (pT3pN0, 55×60×40 mm) showed absent expression of MSH2 and MSH6 protein in immunohistochemistry, consistent with a deficiency of DNA mismatch repair and Lynch syndrome. DNA analysis found an exon 6 sequence variation (c.998G>A) in MSH2 (as detected by semi-automated DNA sequencing and multiplex ligation probe amplification analysis).
Total thyroidectomy and neck dissection showed a 22×20×18 mm papillary thyroid carcinoma (PTC) with lymphovascular and striated muscle invasion and 2/2 lymph node metastases (pT2N1aMx). Progressive radioactive iodine doses and scans and the stimulated and unstimulated thyroglobulin levels are shown in Figure 1A. At 51 years, metastatic PTC was detected in locoregional cervical nodes, and modified radical neck dissection was undertaken.
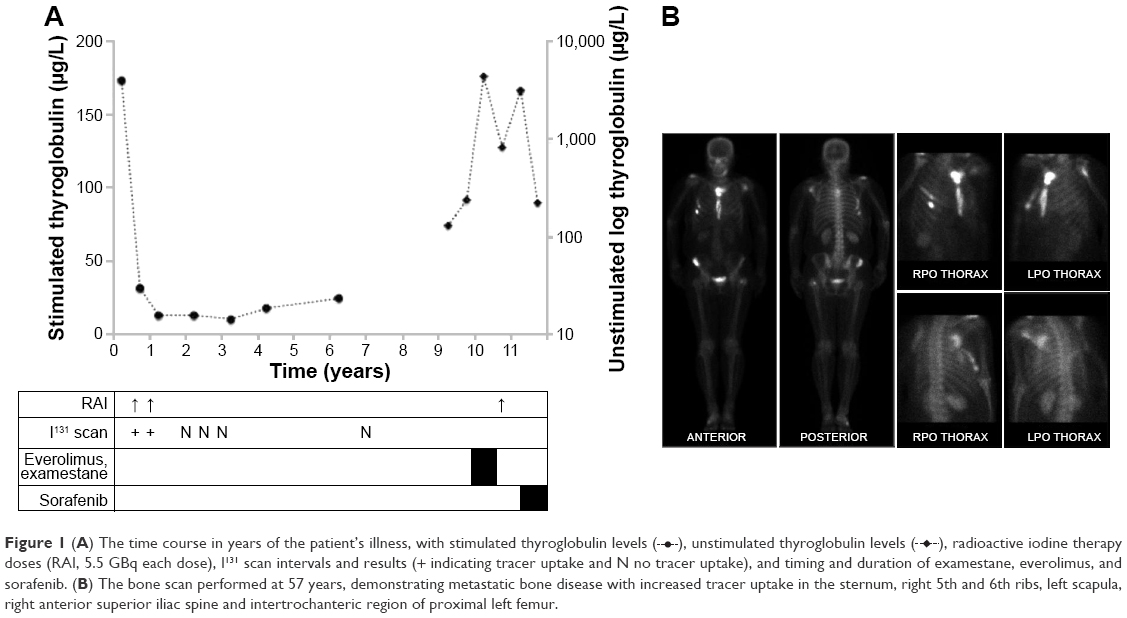 | Figure 1 (A) The time course in years of the patient’s illness, with stimulated thyroglobulin levels ( |
 ), unstimulated thyroglobulin levels (
), unstimulated thyroglobulin levels ( ), radioactive iodine therapy doses (RAI, 5.5 GBq each dose), I131 scan intervals and results (+ indicating tracer uptake and N no tracer uptake), and timing and duration of examestane, everolimus, and sorafenib. (B) The bone scan performed at 57 years, demonstrating metastatic bone disease with increased tracer uptake in the sternum, right 5th and 6th ribs, left scapula, right anterior superior iliac spine and intertrochanteric region of proximal left femur.
), radioactive iodine therapy doses (RAI, 5.5 GBq each dose), I131 scan intervals and results (+ indicating tracer uptake and N no tracer uptake), and timing and duration of examestane, everolimus, and sorafenib. (B) The bone scan performed at 57 years, demonstrating metastatic bone disease with increased tracer uptake in the sternum, right 5th and 6th ribs, left scapula, right anterior superior iliac spine and intertrochanteric region of proximal left femur.Positron emission topography (PET) surveillance at 53 years showed breast fluorodeoxyglucose (FDG)-uptake. Biopsy showed an ER+/PR−/HER2– breast carcinoma. A pT2pN1a grade 2 infiltrating ductal carcinoma was excised and followed by adjuvant radiotherapy and tamoxifen.
At 56 years, a 2-FDG-avid lytic acromial lesion was identified. Biopsy showed poorly differentiated thyroid cancer. Acromial irradiation was undertaken. After 15 months, axillary lymph node tumor burden was evident; biopsy confirmed metastatic breast cancer. Contemporaneous unstimulated thyroglobulin levels were 4,260 μg/L (RR <55 μg/L). Examestane and everolimus were commenced, the latter being trialed at the time for progressive hormone-refractory breast cancer and shown to have anti-thyroid cancer activity.4 Thyroglobulin declined (Figure 1A). Everolimus-associated complications necessitated dose reduction and cessation. Unstimulated thyroglobulin levels rose (Figure 1A). Progressive widespread metastatic disease was evident (Figure 1B). Retrospective molecular analysis of the primary PTC identified a BRAF V600E mutation. Immunohistochemistry on the primary PTC showed normal MSH2 and MSH6 gene expression.
As some of the metastatic burden was thyroid cancer related, 5.6 GBq I131 was administered; the unstimulated thyroglobulin level after 4 weeks was 221 μg/L (Figure 1A). The tyrosine kinase inhibitor sorafenib was then commenced. The patient died after 3 months from disseminated leptomeningeal metastases.
Discussion
This study presents a fascinating case of a young woman with Lynch syndrome-related colonic cancer with synchronous papillary thyroid cancer and metachronous breast cancer with progression to metastatic disease related to both thyroid and breast cancers.
Thyroid cancer generally has an excellent long-term prognosis. Mutations in BRAF, RET/PTC, NTRK1, or RAS have been reported in up to 70% of differentiated thyroid cancers.5 BRAF mutation confers a more aggressive clinical course, as evident in this case.
This is the third case of thyroid cancer described in Lynch syndrome. The first case was a female from a Lynch syndrome pedigree with a germline MSH2 mutation who developed an undifferentiated thyroid cancer as the index cancer. Immunohistochemistry showed that MSH2 expression was absent.1 The second case was a female from a Lynch syndrome pedigree with anaplastic thyroid cancer as the index cancer. Immunochemistry also showed that MSH2 expression was absent.2 Genetic analysis confirmed a germline mutation in case 1 and in family members of case 2.
The present case demonstrated loss of MSH2 and MSH6 staining in her colonic cancer only. Lynch syndrome was confirmed by DNA sequencing of MSH2 that revealed a c.998G>A missense change in exon 6. In this case, the PTC displayed normal mismatch repair protein expression and showed a BRAF mutation, making it unlikely for mismatch repair defects due to the cancer genetics of Lynch syndrome. MSH2 promoter hypermethylation has been shown to occur in colorectal cancers related to Lynch syndrome, with somatic inactivation of the remaining wild-type allele.6
Second, primary tumors, synchronous or metachronous, have been reported in literature.7 It is hypothesized that the synchronous occurrence of two tumors may be a result of a mutagenic trigger that causes independent genetic events7 and that metachronous tumors may be due to persisting effects of genetic and behavioral risk factors.7 It is possible the thyroid gland may be activated by prior hormone-secreting cancers with overstimulation in a reactive autoimmune environment, as hypothesized by Sisti et al.8 Furthermore, a history of thyroid cancer is associated with the increased risk of developing breast cancer, and several studies have evaluated the thyroid hormone and proposed mechanisms for activation of the associated oncogene.9 The most likely explanation for synchronous cancers in the present case seems to be a sporadic thyroid cancer on a background of genetic cancer predisposition.
It should be noted that normal immunohistochemistry does not completely exclude the mismatch repair defects that characterize Lynch syndrome. False negatives may occur due to the following reasons: First, the effectiveness of immunohistochemistry in revealing germline mismatch repair mutations depends on the number of antibodies used. Mismatch repair proteins form heterodimers to maintain function: MSH2 with MSH6; MHL2 with PMS2. Immunohistochemistry with MLH1/MSH2 antibodies has a sensitivity of 85% in detecting a mismatch repair mutation; sensitivity is increased to 92% using PMS2 and MSH6 antibodies.10 Furthermore, if immunohistochemistry is performed with MLH1/MSH2 antibodies only, some MLH1 and MSH2 abnormalities may be missed, as some pathogenic mutations retain protein expression. Second, not all pathogenic mutations result in loss of protein by immunohistochemistry. Immunohistochemistry is effective in screening for mutations due to a truncated or degraded protein, but performs poorly in differentiating abnormal protein due to a missense mutation from normal protein. Greater than 30% of MLH1 mutations are missense mutations that may not be detected by immunohistochemistry. Third, there may be a misleadingly false normal staining pattern due to a second hit that inactivates the wild-type allele resulting in non-functional protein but allowing normal antibody binding.10 Immunohistochemistry with a four antibody panel and microsatellite instability (MSI) testing have similar sensitivity for detecting mismatch repair mutations; however, each may miss cases detectable by the other method. Therefore, if high clinical suspicion synchronous cancers may be linked to the same pathogenic process, normal immunohistochemistry result should be verified with MSI testing.
These few cases raise the importance of considering individual cancer genetics in familial cancer syndromes, particularly when other cancers do not fit the described syndrome. Careful documentation of other malignancies in patients with thyroid cancer and their families would assist in better understanding any potential association of thyroid cancer to known familial cancer syndromes. Appropriate genetic testing of the cancers will clarify whether common genetics explain synchronous or subsequent malignancies in individuals and their families and add to greater understanding of the genetic mechanisms that may explain apparently unrelated cancers.
Disclosure
The authors report no conflicts of interest in this work.
References
Stulp RP, Herkert JC, Karrenbeld A, Mol B, Vos YJ, Sijmons RH. Thyroid cancer in patient with germline MSH2 mutation. Case report and review of Lynch syndrome expanding tumor spectrum. Hered Cancer Clin Pract. 2008;6:15–21. | ||
Broaddus RR, Lynch PM, Lu KH, Luthra R, Michelson SJ. Unusual tumors associated with the hereditary nonpolyposis colorectal cancer syndrome. Modern Pathol. 2004;17:981–989. | ||
Lipton LR, Johnson V, Cummings C, et al. Refining the Amsterdam Criteria and Bethesda Guidelines: testing algorithms for the prediction of mismatch repair mutation status in the familial cancer clinic. J Clin Oncol. 2004;22:4934–4943. | ||
Lim SM, Chang H, Yoon MJ, et al. A multicentre phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann Oncol. 2013;24:3089–3094. | ||
Fagin JA, Mitsiades N. Molecular pathology of thyroid cancer: diagnostic and clinical implications. Best Pract Res Clin Endocrinol Metab. 2008;22:955–969. | ||
Nagasaka T, Rhees J, Kloor M, Gebert J, Naomoto Y, Boland R, Goel A. Somatic hypermethylation of MSH2 is a frequent event in Lynch syndrome colorectal cancers. Cancer Res. 2010;70:3098–3108. | ||
Sisti A, Tassinari J, Nisi G, et al. Synchronous and metachronous malignancies after malignant struma ovarii in the SEER database. In Vivo. 2016;30:713–716. | ||
Sisti G, Di Tommaso M. Commentary on “Study of prevalence of autoimmune thyroid disease in women with breast cancer”. Endocr Pract. 2016;22:114–116. | ||
Liu L, Shi J, Mao F, Wei J, Fu D, Zhang J. Synchronous primary cancers of the thyroid and breast: a case report and review of the literature. Oncol Lett. 2015;9:351–354. | ||
Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. J Mol Diagn. 2008;10:293–300. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.