Back to Journals » Hepatic Medicine: Evidence and Research » Volume 16
THSR Mediated MiR374b Targeting C/EBP β/FOXO1 to Accelerate Thyroid Stimulating Hormone-Induced Hepatic Steatosis
Authors Li J, Ge Y, Chai Y, Kou C, Sun TT, Liu J, Zhang H
Received 6 June 2024
Accepted for publication 6 November 2024
Published 18 November 2024 Volume 2024:16 Pages 91—104
DOI https://doi.org/10.2147/HMER.S481687
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Juyi Li,1,2 Yang Ge,3 Yuwei Chai,1 Chunjia Kou,1 Tian Tian Sun,4 Jia Liu,3,5– 7 Haiqing Zhang1,3,5– 7
1Department of Endocrinology, Shandong Provincial Hospital, Shandong University; Key Laboratory of Endocrine Glucose & Lipids Metabolism and Brain Aging, Ministry of Education, Jinan, Shandong, 250021, People’s Republic of China; 2Department of Endocrinology, Geriatrics Center, The First Affiliated Hospital of Anhui University of Chinese Medicine, Hefei, Anhui, 230031, People’s Republic of China; 3Department of Endocrinology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, 250021, People’s Republic of China; 4Department of Infectious Diseases, Jinan People’s Hospital, Jinan, Shandong, 271100, People’s Republic of China; 5Shandong Key Laboratory of Endocrinology and Lipid Metabolism, Jinan, Shandong, 250021, People’s Republic of China; 6Shandong Institute of Endocrine and Metabolic Diseases, Jinan, Shandong, 250021, People’s Republic of China; 7Shandong Engineering Laboratory of Prevention and Control for Endocrine and Metabolic Diseases, Jinan, Shandong, 250021, People’s Republic of China
Correspondence: Jia Liu; Haiqing Zhang, Department of Endocrinology, Shandong Provincial Hospital, 324 Jingwu Road, Jinan, Shandong Province, 250021, People’s Republic of China, Tel +86 18653155865 ; Tel +86 15168888303, Email [email protected]; [email protected]
Purpose: Thyroid-stimulating hormone (TSH) has been identified as an independent risk factor for non-alcoholic fatty liver disease (NAFLD), TSH binds to the TSH receptor (TSHR) to exert its function. However, the underlying mechanisms by which TSHR influences NAFLD development remain unclear. This study investigates the role of miR374b in NAFLD progression.
Methods: Firstly, a rat model of non-alcoholic fatty liver was constructed and divided into a normal group and a model group. The liver tissue pathology and fat accumulation were detected by Oil Red O staining and hematoxylin-eosin staining. Western blot and Real time PCR were used to detect for the impact of TSHR/miR-374b/C/EBP β/ FoxO1 pathway in the NAFLD model, and the expression of relevant inflammatory factors in each group was detected by ELISA assay. A NAFLD cell model was constructed using HepG2 cells, TSHR overexpression and interference, combined with miR-374b inhibitor and mimics, were transfected simultaneously to demonstrate TSHR/miR-374b/C/EBP β/ The mechanism of FoxO1 adipogenesis in vitro.
Results: TSHR stimulates miR374b secretion in human liver cancer cells (HepG2) and promotes lipid accumulation in the liver. Deficiency of miR374b in HepG2 cells attenuated NAFLD progression. Mechanistically, TSH increases miR374b expression, which then suppresses the transcription of its target genes, CCAAT/enhancer binding protein-b (C/EBP β) and Forkhead Box Protein O1 (FOXO1). This suppression influences the expression of downstream lipid metabolism proteins, including PPARγ, SREBP2, and SREBP1c. Additionally, miR374b directly targets the 3′UTR of C/EBP β and FOXO1, establishing a negative feedback loop in lipid metabolism.
Conclusion: These findings suggest that TSHR-induced upregulation of miR374b accelerates NAFLD progression by modulating lipid metabolism pathways through C/EBP β and FOXO1.
Keywords: NAFLD, miR374b, TSHR
Graphical Abstract:

Introduction
In China, non-alcoholic fatty liver disease (NAFLD) has become the most common chronic liver disease, surpassing both viral hepatitis and alcoholic liver disease.1 Current estimates suggest a prevalence of NAFLD in the general population ranging from 15% to 33%, with rates as high as 23.3% to 59.6% among obese children and adolescents.2
Subclinical hypothyroidism (SCH), characterized by raised thyroid stimulating hormone (TSH) levels, is a prevalent endocrine metabolic disorder and an independent risk factor for NAFLD.3 SCH affects approximately 4–10% of adults, increasing to 20% among women over 60 years old, and is becoming more widespread.4 This condition is a significant contributor to secondary dyslipidemia and hypertension.5,6 TSHR is one of the genes most closely related to GD, and currently K1-70 has been used as a targeted TSHR inhibitor for the treatment of GD eye disease.7 Activation of TSHR in cultured orbital fibroblasts promotes enhanced HA and fat production.8 Despite the established association between increasing serum TSHR levels and the incidence of SCH,9 the mechanisms by which TSHR regulates NAFLD progression remain poorly understood.
MicroRNAs (miRNAs) are known to bind specifically to the 3′ untranslated region (UTR) of target gene mRNAs, forming a complex that suppresses gene translation. Increasing evidence indicates that miRNAs play a critical role in regulating the expression of genes associated with NAFLD, with about 100 miRNAs showing significant expression differences in NAFLD patients.10,11 These miRNAs impact various aspects of NAFLD, including glucose and lipid metabolism,12 oxidative stress,13 cell apoptosis,14 inflammation, endoplasmic reticulum stress,15 and insulin resistance.16 For example, inhibition of miR-33 in the liver significantly slows NAFLD progression,17 while upregulation of miR-122 correlates directly with the severity of liver fibrosis in NAFLD, making it a potential biomarker and therapeutic target.18 MiR-34a influences liver lipid metabolism and steatosis by targeting the transcription factor SIRT1.19 Upregulated levels of miR-378 in the liver may enhance transcription activating factor LXR α, inhibiting key enzymes and effectors in liver lipid metabolism pathways, thereby exacerbating NAFLD.20 Furthermore, extracellular vesicles containing miR-223 from neutrophils and other cells can decrease the expression of the pro-fibrotic gene TAZ in liver cells.21 However, the specific targets of miR374b in SCH remain to be clarified.
Recent research indicates that miR374b targets CCAAT/enhancer-binding protein-b (C/EBP β) and Recombinant Forkhead Box Protein O1 (FOXO1) in adipose tissue, reducing their transcriptional stability and translational output.22 This interaction significantly suppresses the PPAR γ signaling pathway, playing a vital role in lipid metabolism and insulin resistance. It is hypothesized that miR374b could be a critical miRNA in the development and progression of fatty liver disease.
Material and Methods
Animal Model
C57BL/6J mice, purchased from Hangzhou Ziyuan Laboratory Animal Technology Co., Ltd. (Hangzhou, China), underwent a 7-day acclimatization period. After that, 20 male mice were randomized into two groups: 10 mice in the control group and 10 in the model group. The model group was fed a high-fat diet for eight weeks to induce an NAFLD model. High fat feed raw material ratio, maintaining basic feed, 0.435; Lard, 0.175; Sucrose, 0.12; Whole milk powder, 0.1; Casein, 0.13; Experimental animal premix, 0.02; Calcium hydrogen phosphate, 0.02. (XTHF60, Jiangsu Xietong Pharmaceutical Bio-engineering Co., Ltd, China). The Shandong Provincial Hospital Animal Ethics Committee approved animal experiments (Ethical approval No.2021–497) in this study. Experimental animals follow the Guidelines for Ethical Review of Experimental Animal Welfare. The standard number of this guide is GB/T 35892–2018.
Cell Culture
HepG2 cells were purchased from Wuhan Pricella Biotechnology Co., Ltd. (Wuhan, China) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; BIOIND Israel) in a 5% CO2 atmosphere at 37°C.
Oil Red O Staining
HepG2 cells were washed thrice with PBS and fixed with 4% formaldehyde at room temperature for 30 minutes, followed by three additional PBS washes of 5 minutes each. Cells were then stained using an Oil Red O stain kit (Sigma Aldrich, Shanghai, China) for 1 hour after diluting the stock solution with water (3:2 ratio). After staining, cells were washed thrice with PBS (5 minutes per wash) and imaged under a microscope.
TSH/TC/TG Assay
According to the manufacturer’s instructions, measure Thyroid Stimulating Hormone using a mouse/human thyroid stimulating hormone detection kit (Solarbio, China).Total cholesterol and triglyceride levels were measured using the total cholesterol/triglyceride assay kit (Solarbio, China) according to the manufacturer’s instructions.
Dual-Luciferase Reporter Assay
FOXO1/C/EBPβ 3′ UTR-WT-psicheck2 and FOXO1/C/EBPβ 3′ UTR-Mut-psicheck2 plasmids (Limibio Co., Ltd.,China) were transfected into HepG2 cells alongside hsa-miR-374a-5p mimics or miR-NC in 24-well plates. After 48 hours, cells were lysed, and luciferase activities were measured. Firefly luciferase activity was assessed using 20µL of cracking mixture followed by 100 µL of Luc reaction solution. Renilla luciferase activity was then measured using 100µL of termination solution. The ratio of Renilla to firefly luciferase activities was calculated to determine relative luciferase activity.
Quantitative Real-Time PCR
Total RNA was extracted from HepG2 cells and tissues using TRIzol Reagent (Biosharp, China). RNA concentration was measured with a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Reverse transcription was performed using Hifair III first strand cDNA Synthesis SuperMix (Yeasen Biotechnology Co., Ltd., China). qPCR was conducted using Hieff UNICON® Universal Blue qPCR SYBR Master Mix (Yeasen Biotechnology Co. Ltd., China) on a LightCycler480 Real-Time fluorescence quantitative PCR instrument (Roche, Switzerland). The gene primers used in the experiment are shown in Table 1.The thermal cycling conditions were: 1 cycle at 95°C for 2 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 20 seconds, then 95°C for 15 seconds and 60°C for 60 seconds. β-actin was used as an internal control, and each sample was tested in triplicate.
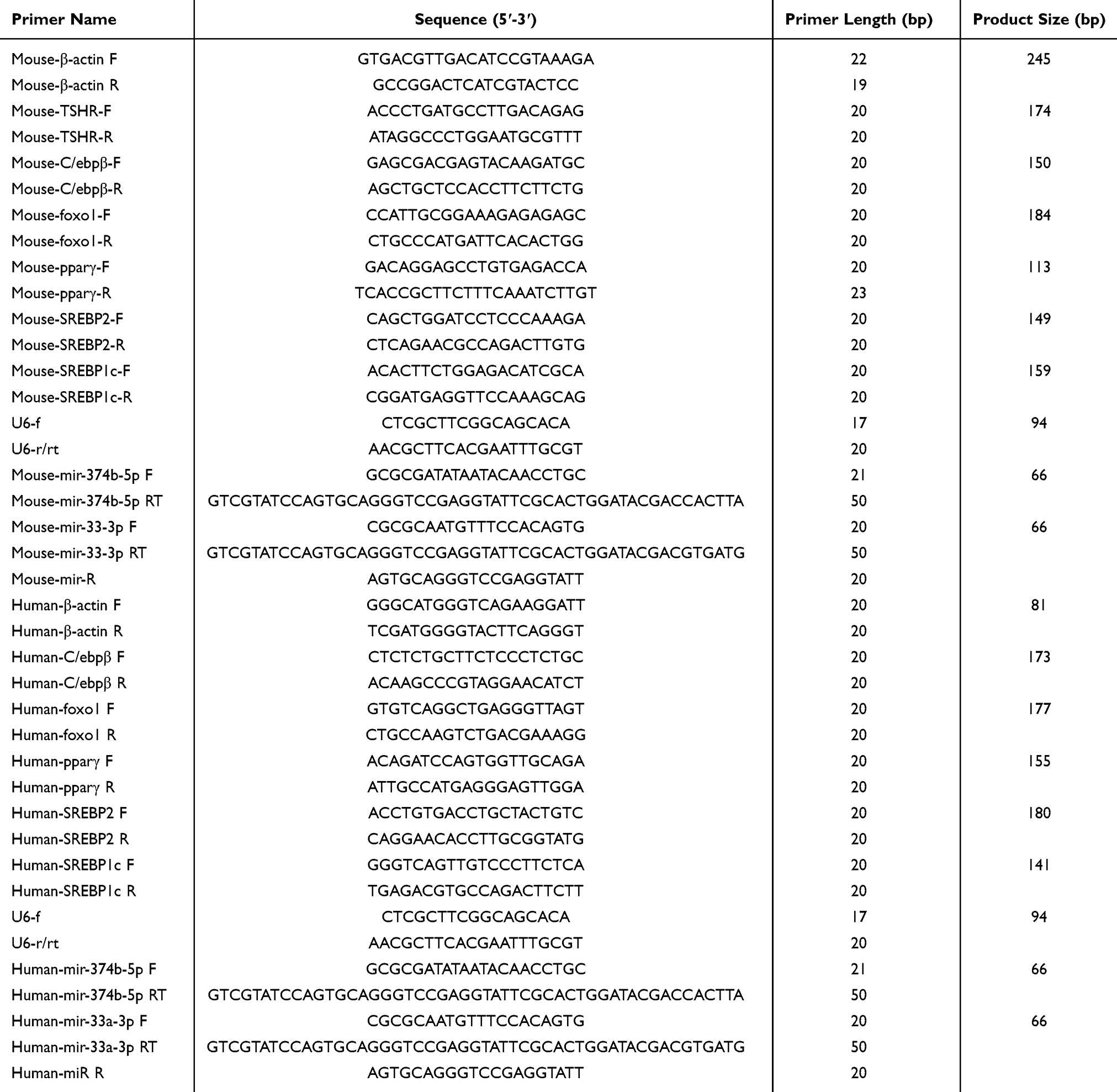 |
Table 1 Specific Primer Sequences for qRT-PCR |
Western Blot
Protein extraction was conducted using a total protein extraction kit (Solarbio, China), and protein concentrations were determined with a BCA protein assay kit (Solarbio, China). For electrophoresis, 20 µg of protein per sample was separated on a 10% SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% skim milk for 1 hour at room temperature, incubated overnight at 4°C with primary antibodies, followed by a 1.5-hour room temperature incubation with corresponding secondary antibodies. Immunoblots were scanned in grayscale, using β-actin as an internal reference protein.Information on the use of the antibodies is provided in Table 2.
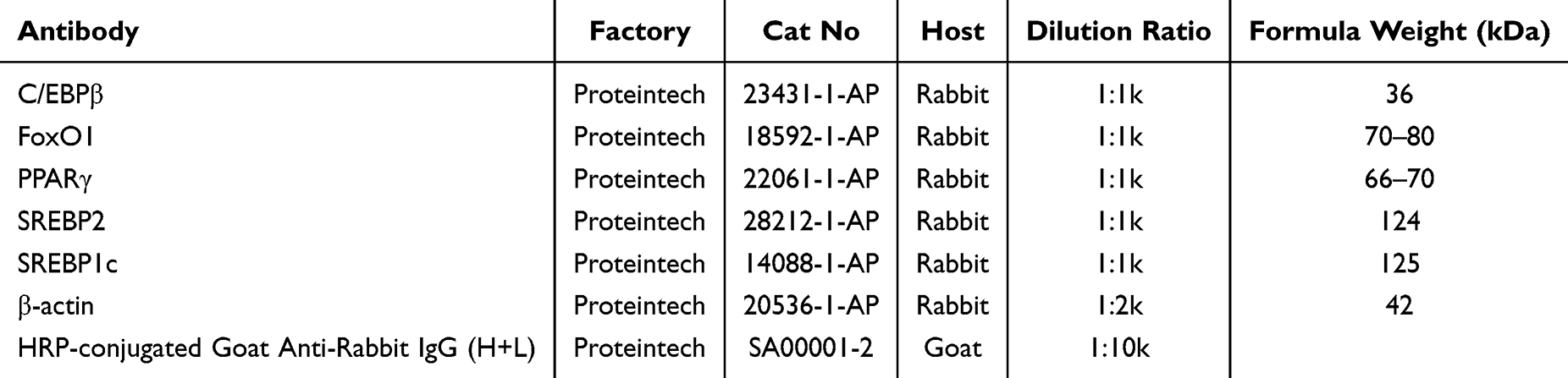 |
Table 2 Information on the Use of the Antibodies |
Statistical Analysis
Statistical analyses were performed using SPSS version 21.0 (IBM, USA). Data were analyzed using the t-test and expressed as mean ± standard deviation (SD). Differences were considered statistically significant at P<0.05 and marked accordingly in the results.
Results
Effect of TSH on Adipose Degeneration in HepG2 Cells
In order to understand the role of TSH in the occurrence and development of NAFLD, ELISA was used to detect the concentration of TSH in the serum of NAFLD mouse models. The results showed that TSH was significantly overexpressed in this group (Figure 1A). In vitro, HepG2 cells were treated with varying concentrations of TSH, and lipid accumulation was assessed using oil red staining. As depicted in Figure 1B, lipid levels were significantly higher in the TSH-treated group than in controls, increasing progressively with rising TSH concentrations. At the same time, levels of total cholesterol (TC) and triglycerides (TG) were elevated in cells treated with TSH (Figure 1C). While TC levels at a 1μM concentration of TSH did not differ significantly from controls, TG levels significantly increased, particularly at a 4μM concentration. Importantly, miR374b expression was found to correlate positively with TSH concentration (Figure 1D), suggesting that TSH induces miR374b expression in HepG2 cells.
 |
Figure 1 Effect of TSH on adipose degeneration in HepG2 cells. (A) Comparison of serum TSH levels between NAFLD mouse model and control group (n=3, * P < 0.05). (B) Oil red O staining of HepG2 cells treated with TSH (0 μM, 1 μM, 2 μM) (n=3), Scale bar = 50 μm. (C) Changes of serum TC and TG levels in each group treated with TSH (0 μM, 1 μM, 2 μM) (n=3, one-way ANOVA and Tukey’s multiple comparison test; ns, nonsignificant; * P < 0.05 versus 0 μM). (D) The expression of miR-374b-5p in each group compared with control (n=3, *** P < 0.001 versus 0 μM, ****P < 0.0001 versus 0 μM). |
TSH-Induced Hepatic Steatosis in HepG2 Cells Dependent on TSHR
To determine if TSH expression depends on TSHR, three siRNAs targeting the TSHR gene in HepG2 cells were designed. Effective knockdown was achieved with sequences TSHR-Si-385 (CGGAAUACCAGGAACUUAATT) and TSHR-Si-656 (GGACAAAGCUGGAUGUGUGUGUGUGUGUTT), which reduced TSHR expression by 70% and 50%, respectively (Figure 2A). TSHR-Si-385 was used for further experiments to knock down TSHR, and recombinant TSH protein was employed for intervention treatments. The Oil Red O staining results, as shown in Figure 2B, showed no significant lipid accumulation in the OE-NC and si NC groups compared to the Control group. The OE-TSHR group showed a significant increase in lipids compared to the OE-NC group, while the si TSHR group showed a decrease in lipid content compared to the si NC group; After TSH treatment of cells, the lipid content of OE-NC+TSH and si-NC+TSH significantly increased; The OE-TSHR+TSH group had the highest lipid content, significantly higher than the lipid content of cells treated with a single treatment; The lipid content of the si TSHR+TSH group was significantly increased compared to the si TSHR group, indicating that TSH promotes liver lipid accumulation in a TSHR dependent manner. Additionally, levels of TC and TG correlated with TSHR expression, increasing in the TSHR overexpression group (Figure 2C). Real-time quantitative PCR analysis demonstrated that both TSH and TSHR overexpression could upregulate miR-374b expression. Notably, even after TSHR knockdown, TSH overexpression still elevated miR-374b levels (Figure 2D), indicating that miR-374b expression is primarily regulated by TSH, with TSHR exerting a regulatory effect.
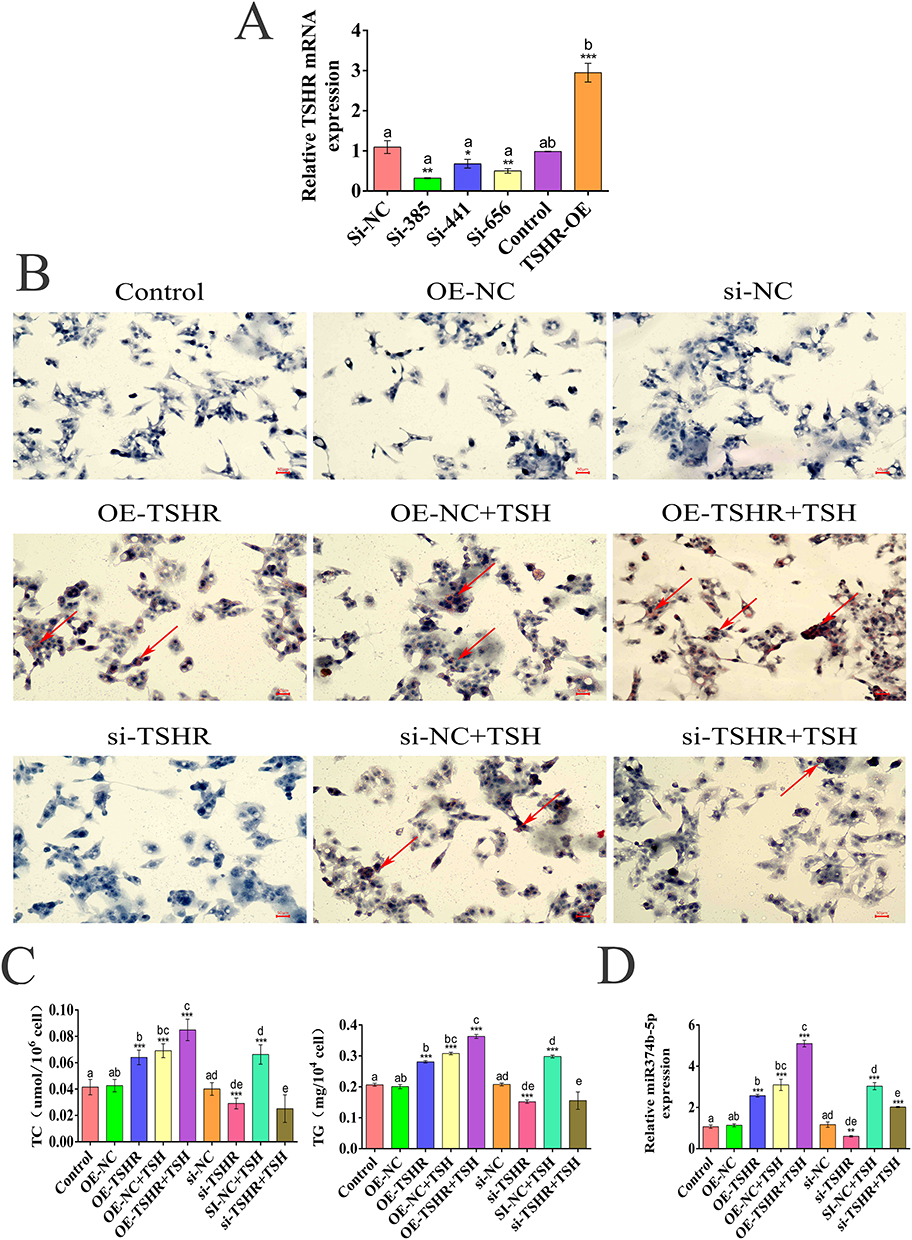 |
Figure 2 TSH-induced hepatic steatosis in HepG2 cells dependent on TSHR. (A) Comparison of TSHR mRNA expression levels between the interference and overexpression TSHR groups and the control group (n=3,* P < 0.05; ** P < 0.01; *** P < 0.01). (B) Oil red O staining of HepG2 cells treated with TSHR combined with TSH (2 μM) (n=3,Red arrow lipid accumulation site,Scale bar = 50 μm. (C) Changes of serum TC and TG levels in each group treated with TSHR-siRNA and TSH (2 μM) (n=3;*** P < 0.001). (D) The expression of miR-374b-5p in each group checked by Realtime-PCR (n=3; **P < 0.01; ***P < 0.001). |
Effect of miR374b on the Regulation of the Transcription Factor C/EBP β and FOXO1 Expression
To delineate the gene regulatory function of miR374b in NAFLD, gene manipulation was conducted on miR374b, with both mimics and inhibitors synthesized and transfected into HepG2 cells. C/EBP β, FoxO1, PPAR γ, SREBP2, and SREBP1c expression levels were assessed using real-time qPCR. Results showed a decrease in the expression of C/EBP β, PPAR γ, and FoxO1 following miR374b upregulation, while miR-33, SREBP2, and SREBP1c expression significantly increased (Figure 3A). In contrast, miR374b knockdown elevated C/EBP β, PPAR γ, and FoxO1 expression and reduced miR-33, SREBP2, and SREBP1c. Western blot analysis corroborated the qPCR findings, confirming the regulatory effects at both protein and mRNA levels (Figure 3B), thus indicating miR374b’s role in modulating the expression of these key metabolic regulators.
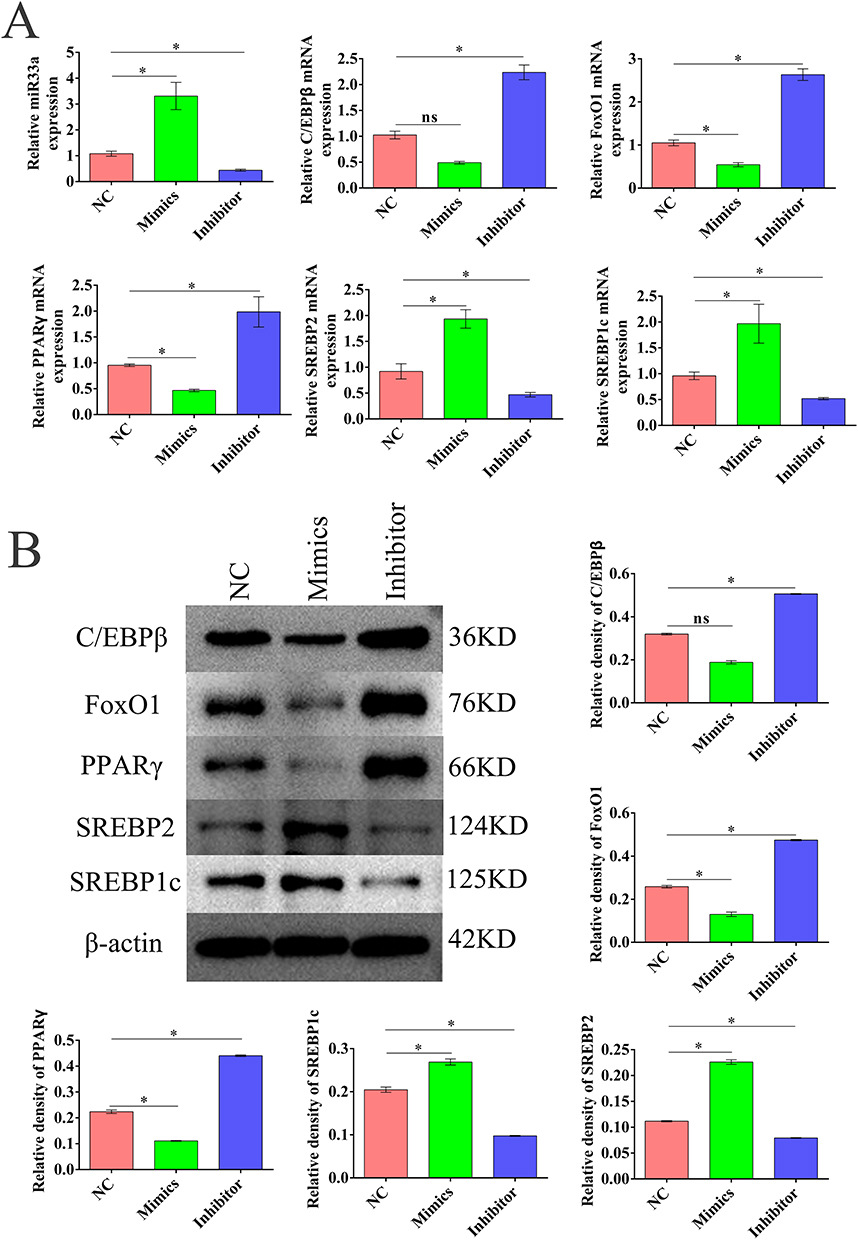 |
Figure 3 Effect of miR374b on the regulation of the transcription factor C/EBP β and FOXO1 expression. (A) The mRNA expression of C/EBP β, FoxO1, PPAR γ, SREBP2, SREBP1c, and miR33a in each group compared with the normal control (n=3,* P < 0.05). (B) Western blot showing the expression of TSHR, C/EBPβ, FOXO1, PPARγ, SREBP2, and SREBP1c in each group compared with normal control (n=3,* P < 0.05). |
MiR374b Regulates NAFLD by Targeting C/EBPβ and FOXO1
To elucidate the role of miR374b in NAFLD, we regulated miR374b expression in a TSH-induced HepG2 cell model by constructing miR374b mimics and inhibitors. Functional validation via oil red staining demonstrated that overexpression of miR374b enhanced lipid accumulation while its knockdown decreased lipid levels (Figure 4A). This effect was exacerbated with TSH intervention, though fat accumulation was significantly reduced in the miR374b knockdown group compared to controls, suggesting that miR374b operates independently of TSH in influencing NAFLD. Biochemical assays confirmed that miR374b overexpression increased TC and TG, which were reduced upon miR374b knockdown (Figure 4B). C/EBP β and FOXO1 were identified as target genes of miR374b, both containing binding sites within their 3′UTR regions, confirmed via dual-luciferase reporter assays (Figure 4C and D). Real-time qPCR analysis revealed that upregulation of miR374b decreased the expression of C/EBP β, FOXO1, and PPAR γ, while miR-33, SREBP2, and SREBP1c expressions were elevated. Conversely, miR374b knockdown led to increased expression of C/EBP β and slight changes in FOXO1 and PPAR γ (Figure 4E). Western blot analysis corroborated these findings (Figure 4F), underscoring miR374b’s role in targeting and suppressing C/EBP β and FOXO1 expression, impacting lipid metabolism in NAFLD.
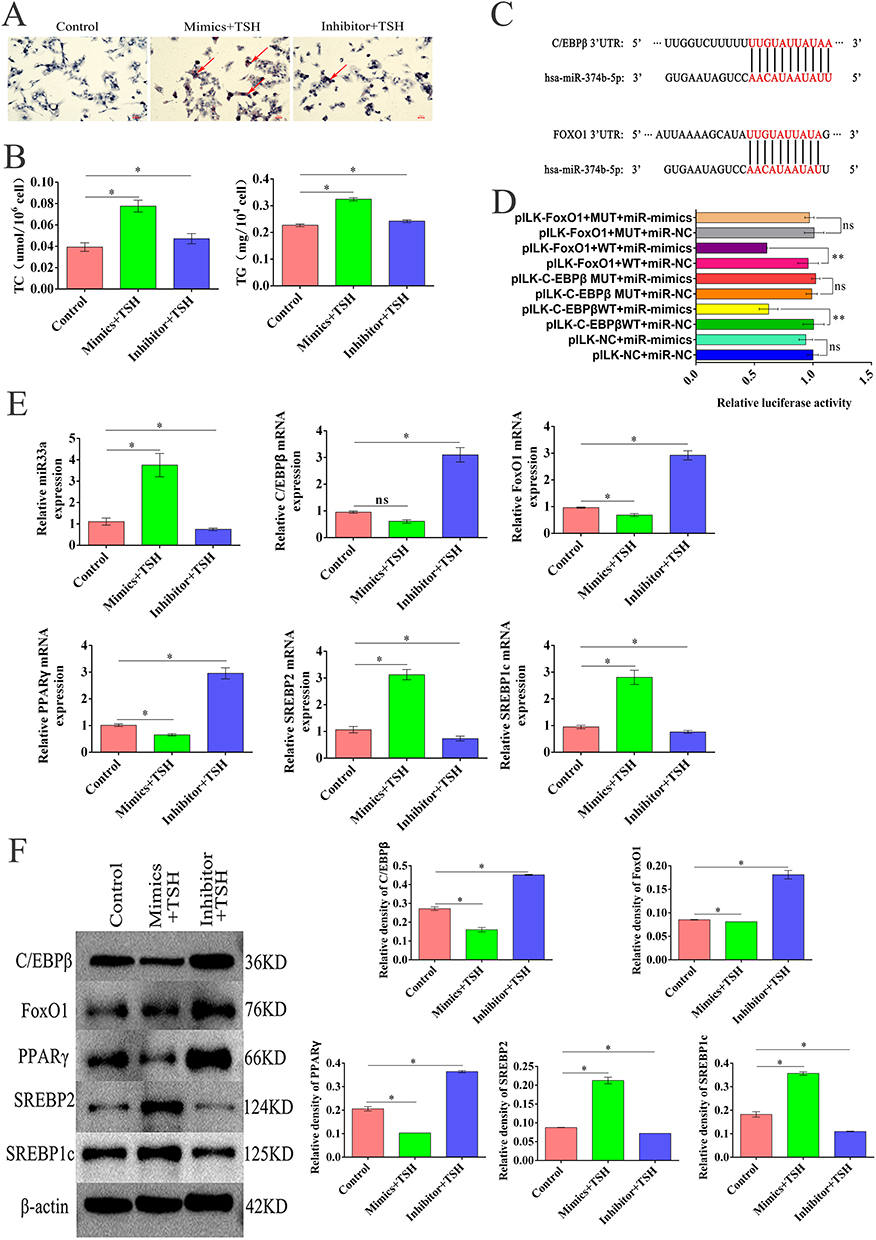 |
Figure 4 MiR374b regulates NAFLD by targeting C/EBP β and FOXO 1. (A) Oil red O staining of HepG2 cells transfected with miR374b mimics and inhibitor (n=3), Scale bar = 50 μm. (B) Changes of serum TC and TG levels in each group treated with miR374b mimics and inhibitors combined with TSH (2 μM) (n=3,* P < 0.05 versus control). (C) Predicted binding sites with C/EBP β and FOXO1. (D) The dual luciferase reporter gene assay confirmed the binding of miR374b to C/EBP β and FOXO1 genes; Error bars indicate the mean ± SD; ** P < 0.01, (E) The mRNA expression of C/EBP β, FoxO1, PPAR γ, SREBP2, SREBP1c, and miR33a in each group compared with normal control (n=3,* P < 0.05). (F) Western blot showed the expression of TSHR, C/EBPβ, FOXO1, PPARγ, SREBP2, and SREBP1c in each group compared with normal control (n=3, * P < 0.05). |
Effect of miR374b on the Regulation of NAFLD in vivo
An NAFLD mouse model was replicated, and fatty lesions in the liver tissues of the model mice were identified via HE pathological staining (Figure 5A). Oil red O staining indicated a significant accumulation of adipocytes in the liver tissues of NAFLD mice (Figure 5B). Quantitative analyses of TC and TG in serum and liver tissues showed elevated levels in the model group compared to the control group. TSH levels were significantly increased in the model group, as illustrated in Figure 5C. qPCR analysis detected upregulated expression of TSHR and miR-374b in the model group (Figure 5D), indicating that TSHR promotes the expression of miR-374b. As a target gene of miR-374b, C/EBP β expression was reduced, leading to downregulation of the transcription factor FOXO1 and its downstream target genes. Western blot analyses corroborated the qPCR findings (Figure 5E). In line with prior studies, an increase in miR-33-3p expression was observed, which may be associated with the regulation of SREBP1. Collectively, these findings suggest that TSH upregulates miR374b expression via TSHR in liver cells, which then influences C/EBP β and modulates critical molecules in lipid metabolism, thereby promoting lipid accumulation in liver cells and inducing hepatic steatosis.
 |
Figure 5 Effect of miR374b on the regulation of NAFLD in vivo. (A) Liver HE staining in NAFLD mouse model and control group (n=3,Red arrow lipid vacuoles, Scale bar = 50 μm). (B) Oil red O staining in NAFLD mouse model and control group (n=3,Red arrow lipid accumulation site, Scale bar = 50 μm). (C) Changes of serum and liver tissues TC and TG levels in NAFLD mouse model compared with negative control (n=6, *P < 0.05). (D) The mRNA expression of C/EBP β, FoxO1, PPAR γ, SREBP2, SREBP1c, and miR33a in each group compared with normal control (n=3, *P < 0.05). (E) Western blot showed the expression of TSHR, C/EBPβ, FOXO1, PPARγ, SREBP2, and SREBP1c in each group compared with normal control (n=3, * P < 0.05). |
Discussion
TSH, a key protein in SCH lesions, is involved in inducing NAFLD through its interaction with the TSH receptor (TSHR).23 TSH regulates thyroid activity and comprehensively activates thyroid metabolism,24 including the synthesis of thyroid-specific proteins such as thyroglobulin (Tg), thyroid peroxidase (TPO), and the sodium-iodide symporter (NIS). Thyroid hormone receptors (TRs), TRα and TRβ, bind with Thyroid hormone (3,5,3′-triiodothyronine, T3) to regulate gene expression associated with TSH.25 Beyond its thyroid-specific roles, TSH influences various hepatic metabolic activities, promoting lipid output, oxidation, and de novo adipogenesis.26 The TSH/TSHR signaling pathway is crucial for the morphogenesis and differentiation of embryonic glands in mice,27 affecting lipid metabolism through modulation of AMPK and PPARα signal transduction.28,29 However, the relationship between TSH and miRNAs has not yet been elucidated.
This study elucidates the novel role of miR374b in the pathogenesis of NAFLD mediated by TSH. The results demonstrate that miR374b significantly influences lipid metabolism and regulates FOXO1 and its downstream target genes in HepG2 cells. MiR374b is involved in the complex regulation of NAFLD lipid metabolism. Notably, miR374b activity is significantly elevated in a NAFLD mouse model; TSH promotes its expression via the TSHR. Moreover, miR374b directly targets C/EBP β and its downstream target genes, playing a critical role in regulating lipid metabolism. Overexpression of miR374b leads to increased fat accumulation, while its downregulation has the opposite effect, highlighting its essential role in maintaining intracellular lipid balance.
The discovery of miR374b’s targeted regulation of C/EBP β, a key factor in triglyceride metabolism and NAFLD pathogenesis,30,31 is particularly significant. C/EBP β as a transcription factor that controls energy metabolism32 can improve lipid accumulation and metabolism in zebrafish larvae,33 while FOXO1 is an important regulatory factor for energy balance in dormant embryos and is crucial for lipid metabolism in animals.34 Increased miR374b levels lead to elevated C/EBP β expression, whereas knocking down miR374b reduces its expression. This study also confirms that miR374b regulates the expression of FOXO1 and its downstream genes. These findings suggest that TSH contributes to NAFLD by influencing miR374b’s targeting of C/EBP β and its downstream gene regulation, presenting a novel therapeutic target for NAFLD management by modulating miR374b activity.
Additionally, this research uncovered that miR374b-induced lipid accumulation correlates negatively with lipid metabolism, thereby exacerbating NAFLD. It also examined the role of the TSHR in NAFLD pathogenesis. Findings demonstrated that TSHR is involved in TSH-induced NAFLD, where TSH binding to TSHR enhances miR374b transcription, subsequently activating C/EBP β to induce lipid production in adipose tissue, highlighting the therapeutic potential of targeting TSHR to mitigate NAFLD onset.The function of TSHR in NAFLD.TSH binds to TSHR, which activates the expression of miR374b. Resulting in a decrease in the expression levels of miR374b target genes C/EBP β and FOXO 1, thereby regulating the expression of downstream lipid metabolism pathway proteins PPAR γ, SREBP2, and SREBP1c.
Despite these insights, the study has limitations that warrant further investigation. Primarily, it relies on the HepG2 cell line, which might not fully mimic the complex cellular dynamics of human lipid metabolism. Future studies should incorporate primary human liver cells to validate these findings. Additionally, while the findings offer valuable in vitro insights, they require validation in clinical settings to confirm their relevance to NAFLD pathogenesis. Moreover, although it was established that miR374b targets C/EBP β and FOXO1, impacting lipid metabolism in NAFLD, the study did not thoroughly explore the detailed molecular mechanisms and downstream lipid metabolism processes. Further investigations are necessary to elucidate the specific signaling pathways and molecular interactions involved.
Conclusion
These findings suggest that TSHR-induced upregulation of miR374b accelerates NAFLD progression by modulating lipid metabolism pathways through C/EBP β and FOXO1, positioning miR374b as a potential therapeutic target for NAFLD.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by grants from the National Natural Science Foundation of China (81770785 and 82370793) and the Scientific Research Program of Higher Education Institutions in Anhui Province (2023AH040103).
Disclosure
Juyi Li was contributed as first author. The authors report no conflicts of interest in this work.
References
1. Ding L, Oligschlaeger Y, Shiri-Sverdlov R, Houben T. Nonalcoholic Fatty Liver Disease. Handb Exp Pharmacol. 2022;270:233–269. doi:10.1007/164_2020_352
2. Duell PB, Welty FK, Miller M, et al. Nonalcoholic Fatty Liver Disease and Cardiovascular Risk: a Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42(6):e168–e185. doi:10.1161/ATV.0000000000000153
3. Fan H, Liu Z, Zhang X, et al. Thyroid Stimulating Hormone Levels Are Associated With Genetically Predicted Nonalcoholic Fatty Liver Disease. J Clin Endocrinol Metab. 2022;107(9):2522–2529. doi:10.1210/clinem/dgac393
4. Gharib H, Tuttle RM, Baskin HJ, et al. Subclinical thyroid dysfunction: a joint statement on management from the American Association of Clinical Endocrinologists, the American Thyroid Association, and the Endocrine Society. Endocr Pract. 2004;10(6):497–501. doi:10.4158/EP.10.6.497
5. Gu Y, Wu X, Zhang Q, et al. High-Normal Thyroid Function Predicts Incident Nonalcoholic Fatty Liver Disease Among Middle-Aged and Older Euthyroid Subjects. J Gerontol a Biol Sci Med Sci. 2022;77(1):197–203. doi:10.1093/gerona/glab037
6. Chen YL, Tian S, Wu J, et al. Impact of Thyroid Function on the Prevalence and Mortality of Metabolic Dysfunction-Associated Fatty Liver Disease. J Clin Endocrinol Metab. 2023;108(7):e434–e443. doi:10.1210/clinem/dgad016
7. Cui X, Wang F, Liu C. A review of TSHR- and IGF-1R-related pathogenesis and treatment of Graves’ orbitopathy. Front Immunol. 2023;14:1062045. doi:10.3389/fimmu.2023.1062045
8. Kumar S, Coenen M, Iyer S, Bahn RS. Forkhead Transcription Factor FOXO1 Is Regulated by Both a Stimulatory Thyrotropin Receptor Antibody and Insulin-Like Growth Factor-1 in Orbital Fibroblasts from Patients with Graves’ Ophthalmopathy. Thyroid. 2015;25(10):1145–1150. doi:10.1089/thy.2015.0254
9. Vidal-Cevallos P, Murua-Beltran Gall S, Uribe M, Chavez-Tapia NC. Understanding the Relationship between Nonalcoholic Fatty Liver Disease and Thyroid Disease. Int J Mol Sci. 2023;24(19):14605. doi:10.3390/ijms241914605
10. Hochreuter MY, Dall M, Treebak JT, Barres R. MicroRNAs in non-alcoholic fatty liver disease: progress and perspectives. Mol Metab. 2022;65:101581. doi:10.1016/j.molmet.2022.101581
11. Mahmoudi A, Butler AE, Jamialahmadi T, Sahebkar A. The role of exosomal miRNA in nonalcoholic fatty liver disease. J Cell Physiol. 2022;237(4):2078–2094. doi:10.1002/jcp.30699
12. Latorre J, Moreno-Navarrete JM, Mercader JM, et al. Decreased lipid metabolism but increased FA biosynthesis are coupled with changes in liver microRNAs in obese subjects with NAFLD. Int J Obes Lond. 2017;41(4):620–630. doi:10.1038/ijo.2017.21
13. Yu Y, Tian T, Tan S, et al. MicroRNA-665-3p exacerbates nonalcoholic fatty liver disease in mice. Bioengineered. 2022;13(2):2927–2942. doi:10.1080/21655979.2021.2017698
14. Qiao JT, Cui C, Qing L, et al. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism. 2018;81:13–24. doi:10.1016/j.metabol.2017.09.010
15. Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69(4):927–947. doi:10.1016/j.jhep.2018.06.008
16. Azzimato V, Chen P, Barreby E, et al. Hepatic miR-144 Drives Fumarase Activity Preventing NRF2 Activation During Obesity. Gastroenterology. 2021;161(6):1982–1997e11. doi:10.1053/j.gastro.2021.08.030
17. Ghareghani P, Shanaki M, Ahmadi S, et al. Aerobic endurance training improves nonalcoholic fatty liver disease (NAFLD) features via miR-33 dependent autophagy induction in high fat diet fed mice. Obes Res Clin Pract. 2018;12(Suppl 2):80–89. doi:10.1016/j.orcp.2017.01.004
18. Long JK, Dai W, Zheng YW, Zhao SP. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol Med. 2019;25(1):26. doi:10.1186/s10020-019-0085-2
19. Wang Z, Zhu Y, Xia L, Li J, Song M, Yang C. Exercise-Induced ADAR2 Protects against Nonalcoholic Fatty Liver Disease through miR-34a. Nutrients. 2022;15(1):121. doi:10.3390/nu15010121
20. Zhang T, Duan J, Zhang L, et al. LXRalpha Promotes Hepatosteatosis in Part Through Activation of MicroRNA-378 Transcription and Inhibition of Ppargc1beta Expression. Hepatology. 2019;69(4):1488–1503. doi:10.1002/hep.30301
21. He Y, Rodrigues RM, Wang X, et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J Clin Invest. 2021;131(3). doi:10.1172/JCI141513
22. Pan S, Zheng Y, Zhao R, Yang X. miRNA-374 regulates dexamethasone-induced differentiation of primary cultures of porcine adipocytes. Horm Metab Res. 2013;45(7):518–525. doi:10.1055/s-0033-1334896
23. Vieira IH, Rodrigues D, Paiva I. The Mysterious Universe of the TSH Receptor. Front Endocrinol. 2022;13:944715. doi:10.3389/fendo.2022.944715
24. Zhou W, Brumpton B, Kabil O, et al. GWAS of thyroid stimulating hormone highlights pleiotropic effects and inverse association with thyroid cancer. Nat Commun. 2020;11(1):3981. doi:10.1038/s41467-020-17718-z
25. Refetoff S. Resistance to thyroid hormone and its molecular basis. Acta Paediatr Jpn. 1994;36(1):1–15. doi:10.1111/j.1442-200x.1994.tb03121.x
26. Walczak K, Sieminska L. Obesity and Thyroid Axis. Int J Environ Res Public Health. 2021;18(18):9434. doi:10.3390/ijerph18189434
27. Postiglione MP, Parlato R, Rodriguez-Mallon A, et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci U S A. 2002;99(24):15462–15467. doi:10.1073/pnas.242328999
28. Yan F, Wang Q, Lu M, et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity. J Hepatol. 2014;61(6):1358–1364. doi:10.1016/j.jhep.2014.06.037
29. Liu YY, Heymann RS, Moatamed F, Schultz JJ, Sobel D, Brent GA. A mutant thyroid hormone receptor alpha antagonizes peroxisome proliferator-activated receptor alpha signaling in vivo and impairs fatty acid oxidation. Endocrinology. 2007;148(3):1206–1217. doi:10.1210/en.2006-0836
30. Lee J, Jung E, Lee J, et al. Isorhamnetin represses adipogenesis in 3T3-L1 cells. Obesity. 2009;17(2):226–232. doi:10.1038/oby.2008.472
31. Kim MS, Baek JH, Lee J, Sivaraman A, Lee K, Chun KH. Deubiquitinase USP1 enhances CCAAT/enhancer-binding protein beta (C/EBPbeta) stability and accelerates adipogenesis and lipid accumulation. Cell Death Dis. 2023;14(11):776. doi:10.1038/s41419-023-06317-7
32. Janardhan HP. The HIF-1 alpha-C/EBP alpha axis. Sci Signal. 2008;1(43):jc2. doi:10.1126/scisignal.143jc2
33. Ajay G, Gokul S, Namasivayam SKR, et al. Serine Threonine-Protein Kinase-Derived IW13 Improves Lipid Metabolism via C/EBP-alpha/SREBP1/FAS Signaling Pathways in HFD-Induced Zebrafish In Vivo Larval Model. Appl Biochem Biotechnol. 2023;195(8):4851–4863. doi:10.1007/s12010-023-04480-3
34. van der Weijden VA, Stotzel M, Iyer DP, et al. FOXO1-mediated lipid metabolism maintains mammalian embryos in dormancy. Nat Cell Biol. 2024;26(2):181–193. doi:10.1038/s41556-023-01325-3
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.