Back to Journals » Medical Devices: Evidence and Research » Volume 12
Thoracic Endovascular Aortic Repair Adverse Events Reported In The Food And Drug Administration Manufacturer And User Facility Device Experience Database
Authors Mansukhani NA, Haleem MS, Eskandari MK ![]()
Received 21 February 2019
Accepted for publication 13 June 2019
Published 14 November 2019 Volume 2019:12 Pages 461—467
DOI https://doi.org/10.2147/MDER.S206269
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Neel A Mansukhani, Meraaj S Haleem, Mark K Eskandari
Division of Vascular Surgery, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA
Correspondence: Mark K Eskandari
Division of Vascular Surgery, 676 N St. Clair St., Suite 650, Chicago, IL 60611, USA
Tel +1 312-926-7775
Fax +1 312-695-4955
Email [email protected]
Purpose: The purpose of this study is to identify adverse events (device- and patient-related) associated with thoracic aortic stent graft systems and their timing post-procedure.
Materials and methods: The Food and Drug Administration’s Manufacturer and User Facility Device Experience (FDA-MAUDE) voluntary database was searched for Thoracic Aortic Endovascular Repair (TEVAR) devices reported over the course of 1 year (January 1, 2014 to December 31, 2014). The data abstracted included the indication for treatment, device used, and adverse events.
Results: During 2014, there were 334 original submissions to the FDA-MAUDE database describing 371 adverse events regarding TEVAR devices that met inclusion criteria for this study. All submissions were from manufacturers, and none were from physicians. The most common pathologies treated were thoracic aortic aneurysm (67.6%) and type B aortic dissection (25.1%). The most frequently reported intraoperative, early postoperative (<30 days), and late postoperative (>30 days) events overall were technical device failure, neurologic complications (stroke, paraplegia), and endoleak, respectively. Of note, there were descriptions of retained deployment materials, late graft infections, and aorto-visceral fistula formation up to 3 years postoperatively.
Conclusion: The MAUDE database is a valuable repository for complications and device failures that are not otherwise in the published literature and submitted by manufacturers relating to this relatively new technology.
Keywords: TEVAR, Food and Drug Administration, MAUDE, complications
Introduction
Thoracic endovascular aortic repair (TEVAR) is a paradigm-shifting technology and has rapidly gained popularity compared to open surgical repair for treatment of both thoracic aortic aneurysm and thoracic aortic dissection.1–3 TEVAR has numerous benefits over traditional open surgical repair of the thoracic aorta including decreased operative time, decreased length of stay, fewer postoperative complications, and improved postoperative morbidity and mortality.1,4,5 Despite widespread adoption of TEVAR and evidence of mid- to long-term safety, long-term complications are not well defined.6,7
The purposes of the FDA-MAUDE are to monitor device performance and detect potential device-related safety issues to contribute to benefit-risk assessments of these products.8 Device users and facilities are mandated by the FDA to submit medical device reports (MDRs) when they become aware of information that suggests that one of their devices may have caused or contributed to a death or serious injury, or if a device has malfunctioned and the malfunction would likely be the cause of or contribute to death or serious injury. This database has been used previously to report on rare medical device-related adverse events.9,10 Submissions of MDRs of suspected device-related deaths, serious injuries, or malfunctions are catalogued by the FDA-MAUDE database.
The majority of currently published outcomes and safety data are derived from industry-sponsored clinical trials,11–16 and single-center reviews.17 Despite overall improved outcomes, TEVAR carries the risk of unique device- and patient-related adverse events compared to traditional open surgery. The purpose of this study is to utilize voluntary reports to the United States Food and Drug Administration’s (FDA) online Manufacturer and User Facility Device Experience (MAUDE) database in order to ascertain the long- and short-term risks and adverse events associated with currently available thoracic aortic stent graft systems.8
Materials And Methods
This is a retrospective review of the Food and Drug Administration’s Manufacturer and User Facility Device Experience (FDA-MAUDE) voluntary database. The FDA-MAUDE database was searched for all currently approved and commercially available TEVAR devices during the study period. Search terms included “TEVAR,” “Bolton RELAY®,” “Cook Zenith TX2®,” “Gore TAG®,” and “Medtronic Valiant®.” The FDA-MAUDE database is publicly available online and contains de-identified data; therefore, this study was exempt from institutional review board approval.
Included in the study were all submissions to the FDA-MAUDE database regarding any currently approved and commercially available TEVAR device from January 1, 2014 to December 31, 2014. Excluded were data submitted to the FDA-MAUDE database that was obtained from published journal articles. Thus, only original case submissions to the FDA-MAUDE database from either manufacturers or users were included in this study. Also excluded were reports in which no device malfunction or adverse event experienced by the patient occurred and reports that did not contain sufficient data about the adverse event. Data abstracted included manufacturer, device name, event date, report date, report number as a unique identifier for each submitted report, indication for treatment, and description and timing of adverse events. Device- and patient-related adverse events were extrapolated from narrative descriptions within individual reports, and recorded as the following complications for analysis: migration, endoleak, infections, technical device failures, aorto-esophageal and aorto-bronchial fistula, dissection/PAU/IMH (PAU/IMH: penetrating aortic ulcer/intramural hematoma) including retrograde dissections, and neurologic complications such as stroke and paraplegia. When one submission to the MAUDE database included multiple adverse events it was noted and recorded; however, the adverse events were logged separately into the data collection database so our data cannot delineate which events occurred most commonly together. Device-related events are those that occurred in either a structural or process-related component of the device, for example, failed deployment or retained components of the device during deployment. Patient-related events are those that occurred either independently or in conjunction with the use of the device, which is illustrated by paraplegia events that may occur due to length of aortic coverage rather than the type of TEVAR device.
Data were collected and stored using Microsoft Excel (Microsoft Corp, Redmond, WA), and analyzed using STATA (Version 14, STATA Corp, College Station, TX). Chi-Squared and Fischer’s Exact tests were used for categorical variables as appropriate. Statistical significance was assumed at P<0.05.
Results
Data Collection
Identified were 523 submissions to the FDA-MAUDE between January 1 and December 31, 2014, for the four approved and commercially available TEVAR devices at the time (Bolton RELAY®, Cook TX2®, Gore TAG®, and Medtronic Valiant®). All submissions were from manufacturers, and none were from physicians. Of those, excluded were 79 reports derived from published case reports in the literature. Also excluded were 108 reports of a user experience without an adverse event towards the patient; these were reported as interesting cases, success stories with off-label device use, individual user’s experience including indications for treatment, device used, and procedure performed, and reviews of a particular device. Also excluded were an additional 2 reports that contained inadequate data pertaining to the adverse event. The remaining 334 original reports describing a total of 371 device- and patient-related adverse events were analyzed (Figure 1).
 |
Figure 1 Study design and data collection. |
Report Characteristics
Only original reports to the FDA-MAUDE were analyzed. None of these reports was submitted by physicians and, in fact, all submissions were reported by manufacturers. There were 16 reports (4.8%) of adverse events using the Bolton RELAY®, 11 reports (3.3%) of adverse events using the Cook TX2®, 123 (36.8%) reports of adverse events using the Gore TAG®, and 184 reports (55.1%) of adverse events using the Medtronic Valiant®. Reports were not limited to users from the United States and reports did not specify whether devices were used for on- or off-label indications. Common pathologies treated were aneurysms (67.6%) and dissections (25.1%).
Adverse Events
Summary Of Reported Events
There were 371 reports of adverse events following TEVAR. The most common adverse events were endoleaks (n=169, 45.55%) of all types. Eleven endoleaks were not specified. There were 114 type 1 endoleaks (21 type 1 not specified, 55 type 1a, 3 combined 1a and 1b, 35 type 1b); 19 type 2, 21 type 3, and 4 type 4 endoleaks. The second most common adverse events were procedure-related dissection, penetrating aortic ulcer, and intramural hematoma (n=67, 18.06%). There were also 54 (14.56%) neurological events. Of these 54 neurological events, 15 (27.78%) were postoperative stroke, 37 (68.52%) were postoperative paraplegia, and 2 (3.70%) were unspecified. There were 18 reported endograft infections (4.85%), 45 reported technical device failures (12.13%), and 11 reported aorto-esophageal and aorto-bronchial fistulae (2.96%). Reported events were categorized by device type (Bolton RELAY®, Cook Zenith TX2®, Gore TAG®, and Medtronic Valiant®) in Table 1. The most common complications reported for the Bolton RELAY® were dissection/IMH/PAU, endoleak, and neurologic complications. The most common complications for the Cook Zenith TX2® were endoleaks, technical device failures, and dissections; interestingly, of the 3 reported dissections associated with the Cook Zenith TX2®, all were retrograde dissections resulting from endograft deployment. The most common complications for the Gore TAG® were endoleaks and neurologic complications. The most common complications for the Medtronic Valiant® were endoleaks, dissections, and technical device failures.
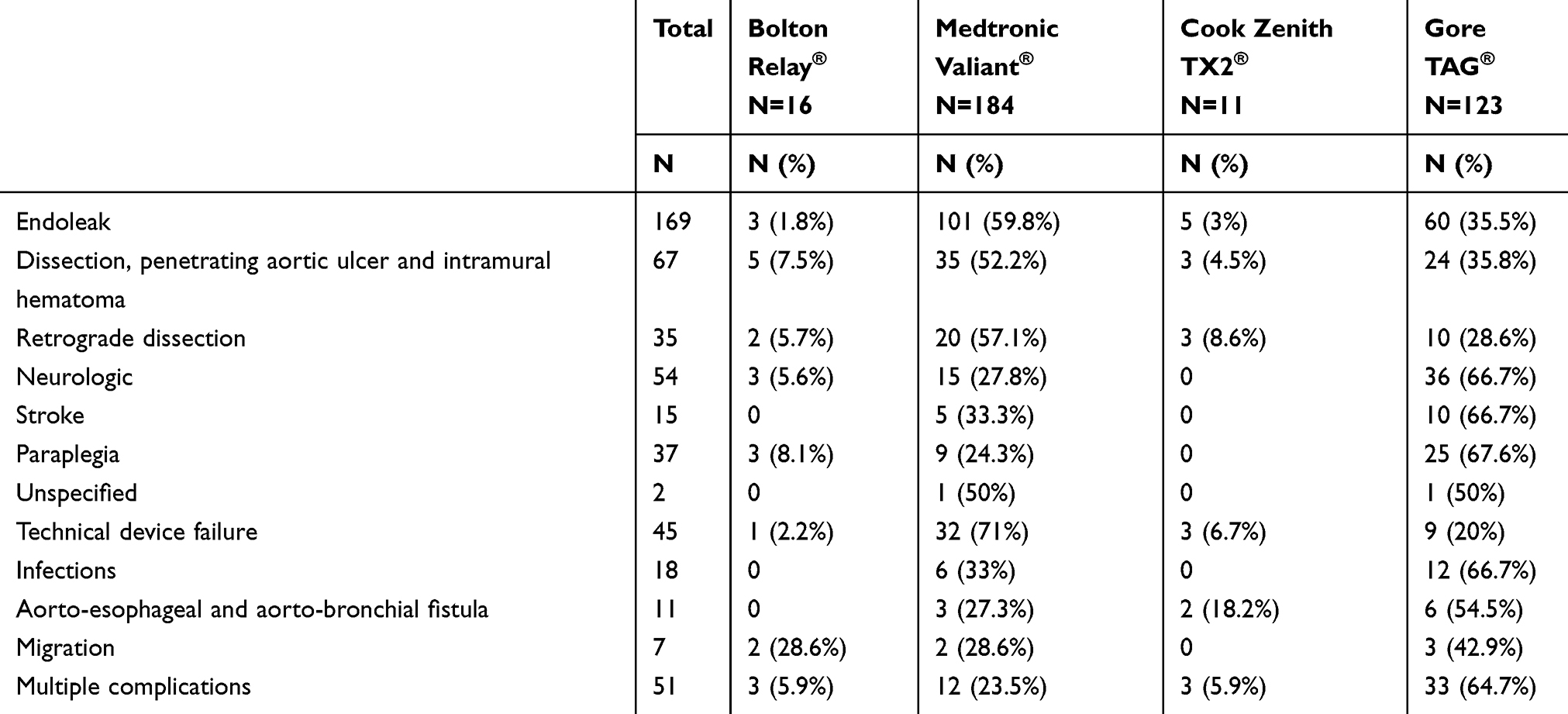 |
Table 1 Reported Device-Related And Patient-Related Events By Device Type |
Multiple complications were most often reported in association with use of the Gore TAG® (n=33/51, 64.7%) compared to the Medtronic Valiant® (n=12/51, 23.5%), Bolton Relay® (n=3/51, 5.9%), and Cook Zenith TX2® (n=3/51, 5.9%). Endoleak was most often reported in association with use of the Medtronic Valiant® (n=101/169, 59.8%) compared to the Gore TAG® (n=60/169, 35.5%), Bolton Relay® (n=3/169, 1.8%), and Cook Zenith TX2® (n=5/169, 3.0%). Technical device failures were most often reported in association with use of the Medtronic Valiant® (n=32/45, 71.0%) compared to the Gore TAG® (n=9/45, 20.0%), Bolton Relay® (n=1/45, 2.2%), and Cook Zenith TX2® (n=3/45, 6.7%). Aorto-esophageal and aorto-bronchial fistula were most often reported in association with use of the Gore TAG® (n=6/11, 54.5%) compared to the Medtronic Valiant® (n=3/11, 27.3%), Bolton Relay® (n=0), and Cook Zenith TX2® (n=2/11, 18.2%). Overall neurological complications (P=0.003), including paraplegia (P=0.008), were reported more often in association with use of the Gore TAG® compared to the Medtronic Valiant®, Bolton Relay®, and Cook Zenith TX2®. There were no significant differences in rates of infection, dissection, penetrating aortic ulcer, intramural hematoma, or stroke between device types.
Reported Events By Timing In Relation To Index Procedure
Adverse events that occurred during the procedure included 45 delivery system failures; of these, 25 resulted in the use of an additional device(s). There were 4 incidences of retained deployment materials. These retained devices were the olive tip or nose cone from various delivery systems. In response to retained deployment material, the reported operator performed a laparoscopic procedure to retrieve the retained material in 1 patient, retrieved the retained material with endovascular techniques in 1 patient, and was not able to retrieve the retained material in 2 patients. There was 1 intraoperative aorto-bronchial fistula precipitated by the deployment of a TEVAR device within a thoracic aneurysm. The thoracic aortic aneurysm ruptured while advancing the TEVAR graft into what was likely an impending aorto-bronchial fistula; this patient expired during the procedure. Finally, there were 6 reports of intraoperative iatrogenic retrograde dissection. Three of the retrograde dissections were attributed to the proximal bare metal components on the Medtronic Valiant® device, and 3 were attributed to other elements of the procedure. Of these 6 iatrogenic retrograde dissections, 1 was managed with deployment of another stent graft, and five reports did not provide details regarding further management.
Early (within 30 days) post-procedure events included endoleak (n=31), infections (n=3), aorto-esophageal and aorto-bronchial fistulae (n=2), aortic dissection, penetrating aortic ulcer, and intramural hematoma (n=13) of which 11 were retrograde dissections, stroke (n=13), and paraplegia (n=24). Late post-procedure events (>30 days) included endoleaks (n=51), infections (n=8), aorto-esophageal/aorto-bronchial fistula (n=6), dissection/PAU/IMH (n=22) of which 12 were retrograde dissections, and 1 stroke.
Device migration occurred more often during the procedure (n=4/7, 57.1%) compared to in the early or late postoperative procedure period (P=0.02). Infections were reported most frequently during the late (>30 days) postoperative period (n=8/18, 44.4%). The latest endograft infection was reported 26 months after the index procedure. Neurological events including stroke and paraplegia were reported most frequently in the early postoperative period (n=13/15, 86.7% of all postoperative strokes, n=24/37, 64.9% of all postoperative paraplegia events). There were no significant differences in the timing of aorto-esophageal and aorto-bronchial fistulae, or aortic dissection/IMH/PAU in relation to the index procedure. However, notably, the latest aorto-esophageal fistula was reported 3 years after the index procedure, and the latest aorto-bronchial fistula was reported 2 years after the index procedure. These data are summarized in detail in Table 2.
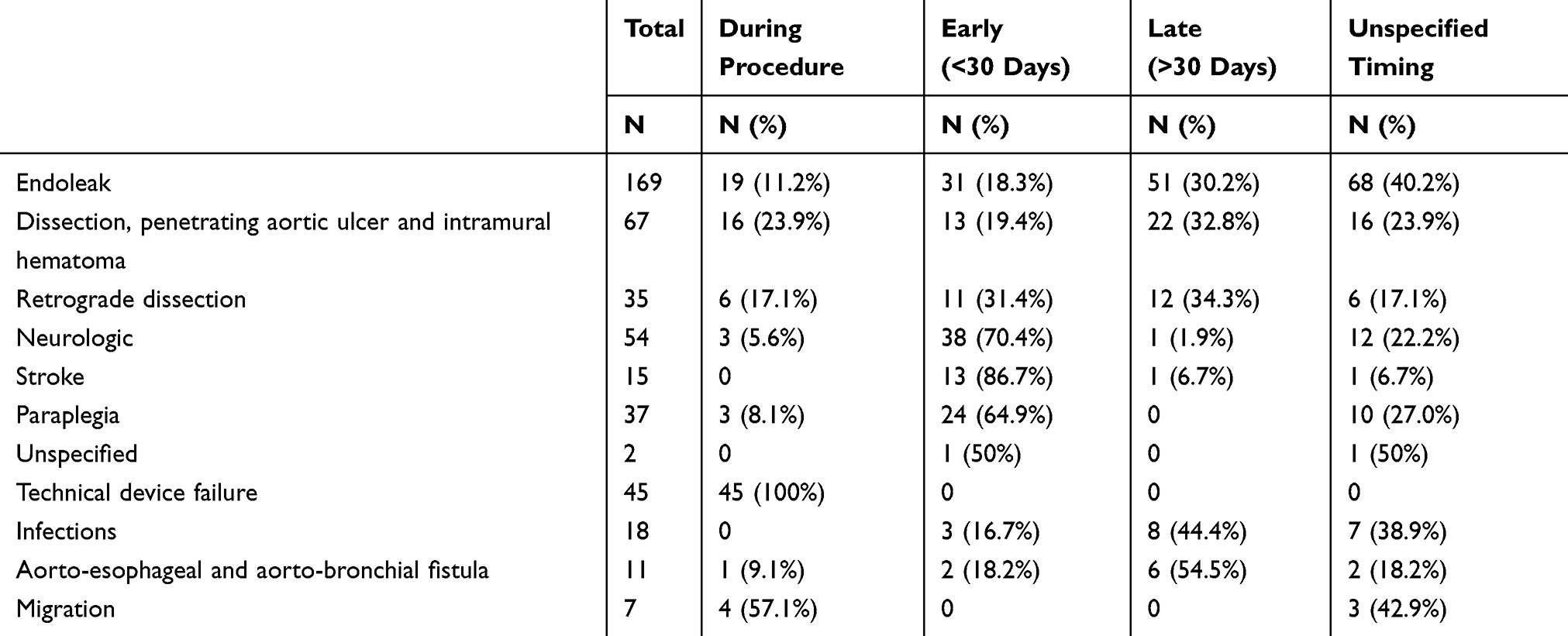 |
Table 2 Reported Device-Related And Patient-Related Events By Timing In Relation To Index Procedure |
Discussion
This study is unique in that it examines publicly reported device- and patient-related adverse events associated with TEVAR for thoracic aortic diseases utilizing the FDA-MAUDE database. This database has been used in the past to report on several devices and treatment modalities including inferior vena cava filters and coronary artery stents,9,10 but has not been previously utilized to examine TEVAR adverse events. Current studies examining the long-term complications of TEVAR are limited to case reports, industry-sponsored trials, single-center retrospective reviews, and individual device registries. The value of the MAUDE database is that it promptly publishes data, and it can capture data missed by trials limited by time or registries limited by location or device. Furthermore, any practitioner or manufacturer can report any device failure or adverse event at any time. The information contained in the MAUDE database is available to the public, thus providing a valuable feedback mechanism whereby many users can learn from the experience of a few users. Importantly, not all device- and patient-related adverse events were captured by short-term industry-sponsored trials or device registries and the MAUDE database can unveil these uncommon events. Although anyone may submit reports to this database, during our study period reports were only submitted by manufacturers/device representatives. This highlights an obvious shortcoming of the MAUDE database in that submissions of events are voluntary. The voluntary nature of the database and the paucity of physician-submitted reports lead us to believe that events are likely underreported. Furthermore, the lack of physician reporting subjects these data to bias in that data reporting comes from non-clinically trained device representatives. This bias must be acknowledged and taken into consideration when interpreting the findings of this study. Physicians can, and should, report adverse events to the FDA directly. Despite these shortcomings, data from the MAUDE database are publicly available, valuable in its own right, and may be used by anyone without fee.
Due to likely underreporting of certain devices, and voluntary good-faith reporting, statistical analyses were not performed to compare events by device type (Table 1) to avoid reporting analyses subject to reporting bias. As such, reported device usage and complications are likely not an accurate reflection of the true market shares of these devices. These data are, therefore, presented as descriptive information with the assumption of underreporting of events and reporting bias.
Technical device failures may occur with or without harm to the patient, but should not be unexpected. Operators should be prepared for events such as retained foreign bodies (such as components of the delivery system, not intended for permanent implantation) and device malfunction requiring off-label use of devices or additional device usage. Fistulization and retrograde dissection may require emergent sternotomy with total aortic arch repair using cardiopulmonary bypass and circulatory arrest. The results of this study are consistent with the report by Canaud et al in that there is no clear evidence to support the concept that proximal bare-metal stents on thoracic endografts are associated with retrograde type A dissections.18 Open surgical repair may be required after thoracic stent-graft infection as well, if the patient can survive this magnitude of repair.
Late (>30 days post-procedure) adverse events are uncommon, but possible. Late events can be catastrophic, as in the case of aorto-esophageal or aorto-bronchial fistula and stent-graft infection. The latest reported stent graft infection was 26 months post-procedure, and the latest reported fistulization between the stent-grafted aorta and another hollow viscus was an aorto-esophageal fistula reported 3 years after the index procedure. Observations from these data suggest that adverse events following TEVAR are possible for several years following the index procedure. Awareness of the possible late complications and time elapsed before such complications arise, the possibility of retained foreign body from the delivery system, and catastrophic injury such as fistulization between the stent-grafted aorta and bronchus/esophagus, or retrograde dissection is crucial for any practitioner performing TEVAR or managing patients following TEVAR. Such awareness is also critical for physicians referring their patients for TEVAR.
There exist multiple endovascular devices to treat these conditions, and each has its own strengths and weaknesses. All thoracic aortic endografts are associated with some complications, and these complications can occur up to years following the index procedure. Although devices continue to evolve, the community of practitioners can learn valuable lessons from the failures of older device types. The severity of adverse events following TEVAR range from asymptomatic to catastrophic and life threatening.
Our study has several limitations. First, this is an observational study using a public database that relies on voluntary reporting. Therefore, we cannot confirm the accuracy of information contained in reports. Second, since reporting is voluntary, device- and patient-related adverse events following TEVAR are likely underreported, so true frequencies of such events in the general population cannot be extrapolated and there is an inherent reporting bias; together these make analyses and conclusions from analyses limited in their generalizability. This database remains valuable since the possibility of device- and patient-related adverse events at certain time points following index procedure is ascertained. Third, Gore and Medtronic obtained FDA approval for the treatment of aortic dissection, which likely contributed to the higher event rates in these devices. There was also likely selection bias where certain grafts were used for certain types of anatomy, which cannot be controlled for in this non-randomized descriptive series. Although not included in this study’s design, future studies can potentially draw correlation between multiple adverse events that happen simultaneously. Finally, these reports are of isolated events without follow-up information and do not distinguish between on- and off-label use of devices. Despite these limitations, this study is valuable because it captures a large number of uncommon adverse events following TEVAR that were not captured by industry-sponsored trials. Knowledge of which adverse events are possible following TEVAR is important for practicing clinicians to have in order to anticipate adverse events and to counsel and care for patients who experience such events.
Conclusion
In conclusion, the MAUDE database is a useful and valuable repository of complications and device failures reported by device manufacturers and not otherwise found in the published literature that can help educate the surgical community. A valuable addition to the MAUDE reporting standards would be a mandatory distinction between on- and off-label use of devices that are reported. TEVAR has lower morbidity and mortality than open repair; however, there do exist uncommon and unique device- and patient-related adverse events associated with TEVAR, which can occur months to years following the procedure. Since the timing and frequency of these long-term adverse events are relatively unknown, it is important to continue reporting these events in order to continually improve this technology and patient safety following TEVAR. Further research of pooled data characterizing uncommon TEVAR device failures and rescue techniques is warranted.
Acknowledgment
The authors would like to acknowledge Ms Janet Goldstein for her administrative and editorial assistance.
Disclosure
MKE has received honoraria from Prairie Education and Research Cooperative (Bard) for service on the LEVANT 2 clinical events committee; from Silk Road Medical, Inc. for service on the Roadster clinical events committee; and from W. L. Gore & Associates as a TEVAR course director. The authors report no other conflicts of interest in this work.
References
1. Desai ND, Burtch K, Moser W, et al. Long-term comparison of thoracic endovascular aortic repair (TEVAR) to open surgery for the treatment of thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2012;144(3):
2. Jones DW, Goodney PP, Nolan BW, et al. National trends in utilization, mortality, and survival after repair of type B aortic dissection in the Medicare population. J Vasc Surg. 2014;60(1):11–9, 9 e1. doi:10.1016/j.jvs.2013.12.047
3. Ultee KH, Soden PA, Chien V, et al. National trends in utilization and outcome of thoracic endovascular aortic repair for traumatic thoracic aortic injuries. J Vasc Surg. 2016;63(5):1232–9 e1. doi:10.1016/j.jvs.2015.11.034
4. Gopaldas RR, Huh J, Dao TK, et al. Superior nationwide outcomes of endovascular versus open repair for isolated descending thoracic aortic aneurysm in 11,669 patients. J Thorac Cardiovasc Surg. 2010;140(5):1001–1010. doi:10.1016/j.jtcvs.2010.08.007
5. Orandi BJ, Dimick JB, Deeb GM, Patel HJ, Upchurch GR
6. Steuer J, Eriksson MO, Nyman R, Bjorck M, Wanhainen A. Early and long-term outcome after thoracic endovascular aortic repair (TEVAR) for acute complicated type B aortic dissection. Eur J Vasc Endovasc Surg. 2011;41(3):318–323. doi:10.1016/j.ejvs.2010.11.024
7. Cheng D, Martin J, Shennib H, et al. Endovascular aortic repair versus open surgical repair for descending thoracic aortic disease: a systematic review and meta-analysis of comparative studies. J Am Coll Cardiol. 2010;55(10):986–1001. doi:10.1016/j.jacc.2009.11.047
8. USFDA. Manufacturer and user facility device experience 2016. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm.
9. Andreoli JM, Lewandowski RJ, Vogelzang RL, Ryu RK. Comparison of complication rates associated with permanent and retrievable inferior vena cava filters: a review of the MAUDE database. J Vasc Interv Radiol. 2014;25(8):1181–1185. doi:10.1016/j.jvir.2014.04.016
10. Mamas MA, Foin N, Abunassar C, Khan MA, Di Mario C, Fraser DG. Stent fracture: insights on mechanisms, treatments, and outcomes from the food and drug administration manufacturer and user facility device experience database. Catheter Cardiovasc Interv. 2014;83(7):E251–E259. doi:10.1002/ccd.25423
11. Makaroun MS, Dillavou ED, Wheatley GH, Cambria RP; Gore TAGI. Five-year results of endovascular treatment with the Gore TAG device compared with open repair of thoracic aortic aneurysms. J Vasc Surg. 2008;47(5):912–918. doi:10.1016/j.jvs.2007.12.006
12. Morales JP, Greenberg RK, Morales CA, et al. Thoracic aortic lesions treated with the Zenith TX1 and TX2 thoracic devices: intermediate- and long-term outcomes. J Vasc Surg. 2008;48(1):54–63. doi:10.1016/j.jvs.2008.02.028
13. Khoynezhad A, Donayre CE, Azizzadeh A, White R; RESCUE Investigators. One-year results of thoracic endovascular aortic repair for blunt thoracic aortic injury (RESCUE trial). J Thorac Cardiovasc Surg. 2015;149(1):155–61 e4. doi:10.1016/j.jtcvs.2014.09.026
14. Bavaria JE, Brinkman WT, Hughes GC, et al. Outcomes of thoracic endovascular aortic repair in acute type B aortic dissection: results from the valiant united states investigational device exemption study. Ann Thorac Surg. 2015;100(3):
15. Fairman RM, Tuchek JM, Lee WA, et al. Pivotal results for the medtronic valiant thoracic stent graft system in the VALOR II trial. J Vasc Surg. 2012;56(5):1222–31 e1. doi:10.1016/j.jvs.2012.04.062
16. Zipfel B, Chiesa R, Kahlberg A, et al. Endovascular repair of traumatic thoracic aortic injury: final results from the relay endovascular registry for thoracic disease. Ann Thorac Surg. 2014;97(3):774–780. doi:10.1016/j.athoracsur.2013.09.034
17. Schaffer JM, Lingala B, Miller DC, Woo YJ, Mitchell RS, Dake MD. Midterm survival after thoracic endovascular aortic repair in more than 10,000 Medicare patients. J Thorac Cardiovasc Surg. 2015;149(3):
18. Canaud L, Ozdemir BA, Patterson BO, Holt PJ, Loftus IM, Thompson MM. Retrograde aortic dissection after thoracic endovascular aortic repair. Ann Surg. 2014;260(2):389–395. doi:10.1097/SLA.0000000000000585
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.