Back to Journals » Infection and Drug Resistance » Volume 12
Therapeutic compounds targeting Lipid II for antibacterial purposes
Authors Malin JJ ![]() , de Leeuw E
, de Leeuw E
Received 9 May 2019
Accepted for publication 29 July 2019
Published 23 August 2019 Volume 2019:12 Pages 2613—2625
DOI https://doi.org/10.2147/IDR.S215070
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Joachim Wink
Jakob J Malin,1,2 Erik de Leeuw3
1University of Cologne, Department I of Internal Medicine, Division of Infectious Diseases, Cologne, Germany; 2Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, Germany; 3Institute of Human Virology and Department of Molecular Biology & Biochemistry of the University of Maryland, Baltimore School of Medicine, Baltimore, MD 21201, USA
Correspondence: Erik de Leeuw
Institute of Human Virology, University of Maryland Baltimore School of Medicine, 725 West Lombard Street Baltimore, MD 21201, USA
Tel +1 410 706 3430
Fax +1 410 706 7583
Email [email protected]
Abstract: Resistance against commonly used antibiotics has emerged in all bacterial pathogens. In fact, there is no antibiotic currently in clinical use against which resistance has not been reported. In particular, rapidly increasing urbanization in developing nations are sites of major concern. Additionally, the widespread practice by physicians to prescribe antibiotics in cases of viral infections puts selective pressure on antibiotics that still remain effective and it will only be a matter of time before resistance develops on a large scale. The biosynthesis pathway of the bacterial cell wall is well studied and a validated target for the development of antibacterial agents. Cell wall biosynthesis involves two major processes; 1) the biosynthesis of cell wall teichoic acids and 2) the biosynthesis of peptidoglycan. Key molecules in these pathways, including enzymes and precursor molecules are attractive targets for the development of novel antibacterial agents. In this review, we will focus on the major class of natural antibacterial compounds that target the peptidoglycan precursor molecule Lipid II; namely the glycopeptides, including the novel generation of lipoglycopeptides. We will discuss their mechanism-of-action and clinical applications. Further, we will briefly discuss additional peptides that target Lipid II such as the lantibiotic nisin and defensins. We will highlight recent developments and future perspectives.
Keywords: antimicrobial peptides, Lipid II, bacterial cell wall, antibiotics
Introduction
Bacterial cell wall assembly
The cell wall of both Gram-negative and -positive bacteria comprises a peptidoglycan layer which is composed of a polymer of alternating amino sugars, N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc). Cross-linking of the polymer chains by pentapeptides achieves mechanical strength and structural integrity of the cell.1 In addition to that it protects the cell from osmotic stress. Inhibition of peptidoglycan biosynthesis inhibits cell growth. This makes the assembly and maintenance of the peptidoglycan polymer a commonly used target for antibiotics. Figure 1 illustrates membrane events in the biosynthesis of the peptidoglycan layer.
 |
Figure 1 Membrane bound processes in the bacterial cell wall biosynthesis cycle. Lipid II binding antibiotics are shown corresponding to the step in the cycle that they inhibit.Note: *varying per species.2 Abbreviations: G, N-acetyl glucosamine; M, N-acetyl muramic acid; MraY, phospho-MurNAc-pentapeptide translocase; MurG, Undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase; UMP, uridine monophosphate; UDP, uridine diphosphate. |
On the cytoplasmic side of the plasma membrane first the soluble precursor UDP-MurNAc-pentapeptide is linked to the membrane carrier bactoprenol-phosphate (C55P) yielding Lipid I. In a second step GlcNac is added by the enzyme MurG to yield Lipid II.3–5 In preparation for building interpeptide bridges between individual Lipid II molecules additional amino acids are added to the pentapeptide by Fem ligases (eg Gly in case of S. aureus).2 Lipid II is then translocated along the membrane to the peripheral side by a not well understoodmechanism.Recent binding studies suggest that this process might be mediated by the flippase enzyme MurJ.6 However, other candidates including RodA and FtsW have been suggested.7,8 On the periplasmic side penicillin-binding proteins (PBPs) catalyze the incorporation of the peptidoglycan unit into the growing cell wall. Class A PBPs obtain insertion of the MurNAc-peptide-GlcNAc subunit into the nascent peptidoglycan layer (transglycosylation) before the peptidoglycan chains are linked together by the formation of peptide crossbridges through the action of both class A - and B PBPs (transpeptidation).9
The remaining complex of lipid anchor and pyrophosphate is shuttled back to the cytosolic side. It can then be reused for following Lipid II synthesis.3 The amount of Lipid II that can be synthesized is limited by the small amount of bactoprenyl phosphate that is available on the cytosolic membrane. About 2×105 molecules C55P per cell have to provide for the enduring synthesis of around 20 peptidoglycan layers in Gram-positive and 1.5 in Gram-negative bacteria.10,11 This is achieved by a high turnover rate of Lipid II which was estimated to be about 1–3 transits per second per prenyl chain.12 The dynamic process of this Lipid II cycle can therefore be regarded as a bottleneck in the bacterial cell wall synthesis. The fact that clustering and dislocating Lipid II leads to dysfunction of the cell wall synthesis suggests that any molecule that binds lipid II with high affinity is a potential antibiotic.4
Lipid II- binding antibiotics
Table 1 Overview of known antibiotic compounds that interact with Lipid II. The stage of development, and the assumed interaction with Lipid II is shown.
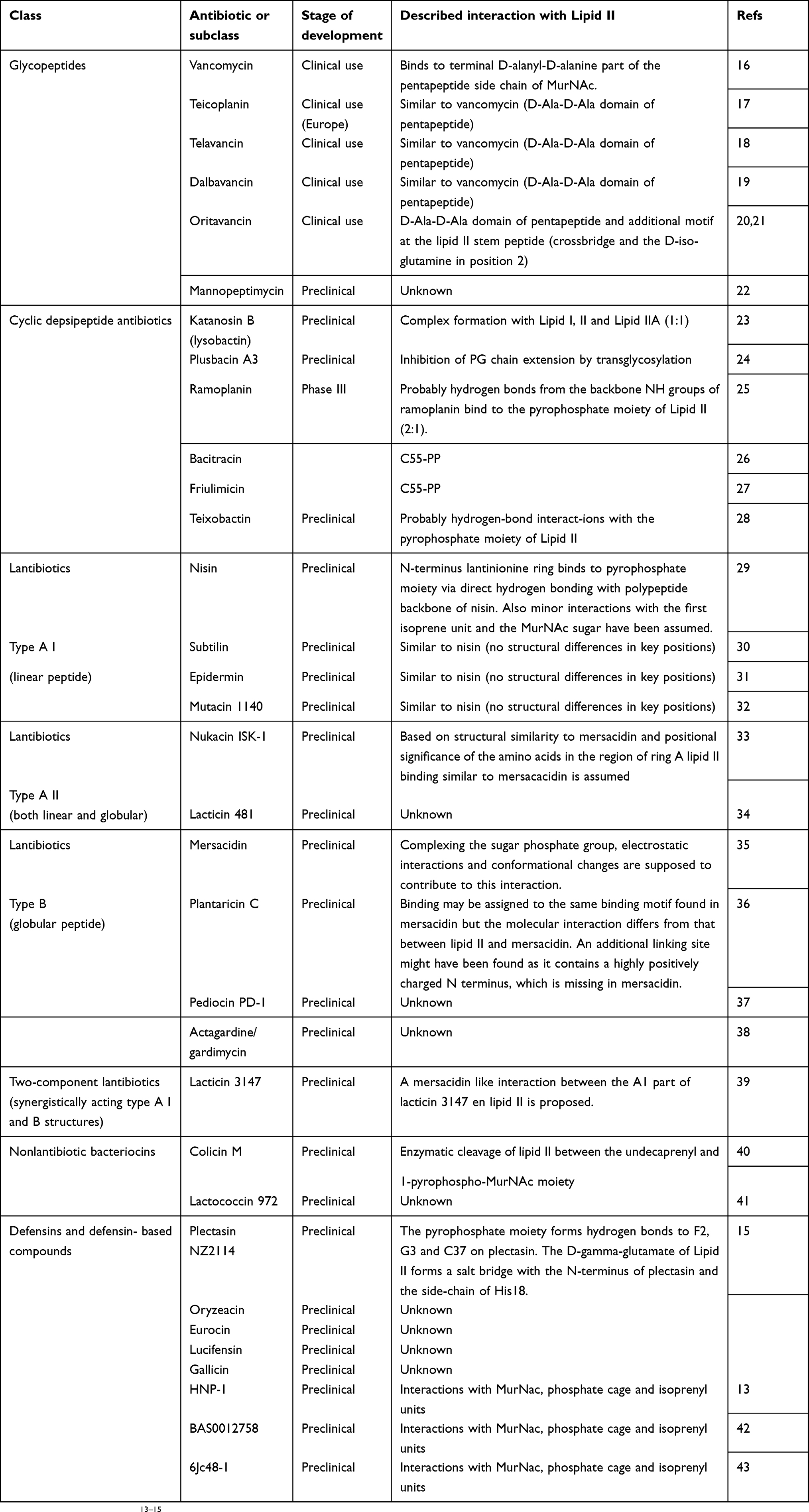 |
Table 1 Summarizes the four main classes of peptide antibiotics that target Lipid II: 1) glycopeptides, including the novel generation of lipoglycopeptides; 2) cyclic depsipeptide antibiotics; and 3) the lantibiotic group and 4) more recently, defensins |
Glycopeptides
All glycopeptides antibiotics have in common that they inhibit the cell wall synthesis by binding to the carboxyl-terminal part of the pentapeptide of lipid II. By binding to the D-Ala-D-Ala domain in growing peptidoglycan chains they disrupt transglycosylation and transpeptidation with destabilization of the cell wall and ultimately cell death.44 Vancomycin, teicoplanin and telavancin are in clinical use today and can be regarded as antibiotics of last resort for the treatment of multidrug-resistant (MDR) Gram-positive pathogens including most of the MRSA strains.44,45 Vancomycin was the first antibiotic identified as targeting Lipid II. It has continuously gained popularity after its clinical introduction in 1988. However, some clinical limitations have been observed such as poor penetration of pulmonary tissues46 and slow bactericidal activity.47,48 In addition to that, vancomycin is associated with an elevated risk of nephrotoxicity, especially when used together with aminoglycosides. Teicoplanin has essentially the same efficacy but fewer adverse effects when compared to vancomycin.49 Despite being marketed throughout Europe, teicoplanin has never been FDA approved for the US market.
Modifications to the basic heptapeptide core of the glycopeptide group have been made in the past years resulting in better pharmacokinetic and antibacterial properties in vitro.44 The (semi-)synthetic lipoglycopeptides telavancin, dalbavancin and oritavancin have an additional lipophilic fatty acyl chain which provides several advantages. By anchoring the compound to the cell membrane the binding affinity to the target site is enhanced. This mechanism also concentrates the compound at its site of action and thereby increases its potency. Lower MIC values and a longer half-life time allow for lower dosage and less frequent applications.44 Besides binding to Lipid II these compounds may provide additional antibiotic activities based on impairing vital membrane functions due to its strong amphipathic character.48,50 All three lipoglycopeptides are promising agents with activity against MDR staphylococci, enterococci and streptococci.45 Telavancin is a semisynthetic derivate of vancomycin that contains a deacylaminoethyl moeiety. Besides the already mentioned advantages obtained from the lipophilic side chain telavancin has an increased activity against MRSA and non-VanA enterococci. In comparison to vancomycin, telavancin provides a stronger binding affinity for the target binding site and better tissue penetration.48,51 Telavancin is now also available on the European market since approval by the European Medicines Agency (EMA) in 2011 for the treatment of hospital-acquired pneumonia caused by MRSA.
Despite these beneficial developments and the extended antimicrobial spectrum of the novel generation glycopeptides there is an increasing concern about dwindling activity of vancomycin and spreading bacterial resistance against glycopeptide antibiotics.52,53 Bacteria carrying the vanA-type gene cluster that encodes an epitope substitution of D-Ala-D-Lactate termini for D-Ala-D-Ala termini are able to evade the attack of glycopeptides including the generation of lipoglycopeptides.54 Although this resistance mechanism is presumed to be extremely cost extensive energetically and causes a fitness reduction in VRSA isolates, the composition of the pentapeptide of lipid II is not functionally critical. Modifications to the pentapeptide composition can therefore easily spread under the selective pressure of glycopeptide usage without any impact on cell wall synthesis.3,55 While VRSA isolates are uncommon, an increasingly growing number of clinical isolates appear with vancomycin-intermediate resistance profiles. These VISA strains cause complicated, hard-to-treat infections and are increasingly associated with poor clinical results.56 The vanA genotype, which is mediated by transposon Tn1546 is the predominant type of resistance that is reported in Europe. It is very common among enterococci which cause ~20,000 infectiond per year in the USA alone and are highly capable of horizontal gene transfer.44,57 This type of resistance is transferable to significant pathogens like MRSA due to its location on conjugative plasmids.58,59 The dissemination of glycopeptide resistance in enterococci can therefore lead to clinical isolates that are resistant to all available antibiotics.52 Five other types of vancomycin resistance (VanB, D, E and G) have been described on phenotypic and genotypic basis in enterococci. The VanA type, however is to date the only one detected in S. aureus. Glycopeptides can directly induce resistance by activating resistance pathways in susceptible isolates. Interestingly, also glycopeptide dependent growth has been described for some VanA and VanB-type enterococci. This is clinically important because long periods of vancomycin treatment can account for this phenomenon.52
Dalbavancin is a semisynthetic derivate of teicoplanin-like antibiotic A-40,926. The acetylglucosamine group of teicoplanin has been removed in order to achieve improved efficacy against VanA enterococci, unfortunately without success.48,60 Oritavancin is the only glycopeotide that is effective against the vanA type isolates because of its ability of binding Lipid II in species with the substitute D-Ala-D-Lac that causes resistance against the other glycopeptide antibiotics. This ability can be explained by binding sites at the Lipid II stem peptide.21 Additional interactions have been described, including the pentaglycine crossbridge or the D-isoglutamine in position 2 of the Lipid II stem peptide of S. aureus and the D-aspartate/D-asparagine cross-bridge in Enterococcus faecium.21,61 However, in vitro induction of moderate level resistance to oritavancin has been observed in Enterococci isolates that express the VanA or VanB phenotypes.62
Clinical trials on (lipo-) glycopeptide antibiotics
For decades vancomycin has been the first line treatment of infections with MRSA including skin and soft-tissue infections, bacterial pneumonia and infectious endocarditis. However, since vancomycin was approved around thirty years ago (and teicoplanin in the European Union), three novel semisynthetic antibiotics targeting Lipid II have been developed that build up the generation of lipoglycopeptides: telavancin, oritavancin and dalbavancin. As described earlier the molecules of lipoglycopeptides differ from that of vancomycin by the presence of a lipophilic fatty acyl side chain that improves target binding, potency and pharmacokinetic properties.63–65
Telavancin, the first developed and approved lipoglycopeptide antibiotic has been evaluated in six randomised controlled trials which will be briefly discussed. The FAST and FAST 2 Study (published in 2005 and 2006) compared clinical outcomes of complicated Gram-positive skin and skin structure infections (cSSTI) treated with either telavancin (FAST: 7.5 mg/kg, FAST 2: 10 mg/kg once daily) or the standard treatment that comprised of vancomycin (1 g twice daily) or ß-lactam antibiotics (nafcillin, oxacillin or cloxacillin in a standard dose). This phase II trials included respectively 167 and 195 patients and showed equal clinical cure rates in both treatment arms (FAST: 80% vs 77%; FAST 2: 96% vs 90% for telavancin vs standard treatment). In the subgroup of patients with MRSA infections (n=48) telavancin was superior to the standard treatment within the FAST trial with cure rates of 82% versus 69% for the standard treatment.66 However, this difference was not statistically significant (p=1.0) and this finding could not be reproduced within the second trial (FAST 2) where cure rates of MRSA infections did not significantly differ in both arms (96% vs 90%). It is important to note, that the amount of patients in the standard treatment arms receiving vancomycin was higher in the FAST 2 Study (93%) compared to the FAST 1 study (75%) which might have contributed to different results.67 The studies also assessed microbiologic eradication rates of MSSA and MRSA after treatment. In contrast to the FAST trial where treatments were equivalent in this aspect, the second trial revealed higher eradication rates in the telavancin group (92% vs 78%, p=0.07) and in the subgroup with proven MRSA infections (92% vs 68%, p=0.04).67 This finding might be due to the use of higher telavancin doses (10 mg/kg) in comparison to the FAST trial. As expected from in vitro studies telavancin had lower minimal inhibitory concentrations (MIC’s) for clinical isolates of S. aureus including MRSA (MIC90≤0.25 vs 1 µg/mL for vancomycin).66–70 In 2008, the results of two methodologic identical randomised controlled phase III trials (ATLAS 1 and 2) were published that confirmed non-inferiority of televancin (10 mg/kg once daily) to vancomycin (1 g twice daily) for the treatment of cSSTI (more recently redefined as acute bacterial skin and skin structure infections, ABSSSI). Of 1867 enrolled patients, 1693 were evaluable for the test of cure outcome. Overall, no differences between both treatments were found concerning clinical cure rates, microbiological eradication and general safety.71 Telavancin was subsequently approved by the FDA (2009) and EMA (2011) for the treatment of cSSTI/ABSSSI. Telavancin has also been assessed for other types of infections. In 2011, data from the ATTAIN studies that included 1503 patients showed non-inferiority of telavancin for the treatment of hospital-acquired pneumonia caused by Gram-positive pathogens including MRSA. This led to an extended approval for this indication in the EU.72 In 2014, the ASSURE study evaluated telavancin for treating uncomplicated S. aureus bloodstream infections including catheter associated S. aureus bacteraemia. The phase II study compared clinical outcomes of treatment with telavancin versus vancomycin. Unfortunately, only 17 out of 60 included patients were clinically evaluable. However, in this proportion clinical cure rates of telavancin were similar to that of vancomycin indicating a potential use of telavancin in gram-positive bloodstream infections.73 The results of the FAST, ATLAS and ATTAIN trials were summarized in a meta-analysis published in 2012 which evaluated the general efficacy and safety of telavancin. Pooled data showed equal clinical cure rates of telavancin in comparison to vancomycin. Microbiologic eradication rates favoured telavancin (90% vs 84%; OR 1.71; 95% CI: 1.08–2.70), but the risk of an increase in serum creatinine slightly favoured vancomycin (OR 2.22; 95% CI 1.38–3.57). The authors therefore concluded that the risk of nephrotoxicity should be taken into account for treatment decisions.74 Safety outcomes of telavancin versus vancomycin were also compared in a post hoc analysis of data from the ATTAIN studies after exclusion of patients with pre-existing acute renal failure and severe renal failure defined as glomerular filtration rate <30 mL/min. In contrast to the previous meta-analysis this study showed no significant differences in renal adverse events among patients treated with either telavancin or vancomycin.75 In summary, telavancin has proven non-inferiority to vancomycin for the treatment of Gram-positive ABSSSI and seems to be a rational alternative for infections with pathogens that are intermediate susceptible for vancomycin or in case of vancomycin intolerance. Although there is some evidence for nephrotoxicity of telavancin, dosing does not need to be guided by serum concentrations which forms a practical advantage to vancomycin.63,64,76
The lipoglycopeptide oritavancin has a prolonged terminal half-life of 245 hrs due to a slow elimination rate.63 This makes it favourable for single or infrequent dosing. In 2011, the SIMPLIFI trial compared clinical response rates at treatment days 21–29 of patients with ABSSSI treated with oritavancin administered either daily (200 mg for 3 to 7 days), in one single dose (1200 mg), or in an infrequent dose (800 mg, and possibly additional 400 mg on day 5). Surprisingly, the test of cure rates in the clinically evaluable population (n=228) were equal in all study groups (72.4%, 81.5% and 77.5%, respectively). Furthermore, similar safety profiles were found in all administration patterns.77 This phase II study was followed by two phase III trials (SOLO I and SOLO II) published in 2014 and 2015, that demonstrated non-inferiority of a single administration of 1200 mg of oritavancin to a 7 to 10 day treatment with vancomycin (1 g twice daily) for treating Gram-positive ABSSSI. There was no significant difference in the primary composite endpoint demonstrating an early treatment response (cessation of spreading or reduction in lesion size, absence of fever and no use of a rescue antibiotic). Cure rates were 79.6% versus 82.9% (SOLO I) and 82.7% versus 80.5% (SOLO II) in the oritavancin and vancomycin treated group, respectively.78–80 Oritavancin was subsequently approved by the authorities in 2014 (FDA) and 2015 (EMA). The lipoglycopeptides telavancin and oritavancin have in common that they provide an additional mechanism of bacterial killing by disrupting the integrity of the bacterial cell membrane. This dual mode of action may contribute to the sustained activity of those agents against vancomycin intermediate susceptible and resistant S. aureus. While both antibiotics provide activity against enterococci carrying the vanB-type resistance gene only oritavancin is active against vancomycin resistant species due to vanA as previously described.64 The pharmacokinetic profile of oritavancin allows for a single dose treatment which makes it an excellent option for outpatient treatment and reducing prolonged hospital stays.
Similar to oritavancin, dalbavancin has an extensive terminal elimination rate (187 hrs) which allows for infrequent dosing intervals. Dalbavancin was firstly evaluated in a phase II proof-of-concept study including 62 patients with ABSSSI receiving either a two dose regimen of dalbavancin (1000 mg at day 1 and 500 mg at day 8), a single dose (1100 mg) or a prospectively defined standard of care treatment. Clinical success rates (defined as cure or improvement) at the follow-up visit were 94.1% among patients treated with two doses of dalbavancin, 61.5% in the one dose only group and 76.2% among patients treated with the standard-of-care.81 The two-dose regimen was subsequently integrated into phase III clinical trials. Two identically designed non-inferiority trials (DISCOVER 1 and 2) compared treatment outcomes of ABSSSI with two doses of dalbavancin (1000 mg +500 mg at day 8) and vancomycin (1 g or 15 mg/kg twice daily) with an option to switch to oral linezolid after day 3 in the vancomycin treated group. A combined analysis of this double-blind randomized controlled phase III trials including 1312 patients from 2014 showed equal results for both arms concerning early clinical responses (primary endpoint, similar to the SOLO trials) and clinical cure rates (secondary endpoint). According to the pooled analysis 79.7% of the dalbavancin group and 97.8% of the control group showed an early clinical response. Clinical cure rates were 90.6% in the dalbavancin group versus 93.8% in the control group.82 The approved two-dose regimen of dalbavancin was challenged in a separate randomized clinical trial comparing efficacies against a single dose of 1500 mg in 698 patients with ABSSSI. The primary endpoint, a 20% reduction in lesion size or more at 48 to 72 hrs was achieved among 81.4% and 48.2% of the single-dose and two-dose treated group, respectively. Clinical outcomes of evaluable patients, including patients with MRSA-positive baseline cultures and adverse events were also similar in both groups.83 These data resulted in an extended approval, including a single-dose treatment of 1500 mg of dalbavancin for the management of ABSSSI. Dalbavancin was furthermore evaluated in phase II randomized controlled trial including 75 patients with catheter associated bloodstream infections caused by S. aureus or coagulase-negative staphylococci (CNS). The primary combined endpoint was a clinically and microbiological treatment response. This was achieved among 87% of patients treated with the two dose regimen of dalbavancin versus 50% among patients treated with vancomycin twice daily for 14 days.84 Hence, dalbavancin might be a good alternative for treating catheter-associated blood stream infections with susceptible organisms. Data from phase I trials assessing pharmacokinetics and distribution of dalbavancin (two-dose regimen) into bone and articular tissues do also suggest that it might be an effective treatment for osteomyelitis because of a long acting tissue exposure, exceeding the MIC of S. aureus for a period of 8 weeks.85 Like oritavancin, dalbavancin is therefore an excellent candidate for use in outpatients that needs a long lasting antimicrobial therapy as in endocarditis and osteomyelitis. However, at this point the only approved indication is the treatment of ABSSSI.
Cyclic depsipeptide antibiotics
This structural diverse group consists of nonribosomal antibiotic peptides (NRPs) that are derived from natural bacterial products, mainly produced by soil bacteria. Structurally, depsipeptides contain one or more ester bonds along with the amide bonds. They have distinct binding motives on Lipid II that include the pyrophosphate-sugar moiety, but not the stem peptide which is targeted by all (lipo-) glycopeptide antibiotics. As development of resistance to glycopeptides is basically caused by changes in the stem peptide, depsipeptides can be considered interesting candidates for further drug development.
Ramoplanin and its analogues (A1-A3) were first discovered in 1984 by an industrial drug screening programme as product of spore forming soil bacteria belonging to the genus of Actinoplanes sp.86 This macrocyclic depsipeptide inhibits the extracellular transglycosylation by binding to Lipid II. Efforts to identify the exact structure of the ramoplanin-Lipid II complex were not successful due to ligand-induced polymerization of the antibiotic-Lipid II complex, resulting in insoluble fibrils without antimicrobial activity.87 However, early studies could show that binding does not depend on the presence of the D-Ala-D-Ala domain and rather involve the pyrophosphate moiety of Lipid II.25 In analogy with lipoglycopeptides, ramoplanins are proposed to exert additional antibacterial effects by interfering with cell membrane integrity.88 Like vancomycin, ramoplanin is not absorbed when administered orally and might be an interesting alternative for vancomycin in treating gastrointestinal infections with Clostridium difficile or intestinal colonization with VRE.89 The latter was demonstrated in one clinical trial with 68 patients receiving ramoplanin (100 mg or 400 mg) or a placebo but the suppression of VRE did not show sustainable effect.90
Katanosin B, which is structurally identical to lysobactin, forms 1:1 complexes with several cell wall precursors including Lipid II.23 Although this agent was first discovered in 1987, the exact binding site has not been described to date. One study from 1988 demonstrated favourable in vitro activity against a wide range of bacteria. However, in vivo studies indicated a relatively high toxicity in mice.91 Efforts to further develop this compound have since been limited in the past years. The cyclic depsipeptides plusbacin A3, bacitracin and friulimicin are also suspected to exert their antibacterial effects by binding to Lipid II or its intermediates, but reported evidence for molecular interactions with Lipid II is limited.92–94
Teixoplanin is novel antibiotic of the cyclic depsipeptide group that has recently been discovered in a screen of uncultured soil bacteria. Binding to Lipid II is most probably mediated by hydrogen-bonds in the pyrophosphate sugar region.95 Teixoplanin is active against a wide range of Gram-positive pathogens, including MRSA and VRE. Like ramoplanin, teixoplanin is also highly active against C. difficile but with an even lower minimum inhibitory concentration (MIC) of 0.005 µg mL.−1
Lantibiotics
Lantibiotics are a large family of peptides that are produced by certain strains of bacilli to kill other bacteria. They block the cell wall synthesis by clustering the Lipid II molecules into patches and therefore relocate it within the membrane.96 Nisin, a type A lantibiotic, has been extensively studied and serves as the paradigm for this family of anti-microbial peptides. Nisin was discovered almost a century ago and initially recognized for its use in the food industry as a preservative before its recognition as an antimicrobial.97–99 Characteristic of lantibiotics, nisin contains lanthionine rings formed by thioether and dehydrated serine and threonine amino acid residues.100,101 Nisin was shown to cluster Lipid II at a 1:2 ratio within the cytoplasmic membrane, but outside the region where it is normally active.11,102 Following its primary interaction with Lipid II, additional nisin molecules are recruited in a larger complex that is capable of membrane perturbation by forming pores in lipid layers.103 The pore is thought to consist of 8 molecules nisin and 4 molecules of lipid II and during the killing these complexes have been shown form even larger clusters.11,104 Together with observations such as the propensity of Lipid II to enhance the pore forming capacity of nisin in lipid layers, an actual model of this interaction suggests that Lipid II acts as a scaffold for the peptide to dock at the membrane surface prior to insertion and pore formation.1,4,29,101,105 Interestingly, resistance to the antibacterial action of nisin was found not to be determined by the level of Lipid II per se, suggesting additional mechanisms-of-action for this peptide.106
Defensins and defensin- based compounds
Antimicrobial peptides such as defensins were believed to kill microorganisms by permeabilizing the cell membranes. Due to their cationic nature, they preferentially target bacterial membranes which contain large amounts of negatively charged phospholipids, whereas the mammalian cell membrane is composed of neutral, zwitterionic phospholipids exclusively. Various models for the selective activity of antimicrobial peptides towards microorganisms have been proposed.107 These include the formation of solvent-permeable pore structures, covering the membrane like detergents or possible inhibition of metabolic processes of microorganisms through intracellular targeting.107 These models agree on the importance of electrostatic interactions in initiating membrane association of cationic peptides.
Disruption of the functional integrity of the bacterial membrane is a common mode of action of many antibacterial compounds and was believed to be the primary mode of bacterial killing by defensins. An early study reported on the bactericidal activity of Human Neutrophil Peptides (HNP-1–3) against E. coli, suggesting a sequential permeabilization of the outer and inner membranes.108 However, we and others reported that linear, unstructured defensins retained their antibacterial activity in a strain-selective manner.109,110 Further we showed that certain defensins were shown to preferentially kill Gram-positive bacteria, whereas other defensins that carry more positive charge kill Gram-negative strains more effectively.111,112 Based on these observations, we were among the first to report on the functional interaction between Lipid II and Human Neutrophil Peptide 1 (HNP-1), a member of a major family of natural antimicrobial peptides called defensins that protect the host’s epithelial surfaces against microbial invasion.13,113 Several studies on defensins from other species, including fungi,15 invertebrates14 and humans114 have firmly established Lipid II as a target for this class of natural antimicrobial peptides. In their landmark report, Schneider et al reported on the Lipid II-binding fungal defensin plectasin. In this study, interactions between plectasin and the solvent-exposed pyrophosphate region of Lipid II were described.15 These interactions involved residues Phe2, Gly3, Cys4 and Cys37 as well as the N-terminus and His18 side-chain of this defensin. Human defensin peptides themselves are unlikely to be developed into therapeutics because of their well-described functional versatility.115–119 A derivative of the fungal defensin plectasin, termed NZ2114, has shown most promise to date, with in vivo efficacy in infectious models as well as applications to combat biofilm formation.120–122 Similar to the plectasin study, we identified C-terminal hydrophobic residues Ile20 and Leu25 of Human Neutrophil Peptide-1 to be mainly involved in Lipid II binding. However, since HNP-1 and other defensins show little promise as therapeutics, we identified small molecules that bind to Lipid II via a unique mechanism. Using functional and structural approaches, these small molecules bind to three distinct sites of the Lipid II molecule and are excellent lead candidates for the development of next-generation drugs for this target.13,42,43,123,124
Future perspective
The discovery of the glycopeptide vancomycin and development of its next-generation derivatives has been an important milestone in the treatment of multi-drug resistant Gram-positive infections. Currently, vancomycin still serves as a first-line treatment for many infections caused by Gram-positive pathogens including methicillin-resistant S. aureus (MRSA), coagulase-negative Staphylococci (CNS) and Enterococci. In general, the clinical use of vancomycin has important drawbacks that might increase the use of next-generation glycopeptides in the future. Besides its infamous nephrotoxicity, the IV-only route of administration every 12 hrs and the need for a trough concentration-guided dosing are unpractical. Especially in cases of complex infections that require prolonged treatments like osteomyelitis or endocarditis this fact forms a relevant disadvantage that hinders early release from hospital and treatments in an out-patient setting. The novel class of lipoglycopeptides showed non-inferiority in clinical studies and can solve some of these problems. However, an oral alternative with a favorable safety profile is urgently needed. In addition to the practical issues with the available glycopeptides, it will only be a matter of time before resistance against this class of antibacterials will reach a breaking point.125 Though still low, resistance to vancomycin in staphylococci is increasing and a number of vancomycin insensitive strains have been described.56 Vancomycin resistance is a particular acute issue for vancomycin-resistant and multi-drug resistant (MDR) Enterococci.126 In case of MDR Enterococci, resistance to alternatives of vancomycin such as oxazolidinone-linezolid, daptomycin, tigecycline and the last resort formulation quinupristin/dalfopristin, can lead to serious limitations for available treatments options.127,128 As all compounds of the (lipo-)glycopeptide group basically share a similar mode of action, a high degree of cross-resistance can be expected. Still, Lipid II and the peptidoglycan synthesis remain attractive and underexplored targets for antibiotic drug discovery when focusing on compounds with a distinct mode of interaction to that of already approved compounds. In this light, small molecules that interfere with peptidoglycan synthesis129 or that bind to Lipid II directly,42,43,123 hold promise for the development of novel classes of orally available drugs.
Acknowledgments
This work was supported by Center for Biomolecular Therapeutics (PR155EDL1; UMB), a UM Ventures Seed Grant and Center for Maryland Advanced Ventures Grant to EdL. The funders had no role in study design, data collection and analysis, or preparation of the manuscript.
Disclosure
EdL is an inventor on US patents #8,796,323, #9,351,963 and PCT/US11/59432. The authors report no other conflicts of interest in this work.
References
1. Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nat Rev Drug Discov. 2006;5(4):321–332. doi:10.1038/nrd2004
2. van Heijenoort J. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol Mol Biol Rev. 2007;71(4):620–635. doi:10.1128/MMBR.00016-07
3. de Kruijff B, van Dam V, Breukink E. Lipid II: a central component in bacterial cell wall synthesis and a target for antibiotics. Prostaglandins Leukot Essent Fatty Acids. 2008;79(3–5):117–121. doi:10.1016/j.plefa.2008.09.020
4. Hsu ST, Breukink E, Tischenko E, et al. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol. 2004;11(10):963–967. doi:10.1038/nsmb830
5. Okesola AO. Community-acquired methicillin-resistant Staphylococcus aureus–a review of literature. Afr J Med Med Sci. 2011;40(2):97–107.
6. Bolla JR, Sauer JB, Wu D, et al. Direct observation of the influence of cardiolipin and antibiotics on lipid II binding to MurJ. Nat Chem. 2018;10(3):363–371. doi:10.1038/nchem.2919
7. Ruiz N. Lipid Flippases for Bacterial Peptidoglycan Biosynthesis. Lipid Insights. 2015;8(Suppl 1):21–31. doi:10.4137/LPI.S31783
8. Mohammadi T, van Dam V, Sijbrandi R, et al. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011;30(8):1425–1432. doi:10.1038/emboj.2011.61
9. Zapun A, Noirclerc-Savoye M, Helassa N, Vernet T. Peptidoglycan assembly machines: the biochemical evidence. Microb Drug Resist. 2012;18(3):256–260. doi:10.1089/mdr.2011.0236
10. Ganchev DN, Hasper HE, Breukink E, de Kruijff B. Size and orientation of the lipid II headgroup as revealed by AFM imaging. Biochemistry. 2006;45(19):6195–6202. doi:10.1021/bi051913e
11. Hasper HE, Kramer NE, Smith JL, et al. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science. 2006;313(5793):1636–1637. doi:10.1126/science.1129818
12. Bugg TD, Brandish PE. From peptidoglycan to glycoproteins: common features of lipid-linked oligosaccharide biosynthesis. FEMS Microbiol Lett. 1994;119(3):255–262. doi:10.1111/j.1574-6968.1994.tb06898.x
13. de Leeuw E, Li C, Zeng P, et al. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010;584(8):1543–1548. doi:10.1016/j.febslet.2010.03.004
14. Schmitt P, Wilmes M, Pugniere M, et al. Insight into invertebrate defensin mechanism of action: oyster defensins inhibit peptidoglycan biosynthesis by binding to lipid II. J Biol Chem. 2010;285(38):29208–29216. doi:10.1074/jbc.M110.143388
15. Schneider T, Kruse T, Wimmer R, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science. 2010;328(5982):1168–1172. doi:10.1126/science.1185723
16. Knox JR, Pratt RF. Different modes of vancomycin and D-alanyl-D-alanine peptidase binding to cell wall peptide and a possible role for the vancomycin resistance protein. Antimicrob Agents Chemother. 1990;34(7):1342–1347. doi:10.1128/aac.34.7.1342
17. Somma S, Gastaldo L, Corti A. Teicoplanin, a new antibiotic from Actinoplanes teichomyceticus nov. sp. Antimicrob Agents Chemother. 1984;26(6):917–923. doi:10.1128/aac.26.6.917
18. King A, Phillips I, Kaniga K. Comparative in vitro activity of telavancin (TD-6424), a rapidly bactericidal, concentration-dependent anti-infective with multiple mechanisms of action against Gram-positive bacteria. J Antimicrob Chemother. 2004;53(5):797–803. doi:10.1093/jac/dkh156
19. Woodford N. Novel agents for the treatment of resistant Gram-positive infections. Expert Opin Investig Drugs. 2003;12(2):117–137. doi:10.1517/13543784.12.2.117
20. Schwalbe RS, McIntosh AC, Qaiyumi S, et al. In vitro activity of LY333328, an investigational glycopeptide antibiotic, against enterococci and staphylococci. Antimicrob Agents Chemother. 1996;40(10):2416–2419.
21. Munch D, Engels I, Muller A, et al. Structural variations of the cell wall precursor lipid II and their influence on binding and activity of the lipoglycopeptide antibiotic oritavancin. Antimicrob Agents Chemother. 2015;59(2):772–781. doi:10.1128/AAC.02663-14
22. Singh MP, Petersen PJ, Weiss WJ, et al. Mannopeptimycins, new cyclic glycopeptide antibiotics produced by Streptomyces hygroscopicus LL-AC98: antibacterial and mechanistic activities. Antimicrob Agents Chemother. 2003;47(1):62–69. doi:10.1128/aac.47.1.62-69.2003
23. Lee W, Schaefer K, Qiao Y, et al. The Mechanism of Action of Lysobactin. J Am Chem Soc. 2016;138(1):100–103. doi:10.1021/jacs.5b11807
24. O’Connor RD, Singh M, Chang J, et al. Dual Mode of Action for Plusbacin A3 in Staphylococcus aureus. J Phys Chem B. 2017;121(7):1499–1505. doi:10.1021/acs.jpcb.6b11039
25. Reynolds PE, Somner EA. Comparison of the target sites and mechanisms of action of glycopeptide and lipoglycodepsipeptide antibiotics. Drugs Exp Clin Res. 1990;16(8):385–389.
26. Stone KJ, Strominger JL. Mechanism of action of bacitracin: complexation with metal ion and C 55 -isoprenyl pyrophosphate. Proc Natl Acad Sci U S A. 1971;68(12):3223–3227. doi:10.1073/pnas.68.12.3223
27. Vertesy L, Ehlers E, Kogler H, et al. Friulimicins: novel lipopeptide antibiotics with peptidoglycan synthesis inhibiting activity from Actinoplanes friuliensis sp. nov. II. Isolation and structural characterization. J Antibiot (Tokyo). 2000;53(8):816–827. doi:10.7164/antibiotics.53.816
28. Zong Y, Sun X, Gao H, et al. Developing Equipotent Teixobactin Analogues against Drug-Resistant Bacteria and Discovering a Hydrophobic Interaction between Lipid II and Teixobactin. J Med Chem. 2018;61(8):3409–3421. doi:10.1021/acs.jmedchem.7b01241
29. Breukink E, Wiedemann I, van Kraaij C, et al. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science. 1999;286(5448):2361–2364. doi:10.1126/science.286.5448.2361
30. Nakano MM, Zuber P. Molecular biology of antibiotic production in Bacillus. Crit Rev Biotechnol. 1990;10(3):223–240. doi:10.3109/07388559009038209
31. Schnell N, Entian KD, Schneider U, et al. Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature. 1988;333(6170):276–278. doi:10.1038/333276a0
32. Smith L, Hasper H, Breukink E, et al. Elucidation of the antimicrobial mechanism of mutacin 1140. Biochemistry. 2008;47(10):3308–3314. doi:10.1021/bi701262z
33. Sashihara T, Kimura H, Higuchi T, et al. A novel lantibiotic, nukacin ISK-1, of Staphylococcus warneri ISK-1: cloning of the structural gene and identification of the structure. Biosci Biotechnol Biochem. 2000;64(11):2420–2428. doi:10.1271/bbb.64.2420
34. Demel RA, Peelen T, Siezen RJ, De Kruijff B, Kuipers OP, Nisin Z. mutant nisin Z and lacticin 481 interactions with anionic lipids correlate with antimicrobial activity A monolayer study. Eur J Biochem. 1996;235(1–2):267–274. doi:10.1111/j.1432-1033.1996.00267.x
35. Brötz H, Bierbaum G, Markus A, Molitor E, Sahl HG. Mode of action of the lantibiotic mersacidin: inhibition of peptidoglycan biosynthesis via a novel mechanism? Antimicrob Agents Chemother. 1995;39(3):714–719. doi:10.1128/aac.39.3.714
36. Bauer R, Dicks LM. Mode of action of lipid II-targeting lantibiotics. Int J Food Microbiol. 2005;101(2):201–216. doi:10.1016/j.ijfoodmicro.2004.11.007
37. Montville TJ, Chen Y. Mechanistic action of pediocin and nisin: recent progress and unresolved questions. Appl Microbiol Biotechnol. 1998;50(5):511–519.
38. Somma S, Merati W, Parenti F. Gardimycin, a new antibiotic inhibiting peptidoglycan synthesis. Antimicrob Agents Chemother. 1977;11(3):396–401. doi:10.1128/aac.11.3.396
39. Wiedemann I, Bottiger T, Bonelli RR, et al. The mode of action of the lantibiotic lacticin 3147–a complex mechanism involving specific interaction of two peptides and the cell wall precursor lipid II. Mol Microbiol. 2006;61(2):285–296. doi:10.1111/j.1365-2958.2006.05223.x
40. El Ghachi M, Bouhss A, Barreteau H, et al. Colicin M exerts its bacteriolytic effect via enzymatic degradation of undecaprenyl phosphate-linked peptidoglycan precursors. J Biol Chem. 2006;281(32):22761–22772. doi:10.1074/jbc.M602834200
41. Martinez B, Suarez JE, Rodriguez A. Lactococcin 972: a homodimeric lactococcal bacteriocin whose primary target is not the plasma membrane. Microbiology. 1996;142(Pt 9):2393–2398. doi:10.1099/00221287-142-9-2393
42. Varney KM, Bonvin AM, Pazgier M, et al. Turning defense into offense: defensin mimetics as novel antibiotics targeting lipid II. PLoS Pathog. 2013;9(11):e1003732. doi:10.1371/journal.ppat.1003732
43. Chauhan J, Cardinale S, Fang L, et al. Towards Development of Small Molecule Lipid II Inhibitors as Novel Antibiotics. PLoS One. 2016;11(10):e0164515. doi:10.1371/journal.pone.0164515
44. Zhanel GG, Calic D, Schweizer F, et al. New lipoglycopeptides: a comparative review of dalbavancin, oritavancin and telavancin. Drugs. 2010;70(7):859–886. doi:10.2165/11534440-000000000-00000
45. Cornaglia G, Rossolini GM. Forthcoming therapeutic perspectives for infections due to multidrug-resistant Gram-positive pathogens. Clin Microbiol Infect. 2009;15(3):218–223. doi:10.1111/j.1469-0691.2009.02740.x
46. Stein GE, Wells EM. The importance of tissue penetration in achieving successful antimicrobial treatment of nosocomial pneumonia and complicated skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus: vancomycin and linezolid. Curr Med Res Opin. 2010;26(3):571–588. doi:10.1185/03007990903512057
47. Rybak MJ, Lomaestro BM, Rotschafer JC, et al. Therapeutic monitoring of vancomycin in adults summary of consensus recommendations from the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Pharmacotherapy. 2009;29(11):1275–1279.
48. Zhanel GG, Trapp S, Gin AS, et al. Dalbavancin and telavancin: novel lipoglycopeptides for the treatment of Gram-positive infections. Expert Rev Anti Infect Ther. 2008;6(1):67–81. doi:10.1586/14787210.6.1.67
49. Cavalcanti AB, Goncalves AR, Almeida CS, Bugano DD, Silva E. Teicoplanin versus vancomycin for proven or suspected infection. Cochrane Database Syst Rev. 20106:CD007022.
50. Schneider T, Sahl HG. An oldie but a goodie - cell wall biosynthesis as antibiotic target pathway. Int J Med Microbiol. 2010;300(2–3):161–169. doi:10.1016/j.ijmm.2009.10.005
51. Charneski L, Patel PN, Sym D. Telavancin: a novel lipoglycopeptide antibiotic. Ann Pharmacother. 2009;43(5):928–938. doi:10.1345/aph.1G417
52. Courvalin P. Vancomycin resistance in gram-positive cocci. Clin Infect Dis. 2006;42(Suppl 1):S25–S34. doi:10.1086/491711
53. Gould IM. Is vancomycin redundant for serious staphylococcal infection? Int J Antimicrob Agents. 2010;36(Suppl 2):S55–S57. doi:10.1016/j.ijantimicag.2010.11.005
54. Hughes D. Exploiting genomics, genetics and chemistry to combat antibiotic resistance. Nat Rev Genet. 2003;4(6):432–441. doi:10.1038/nrg1084
55. Jovetic S, Zhu Y, Marcone GL, Marinelli F, Tramper J. beta-Lactam and glycopeptide antibiotics: first and last line of defense? Trends Biotechnol. 2010;28(12):596–604. doi:10.1016/j.tibtech.2010.09.004
56. McGuinness WA, Malachowa N, DeLeo FR. Vancomycin Resistance in Staphylococcus aureus. Yale J Biol Med. 2017;90(2):269–281.
57. Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10(4):266–278. doi:10.1038/nrmicro2761
58. Whitener CJ, Park SY, Browne FA, et al. Vancomycin-resistant Staphylococcus aureus in the absence of vancomycin exposure. Clin Infect Dis. 2004;38(8):1049–1055. doi:10.1086/382357
59. Chang S, Sievert DM, Hageman JC, et al. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N Engl J Med. 2003;348(14):1342–1347. doi:10.1056/NEJMoa025025
60. Kim A, Kuti JL, Nicolau DP. Review of dalbavancin, a novel semisynthetic lipoglycopeptide. Expert Opin Investig Drugs. 2007;16(5):717–733. doi:10.1517/13543784.16.5.717
61. Patti GJ, Kim SJ, Yu TY, et al. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J Mol Biol. 2009;392(5):1178–1191. doi:10.1016/j.jmb.2009.06.064
62. Arthur M, Depardieu F, Reynolds P, Courvalin P. Moderate-level resistance to glycopeptide LY333328 mediated by genes of the vanA and vanB clusters in enterococci. Antimicrob Agents Chemother. 1999;43(8):1875–1880.
63. Van Bambeke F. Lipoglycopeptide antibacterial agents in gram-positive infections: a comparative review. Drugs. 2015;75(18):2073–2095. doi:10.1007/s40265-015-0505-8
64. Klinker KP, Borgert SJ. Beyond Vancomycin: The Tail of the Lipoglycopeptides. Clin Ther. 2015;37(12):2619–2636. doi:10.1016/j.clinthera.2015.11.007
65. Guskey MT, Tsuji BT. A comparative review of the lipoglycopeptides: oritavancin, dalbavancin, and telavancin. Pharmacotherapy. 2010;30(1):80–94. doi:10.1592/phco.30.1.80
66. Stryjewski ME, O’Riordan WD, Lau WK, et al. Telavancin versus standard therapy for treatment of complicated skin and soft-tissue infections due to gram-positive bacteria. Clin Infect Dis. 2005;40(11):1601–1607. doi:10.1086/429914
67. Stryjewski ME, Chu VH, O’Riordan WD, et al. Telavancin versus standard therapy for treatment of complicated skin and skin structure infections caused by gram-positive bacteria: FAST 2 study. Antimicrob Agents Chemother. 2006;50(3):862–867. doi:10.1128/AAC.50.3.862-867.2006
68. Leonard SN, Szeto YG, Zolotarev M, Grigoryan IV. Comparative in vitro activity of telavancin, vancomycin and linezolid against heterogeneously vancomycin-intermediate Staphylococcus aureus (hVISA). Int J Antimicrob Agents. 2011;37(6):558–561. doi:10.1016/j.ijantimicag.2011.02.007
69. Leuthner KD, Cheung CM, Rybak MJ. Comparative activity of the new lipoglycopeptide telavancin in the presence and absence of serum against 50 glycopeptide non-susceptible staphylococci and three vancomycin-resistant Staphylococcus aureus. J Antimicrob Chemother. 2006;58(2):338–343. doi:10.1093/jac/dkl235
70. Laohavaleeson S, Kuti JL, Nicolau DP. Telavancin: a novel lipoglycopeptide for serious gram-positive infections. Expert Opin Investig Drugs. 2007;16(3):347–357. doi:10.1517/13543784.16.3.347
71. Stryjewski ME, Graham DR, Wilson SE, et al. Telavancin versus vancomycin for the treatment of complicated skin and skin-structure infections caused by gram-positive organisms. Clin Infect Dis. 2008;46(11):1683–1693. doi:10.1086/587896
72. Rubinstein E, Lalani T, Corey GR, et al. Telavancin versus vancomycin for hospital-acquired pneumonia due to gram-positive pathogens. Clin Infect Dis. 2011;52(1):31–40. doi:10.1093/cid/ciq031
73. Stryjewski ME, Lentnek A, O’Riordan W, et al. A randomized Phase 2 trial of telavancin versus standard therapy in patients with uncomplicated Staphylococcus aureus bacteremia: the ASSURE study. BMC Infect Dis. 2014;14:289. doi:10.1186/1471-2334-14-289
74. Polyzos KA, Mavros MN, Vardakas KZ, et al. Efficacy and safety of telavancin in clinical trials: a systematic review and meta-analysis. PLoS One. 2012;7(8):e41870. doi:10.1371/journal.pone.0041870
75. Torres A, Rubinstein E, Corey GR, Stryjewski ME, Barriere SL. Analysis of Phase 3 telavancin nosocomial pneumonia data excluding patients with severe renal impairment and acute renal failure. J Antimicrob Chemother. 2014;69(4):1119–1126. doi:10.1093/jac/dkt490
76. Bosso JA, Nappi J, Rudisill C, et al. Relationship between vancomycin trough concentrations and nephrotoxicity: a prospective multicenter trial. Antimicrob Agents Chemother. 2011;55(12):5475–5479. doi:10.1128/AAC.00168-11
77. Dunbar LM, Milata J, McClure T, Wasilewski MM, Team SS. Comparison of the efficacy and safety of oritavancin front-loaded dosing regimens to daily dosing: an analysis of the SIMPLIFI trial. Antimicrob Agents Chemother. 2011;55(7):3476–3484. doi:10.1128/AAC.00029-11
78. Corey GR, Kabler H, Mehra P, et al. Single-dose oritavancin in the treatment of acute bacterial skin infections. N Engl J Med. 2014;370(23):2180–2190. doi:10.1056/NEJMoa1310422
79. Corey GR, Good S, Jiang H, et al. Single-dose oritavancin versus 7–10 days of vancomycin in the treatment of gram-positive acute bacterial skin and skin structure infections: the SOLO II noninferiority study. Clin Infect Dis. 2015;60(2):254–262. doi:10.1093/cid/ciu778
80. Corey GR, Arhin FF, Wikler MA, et al. Pooled analysis of single-dose oritavancin in the treatment of acute bacterial skin and skin-structure infections caused by Gram-positive pathogens, including a large patient subset with methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents. 2016;48(5):528–534. doi:10.1016/j.ijantimicag.2016.07.019
81. Seltzer E, Dorr MB, Goldstein BP, et al. Once-weekly dalbavancin versus standard-of-care antimicrobial regimens for treatment of skin and soft-tissue infections. Clin Infect Dis. 2003;37(10):1298–1303. doi:10.1086/379015
82. Boucher HW, Wilcox M, Talbot GH, et al. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370(23):2169–2179. doi:10.1056/NEJMoa1310480
83. Dunne MW, Puttagunta S, Giordano P, et al. A Randomized Clinical Trial of Single-Dose Versus Weekly Dalbavancin for Treatment of Acute Bacterial Skin and Skin Structure Infection. Clin Infect Dis. 2016;62(5):545–551. doi:10.1093/cid/civ982
84. Raad I, Darouiche R, Vazquez J, et al. Efficacy and safety of weekly dalbavancin therapy for catheter-related bloodstream infection caused by gram-positive pathogens. Clin Infect Dis. 2005;40(3):374–380. doi:10.1086/427283
85. Dunne MW, Puttagunta S, Sprenger CR, et al. Extended-duration dosing and distribution of dalbavancin into bone and articular tissue. Antimicrob Agents Chemother. 2015;59(4):1849–1855. doi:10.1128/AAC.04550-14
86. de la Cruz M, Gonzalez I, Parish CA, et al. Production of Ramoplanin and Ramoplanin Analogs by Actinomycetes. Front Microbiol. 2017;8:343. doi:10.3389/fmicb.2017.00343
87. Hamburger JB, Hoertz AJ, Lee A, et al. A crystal structure of a dimer of the antibiotic ramoplanin illustrates membrane positioning and a potential Lipid II docking interface. Proc Natl Acad Sci U S A. 2009;106(33):13759–13764. doi:10.1073/pnas.0904686106
88. Cheng M, Huang JX, Ramu S, Butler MS, Cooper MA. Ramoplanin at bactericidal concentrations induces bacterial membrane depolarization in Staphylococcus aureus. Antimicrob Agents Chemother. 2014;58(11):6819–6827. doi:10.1128/AAC.00061-14
89. Montecalvo MA. Ramoplanin: a novel antimicrobial agent with the potential to prevent vancomycin-resistant enterococcal infection in high-risk patients. J Antimicrob Chemother. 2003;51(Suppl 3):iii31–iii35. doi:10.1093/jac/dkg274
90. Wong MT, Kauffman CA, Standiford HC, et al. Effective suppression of vancomycin-resistant Enterococcus species in asymptomatic gastrointestinal carriers by a novel glycolipodepsipeptide, ramoplanin. Clin Infect Dis. 2001;33(9):1476–1482. doi:10.1086/322687
91. Bonner DP, O’Sullivan J, Tanaka SK, Clark JM, Whitney RR. Lysobactin, a novel antibacterial agent produced by Lysobacter sp. II. Biological properties. J Antibiot (Tokyo). 1988;41(12):1745–1751. doi:10.7164/antibiotics.41.1745
92. Economou NJ, Cocklin S, Loll PJ. High-resolution crystal structure reveals molecular details of target recognition by bacitracin. Proc Natl Acad Sci U S A. 2013;110(35):14207–14212. doi:10.1073/pnas.1308268110
93. Kim SJ, Singh M, Wohlrab A, et al. The isotridecanyl side chain of plusbacin-A3 is essential for the transglycosylase inhibition of peptidoglycan biosynthesis. Biochemistry. 2013;52(11):1973–1979. doi:10.1021/bi4000222
94. Schneider T, Gries K, Josten M, et al. The lipopeptide antibiotic Friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob Agents Chemother. 2009;53(4):1610–1618. doi:10.1128/AAC.01040-08
95. Ling LL, Schneider T, Peoples AJ, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517(7535):455–459. doi:10.1038/nature14098
96. Smith L, Hillman J. Therapeutic potential of type A (I) lantibiotics, a group of cationic peptide antibiotics. Curr Opin Microbiol. 2008;11(5):401–408. doi:10.1016/j.mib.2008.09.008
97. Rogers LA, Whittier EO. Limiting Factors in the Lactic Fermentation. J Bacteriol. 1928;16(4):211–229.
98. Delves-Broughton J, Blackburn P, Evans RJ, Hugenholtz J. Applications of the bacteriocin, nisin. Antonie Van Leeuwenhoek. 1996;69(2):193–202.
99. Cotter PD, Hill C, Ross RP. Bacteriocins: developing innate immunity for food. Nat Rev Microbiol. 2005;3(10):777–788. doi:10.1038/nrmicro1273
100. Karakas Sen A, Narbad A, Horn N, et al. Post-translational modification of nisin. The involvement of NisB in the dehydration process. Eur J Biochem. 1999;261(2):524–532. doi:10.1046/j.1432-1327.1999.00303.x
101. Wiedemann I, Breukink E, van Kraaij C, et al. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J Biol Chem. 2001;276(3):1772–1779. doi:10.1074/jbc.M006770200
102. Medeiros-Silva J, Jekhmane S, Paioni AL, et al. High-resolution NMR studies of antibiotics in cellular membranes. Nat Commun. 2018;9(1):3963. doi:10.1038/s41467-018-06314-x
103. Medeiros-Silva J, Jekhmane S, Breukink E, Weingarth M. Towards the native binding modes of Lipid II targeting antibiotics. Chembiochem. 2019. doi:10.1002/cbic.201800796
104. Scherer KM, Spille JH, Sahl HG, Grein F, Kubitscheck U. The lantibiotic nisin induces lipid II aggregation, causing membrane instability and vesicle budding. Biophys J. 2015;108(5):1114–1124. doi:10.1016/j.bpj.2015.01.020
105. Oppedijk SF, Martin NI, Breukink E. Hit ‘em where it hurts: the growing and structurally diverse family of peptides that target lipid-II. Biochim Biophys Acta. 2016;1858(5):947–957. doi:10.1016/j.bbamem.2015.10.024
106. Kramer NE, Smid EJ, Kok J, et al. Resistance of Gram-positive bacteria to nisin is not determined by lipid II levels. FEMS Microbiol Lett. 2004;239(1):157–161. doi:10.1016/j.femsle.2004.08.033
107. Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3(3):238–250. doi:10.1038/nrmicro1098
108. Lehrer RI, Barton A, Daher KA, et al. Interaction of human defensins with Escherichia coli. Mechanism of bactericidal activity. J Clin Invest. 1989;84(2):553–561. doi:10.1172/JCI114198
109. Hadjicharalambous C, Sheynis T, Jelinek R, et al. Mechanisms of alpha-defensin bactericidal action: comparative membrane disruption by Cryptdin-4 and its disulfide-null analogue. Biochemistry. 2008;47(47):12626–12634. doi:10.1021/bi800335e
110. de Leeuw E, Burks SR, Li X, Kao JP, Lu W. Structure-dependent functional properties of human defensin 5. FEBS Lett. 2007;581(3):515–520. doi:10.1016/j.febslet.2006.12.036
111. Ericksen B, Wu Z, Lu W, Lehrer RI. Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob Agents Chemother. 2005;49(1):269–275. doi:10.1128/AAC.49.1.269-275.2005
112. Zou G, de Leeuw E, Li C, et al. Toward understanding the cationicity of defensins: ARG and LYS versus their noncoded analogs. J Biol Chem. 2007. doi:10.1074/jbc.M611003200.
113. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3(9):710–720. doi:10.1038/nri1180
114. Sass V, Schneider T, Wilmes M, et al. Human beta-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect Immun. 2010;78(6):2793–2800. doi:10.1128/IAI.00688-09
115. Maisetta G, Batoni G, Esin S, et al. Activity of human beta-defensin 3 alone or combined with other antimicrobial agents against oral bacteria. Antimicrob Agents Chemother. 2003;47(10):3349–3351. doi:10.1128/aac.47.10.3349-3351.2003
116. Pazger M, Wei G, Ericksen B, et al. Sometimes It Takes Two to Tango: Contributions of dimerization to functions of human alpha-defensin HNP1 Peptide. J Biol Chem. 2012;287(12):8944–8953. doi:10.1074/jbc.M111.332205
117. Pazgiera M, Hoover DM, Yang D, Lu W, Lubkowski J. Human beta-defensins. Cell Mol Life Sci. 2006;63(11):1294–1313. doi:10.1007/s00018-005-5540-2
118. Wei G, de Leeuw E, Pazgier M, et al. Through the looking glass, mechanistic insights from enantiomeric human defensins. J Biol Chem. 2009;284(42):29180–29192. doi:10.1074/jbc.M109.018085
119. Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu Rev Immunol. 2004;22:181–215. doi:10.1146/annurev.immunol.22.012703.104603
120. Andes D, Craig W, Nielsen LA, Kristensen HH. In vivo pharmacodynamic characterization of a novel plectasin antibiotic, NZ2114, in a murine infection model. Antimicrob Agents Chemother. 2009;53(7):3003–3009. doi:10.1128/AAC.01584-08
121. Jiao J, Mao R, Teng D, et al. In vitro and in vivo antibacterial effect of NZ2114 against Streptococcus suis type 2 infection in mice peritonitis models. AMB Express. 2017;7(1):44. doi:10.1186/s13568-017-0347-8
122. Klein K, Gronnemose RB, Alm M, et al. Controlled Release of Plectasin NZ2114 from a Hybrid Silicone-Hydrogel Material for Inhibition of Staphylococcus aureus Biofilm. Antimicrob Agents Chemother. 2017;61(7). doi:10.1128/AAC.00604-17.
123. de Leeuw EP. Efficacy of the small molecule inhibitor of Lipid II BAS00127538 against acinetobacter baumannii. Drug Des Devel Ther. 2014;8:1061–1064. doi:10.2147/DDDT.S68020
124. Fletcher S, Yu W, Huang J, et al. Structure-activity exploration of a small-molecule Lipid II inhibitor. Drug Des Devel Ther. 2015;9:2383–2394. doi:10.2147/DDDT.S79504
125. Srinivasan A, Dick JD, Perl TM. Vancomycin resistance in staphylococci. Clin Microbiol Rev. 2002;15(3):430–438. doi:10.1128/cmr.15.3.430-438.2002
126. O’Driscoll T, Crank CW. Vancomycin-resistant enterococcal infections: epidemiology, clinical manifestations, and optimal management. Infect Drug Resist. 2015;8:217–230. doi:10.2147/IDR.S54125
127. Bender JK, Fleige C, Klare I, et al. Detection of a cfr(B) variant in German Enterococcus faecium clinical isolates and the impact on linezolid resistance in enterococcus spp. PLoS One. 2016;11(11):e0167042. doi:10.1371/journal.pone.0167042
128. Fiedler S, Bender JK, Klare I, et al. Tigecycline resistance in clinical isolates of Enterococcus faecium is mediated by an upregulation of plasmid-encoded tetracycline determinants tet(L) and tet(M). J Antimicrob Chemother. 2016;71(4):871–881. doi:10.1093/jac/dkv420
129. Derouaux A, Turk S, Olrichs NK, et al. Small molecule inhibitors of peptidoglycan synthesis targeting the lipid II precursor. Biochem Pharmacol. 2011;81(9):1098–1105. doi:10.1016/j.bcp.2011.02.008
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.