Back to Journals » International Journal of Women's Health » Volume 17
The Sensitive Genes for Cervical Cancer: Two-Sample Mendelian Randomization with Experimental Validation
Authors Zhang R, Chai S, Chen Q, Lai J, Cai C
Received 31 January 2025
Accepted for publication 14 May 2025
Published 26 May 2025 Volume 2025:17 Pages 1511—1532
DOI https://doi.org/10.2147/IJWH.S516444
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Matteo Frigerio
Rong Zhang,1– 4,* Shengjun Chai,1– 4,* Qihang Chen,1– 5 Jiaming Lai,1– 4,6 Chunmei Cai1– 4
1Research Center for High Altitude Medicine, Qinghai University Medical College, Xining, Qinghai, People’s Republic of China; 2Key Laboratory of the Ministry of High Altitude Medicine, Qinghai University Medical College, Xining, Qinghai, People’s Republic of China; 3Key Laboratory of Applied Fundamentals of High Altitude Medicine, (Qinghai-Utah Joint Key Laboratory of Plateau Medicine), Qinghai University Medical College, Xining, Qinghai, People’s Republic of China; 4Laboratory for High Altitude Medicine of Qinghai Province, Qinghai University Medical College, Xining, Qinghai, People’s Republic of China; 5Department of Biotechnology, College of Ecological and Environmental Engineering, Qinghai University, Xining, People’s Republic of China; 6Physical Engineering Department, The Fifth People’s Hospital of Qinghai Provincial (Provincial Cancer Hospital), Xining, Qinghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chunmei Cai, Email [email protected]
Background: Cervical cancer, the fourth leading cause of female cancer mortality globally, faces treatment limitations due to drug resistance and few therapeutic options. This study seeks to identify novel therapeutic targets to address this urgent clinical need.
Methods: Our team identified differentially expressed genes (DEGs) in cervical cancer using gene expression omnibus (GEO) datasets. Subsequently, Mendelian randomization (MR) analysis identified causal gene–cancer relationships, followed by enrichment analysis and The Cancer Genome Atlas (TCGA) validation. Finally, we further validated the functions of the selected target genes in cervical cancer cells and analyzed their Gene Set Enrichment Analysis (GSEA) results, drug sensitivity, and prognostic value.
Results: We identified 2,801 upregulated and 1,646 downregulated DEGs. MR analysis identified 21 key cervical cancer-associated genes (14 upregulated, 7 downregulated), with TCGA validation confirming significant differential expression patterns. Among them, few studies have examined these core genes, particularly MERTK and SERPINF1, in cervical cancer. Experiments showed that MERTK and SERPINF1 play a role in cervical cancer. These genes help cancer cells grow, spread, and invade surrounding tissue. Mechanistically, MERTK regulates immune infiltration, whereas SERPINF1 modulates chromosomal activity. Clinically, SERPINF1 enhances overall survival (OS), disease-specific survival (DSS), and progression-free interval (PFI) in individuals with cervical cancer. Moreover, we discovered that several commonly used drugs for cervical cancer treatment, like paclitaxel, showed high efficacy against MERTK and SERPINF1.
Conclusion: Our study uncovers MERTK and SERPINF1 as critical regulators of cervical cancer progression and survival, offering mechanistic insights into their roles in tumor behavior and the immune microenvironment. These findings provide a foundation for precision therapies, with SERPINF1 restoration and MERTK inhibition as promising strategies. Clinical translation of these targets could address current treatment limitations.
Keywords: Mendelian randomization, cervical cancer, MERTK, SERPINF1, experimental validation
Introduction
According to GLOBOCAN data, there were approximately 661,021 new cervical cancer cases worldwide (the age-standardized rate (ASR): 14.1%) and 348,189 deaths (ASR: 7.1%) in recent years.1,2 Low-income countries showed higher rates than high-income nations (19.3 vs 12.1 per 100,000 incidence; 12.4 vs 4.8 mortality).1 Between 1990 and 2021, cases increased 62.9% (400,000 to 600,000), while deaths rose 40.2% (200,000 to 280,000).3 However, age-standardized rates declined annually (the age-standardized incidence rate (ASIR): −0.5%; age-standardized mortality rate (ASMR): −1.3%) during this period.3 Countries with strong screening programs (Sweden, UK, Australia, US) maintained low rates (~10/100,000) in women under 35.4 Conversely, nations with limited screening (Colombia, India, Uganda) saw rates climb sharply with age (peaking at 50–190/100,000).4 Following 5–9 years of human papillomavirus (HPV) vaccination, cervical intraepithelial neoplasia grade 2 or worse (CIN2+) cases fell substantially (15–19y: 51%; 20–24y: 31%).5 As CIN2+ is cancer’s direct precursor, this decline predicts future cervical cancer reductions.
Current cervical cancer treatment faces several significant challenges. The two main subtypes – squamous cell carcinoma (SCC, 75%) and adenocarcinoma (AC, 20%) – demonstrate different clinical behaviors.6–8 AC patients show worse survival outcomes than SCC patients regardless of receiving radiotherapy or chemoradiotherapy.9,10 Drug resistance remains a primary cause of mortality in cervical cancer cases. Studies reveal long non-coding RNA (lncRNA) and circular RNA (circRNA)-mediated competing endogenous RNA (ceRNA) networks promote resistance to cisplatin, paclitaxel and 5-fluorouracil through multiple pathways, such as miR-5047/vascular endothelial growth factor (VEGFA), miR-432-5p/myeloid cell leukemia sequence 1 (MCL1), miR-206/Cyclin D2 (CCND2), and miR-130a-3p-YTH Domain-Containing Family Protein 2(YTHDF2) pathways.11–13 Targeted therapies can help overcome this chemoresistance – bevacizumab improves cisplatin sensitivity by inhibiting aldehyde dehydrogenase 1 (ALDH1).14 However, resistance to targeted agents also occurs, such as PD173074 resistance via fibroblast growth factor receptors (FGFRs) overexpression.15 Many targeted drug resistance mechanisms remain poorly understood.16 Approved therapies like bevacizumab, immunotherapies and antibody-drug conjugates represent major advances.17 Unfortunately, bevacizumab’s high cost restricts access in low-resource settings, programmed cell death protein 1 (PD-1) inhibitors like pembrolizumab show promise but require more clinical validation.18 These challenges – including cost, resistance and limited availability – highlight the need for better biomarkers and novel therapies.
Growing evidence underscores the importance of genetic factors in cervical cancer development and progression. Research shows advanced cases display distinct genetic changes in keratinization and glycosylation pathways involving TP53 compared to early-stage tumors.19 The disease also features alterations in 15 homologous recombination-directed repair (HDR) genes and multiple mismatch repair (MMR) genes, with MMR variants more common in AC and HDR changes predominant in SCC.20 Genome-wide association studies (GWAS) studies have pinpointed several susceptibility loci linked to HPV infection, including HLA-DRB1, complement component 4B (C4B), and HLA-DRB6.21–23 Comprehensive genomic analyses have further classified cervical cancers into molecular subtypes.24 While Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases provide mutation profiles, gene-phenotype associations (eg, uncoupling protein 2(UCP2), leptin receptor (LEPR) with prognosis) often remain correlative rather than proven.25–27 This highlights the urgent need for validated genetic biomarkers to address disease heterogeneity. Currently, limited genetic data and insufficient Mendelian randomization (MR) validation studies hinder clinical translation of these genetic findings.
Cervical cancer treatment faces significant therapeutic challenges and translational barriers in clinical genetics. To address this, we applied two-sample MR to establish causal gene expression-cancer risk relationships. Our integrated approach combines MR analysis with functional validation to identify promising therapeutic targets. We focused on MERTK and SERPINF1, two understudied genes implicated in immune regulation and tumor suppression. This study establishes a novel “genetics-to-function” pipeline bridging causal inference with mechanistic validation. Unlike correlation-based studies, our method provides clinically actionable insights for precision oncology while overcoming traditional biomarker limitations. The integrated framework accelerates target discovery by merging unbiased genetic evidence with experimental confirmation.
Materials and Methods
Ethics
All data used in this study were legally obtained from public databases in anonymized form. In accordance with Items 1 and 2 of Article 32 of China’s “Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects” (effective February 18, 2023), this study qualifies for exemption from ethical review.
Data Collection
As of March 20, 2024, Gene expression datasets and clinical phenotype data related to the keywords “cervical cancer”, “Homo sapiens”, and “gene expression” were obtained through the analysis of microarray datasets. All relevant gene expression data and corresponding platform probe annotations can be downloaded from the GEO datasets (https://www.ncbi.nlm.nih.gov/geo/). The selection criteria for cervical cancer-related datasets include (i) a minimum of six samples, with at least three cases and three controls; (ii) samples that have not been subjected to chemical treatment or genetic modification; and (iii) the availability of raw data or array gene expression profile analyses in the GEO database. Ultimately, three gene expression microarray datasets containing cervical cancer samples were included GSE122697, GSE127265, and GSE166466.
Detection of DEGs
These three datasets were preprocessed and normalized using R language (version 4.2.2), with individual dataset corrections applied. The merged dataset, consisting of 15 normal and 34 cervical cancer samples, was subjected to batch correction and differential expression analysis. The threshold of |log2FoldChange| >0.585 (1.5-fold change) with both raw P-value <0.05 and false discovery rate <0.05 was selected to optimally balance sensitivity and specificity in detecting biologically relevant DEGs while maintaining statistical rigor. These significant DEGs were subsequently visualized using the “pheatmap” package in R, which generated: (1) a volcano plot displaying the top 50 most significantly upregulated and downregulated DEGs and (2) a heatmap demonstrating the expression patterns of DEGs across all samples. All data normalization and standardization were conducted based on gene expression matrices and annotation files obtained from the GEO database. Principal Component Analysis (PCA) was performed using the “prcomp” function to mitigate batch effects and enhance visualization. PCA corrects the data set into PCA ridge map to realize the “dimension reduction” processing of diversified data, thereby aiding further identification and validation of key genes that distinguish cervical cancer samples from controls.
Expression Quantitative Trait Loci (eQTLs) Analysis of Exposure Data
EQTL datasets were utilized to investigate which Mendelian genetic variants were associated with the expression levels of these DEGs. The most comprehensive meta-analysis of eQTL data to date was conducted by Westra et al, covering peripheral blood eQTL data from 5311 Europeans.28 The eQTL data used in this study were retrieved from the GWAS Catalog website (https://gwas.mrcieu.ac.uk/). All SNPs used for MR analysis must satisfy three fundamental assumptions: (1) Relevance Assumption: The instrumental variable (SNP) must show significant association with the exposure factor. (2) Independence Assumption: The instrumental variable must be independent of confounding factors (3) Exclusion Restriction Assumption: The instrumental variable can only influence the outcome through the exposure factor, with no alternative direct or indirect pathways. R language was used to include SNPs strongly associated (P < 5 × 10^-8) with DEGs. SNPs with weak associations (F-statistic < 10) or limited explanatory power for phenotypic variation were systematically excluded to satisfy the instrumental variable relevance assumption in MR analysis. To meet the independence assumption, we removed SNPs potentially associated with the outcome (P < 5 × 10^-5). Stringent quality control measures included: (1) setting linkage disequilibrium parameters at r² <0.001, and (2) requiring all candidate SNPs to be located within ±1000 kb of DEG genomic loci, thereby ensuring compliance with MR’s exclusion restriction criteria.
Ascertainment of Outcome Data
The outcome data were derived from the genetic association database provided by the GWAS Catalog (https://gwas.mrcieu.ac.uk/) in the form of GWAS summary datasets (IEU). The specific GWAS ID utilized in this study was ebi-a-GCST90018817, which yielded 909 cervical cancer samples and 238,249 control cases, including a total of 24,138,337 SNPs. All GWAS summary statistics cited in this study are publicly accessible and available for download.
Two-Sample MR Analysis
The “TwoSampleMR” software package was employed to conduct MR analysis using the Inverse Variance Weighted (IVW) method to investigate the causal association between specific genes and cervical cancer. Additional MR-Egger, simple mode, weighted median, and weighted mode methods were also used to estimate causal effects.29,30 The identification of disease-associated genes followed a three-step criterion: (i) Initially, genes with p-values less than 0.05 in the IVW method were selected; (ii) Genes were further refined based on the consistency of the direction of results (odds ratios) across a minimum of three different MR analysis methods; (iii) Pleiotropic genes with p-values less than 0.05 were excluded. Throughout this process, cross-analysis identified co-expressed genes between disease-associated genes and DEGs, including both upregulated and downregulated genes. Subsequently, MR analysis was conducted on all intersecting genes to determine their causal relationships with the disease. Multiple auxiliary tests were employed to determine the usability of the cross-analysis results. Heterogeneity analysis was used to assess the variability of the instrumental variables, pleiotropy testing was conducted to detect the pleiotropic effects of the genetics, and MR leave-one-out sensitivity analysis was performed to evaluate whether any single SNP had a significant impact on the results. The findings from these analyses were then visualized using funnel plots, scatter plots, and forest plots, respectively. The P-values for both the heterogeneity analysis and pleiotropy testing of these genes were >0.05, and the MR leave-one-out sensitivity analysis indicated that removing any single SNP had little effect on the MR results. Therefore, our findings are robust and reliable.
GO and KEGG Enrichment Analyses
The “clusterProfiler” R package, with a filtering criterion of a P-value of less than 0.05, was employed for functional annotation of co-expressed genes using GO terms and KEGG pathways. GO analysis categorizes gene functions into three major classifications: molecular function (MF), biological process (BP), and cellular component (CC). It evaluates the function of co-expressed genes by statistically assessing the enrichment level of gene sets in GO terms. KEGG primarily investigates the involvement of co-expressed genes across various pathways. Finally, the importance of pathways is represented by the number of enriched genes and their P-values.
Immune Cell Analysis
CIBERSORT was used to analyze the infiltration levels of 22 immune cell types in cervical cancer. Specifically, the proportion of each immune cell type in every sample was evaluated, and differences in their abundance between healthy and disease groups were compared. Additionally, we examined the correlation between co-expressed genes in cervical cancer and the infiltration of these immune cells.
External Validation of the TCGA Database
R software (version 4.2.2) was utilized to verify the differences in co-expressed genes between the tumor and normal tissue samples of patients with cervical cancer, retrieved from the TCGA database that was as an external validation cohort. These findings from the TCGA database were then compared with MR analysis results.
Construction of the PPI Network and Nomogram Models
The STRING website (https://string-db.org/) and the GeneMANIA database (https://GeneMANIA.org/) were employed to construct the PPI network. The STRING platform focuses on utilizing the latest data to predict the interactions among co-expressed genes. Subsequently, Cytoscape tool (version 3.10.2) is utilized to extract core targets from the PPI network and visualize these interactions. GeneMANIA is primarily used to explore the interactions between co-expressed genes and other proteins, hypothesizing gene functions and determining functional priorities. To clarify the role of cross genes in clinical applications, the “rms” software package was used to construct a nomogram model with receiver operating characteristic (ROC) analysis. The nomogram was employed to investigate the predictive priority of several key targets in cervical cancer, while ROC curves were used to examine the diagnostic value of core genes in cervical cancer. Additionally, calibration curves were applied to determine the consistency between predicted values and actual observations.
Cell Culture
Human cervical cancer cell lines were purchased from China Xiamen Immocell Biotechnology, including SiHa (#IM-H012) and HeLa (#IM-H010). All cells were cultured in DMEM medium (#C11995500BT, Gibco) supplemented with 10% fetal bovine serum (FBS) (#10099141C, Gibco), penicillin (100 U/mL), and streptomycin (100 μg/mL) (#10378016, Gibco). The incubator was maintained at a temperature of 37°C with a 5% CO2 (Thermo Fisher Scientific).
Cell Transfection
Seed cells to be 60–80% confluent at transfection. Dilute Lipofectamine RNAiMAX Reagent (Invitrogen, USA) in Opti-MEM Medium (#11058021, Gibco), adjusting the final concentration of Lipofectamine RNAiMAX to 6%. Dilute small interfering RNAs (siRNAs) targeting MERTK (si-MERTK) and SERPINF1 (si-SERPINF1) (Sangon Biotech, China) in Opti-MEM Medium, adjusting the siRNA concentration to 0.2 pmol/μL. Add diluted siRNA to diluted Lipofectamine RNAiMAX Reagent (1:1 ratio). Incubate for 5 minutes at room temperature. Add siRNA-lipid complex to cells. siRNA sequences were as follows:
Control siRNA sense: 5′- UUCUCCGAACGUGUCACGUdTdT-3′
MERTK siRNA sense: 5′-CCAUCUACAUCGAAGUACAdTdT-3′
SERPINF1 siRNA sense: 5′- CCCGGAUCGUCUUUGAGAAdTdT-3′
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from cervical cancer cells using Trizol reagent (Invitrogen, USA). Following the addition of HiScript II Q RT SuperMix for qPCR (+gDNA wiper) (#R223-01, Vazyme) to the RNA, cDNA reverse transcription was completed in two steps. Finally, 2× RealStar Green Power Mixture (#A311-10 Genstar) was utilized to perform qRT-PCR. Expression data were normalized to the endogenous reference gene RPL13A to control for variability in expression levels. Relative quantification calculations were performed using the 2−ΔΔCT method. Primer sequences used in this study were as follows:
RPL13A-F: 5′- GCCATCGTGGCTAAACAGGTA-3′
RPL13A-R: 5′- GTTGGTGTTCATCCGCTTGC-3′
MERTK-F: 5′- CAAACTGACCACACACCGCT-3′
MERTK-R: 5′- CTGGTACCCACTGGCGTGAG-3′
SERPINF1-F: 5′- AGTTATGAAGGCGAAGTCACCAAG-3′
SERPINF1-R: 5′- CCATCCTCGTTCCACTCAAAGC-3′
Cell Proliferation Assay
Cells were seeded at a density of 1 × 104 cells per well in a 96-well plate (Corning, USA). The transfection reagents were added to the adherent cells. MTS assays were then performed on days 0, 1, 2, 3, 4, and 5 as follows: thawing CellTiter96 AQueous One Solution Reagent (#G3580, Promega, USA), and 20 μL of the reagent was added to each well containing 100 μL of culture medium. The plates were incubated at 37°C, 5% CO2 for 1–4 hours, and the absorbance was measured at 490 nm using a microplate spectrophotometer (Tecan, Switzerland).
Wound Healing Assay
1 × 106 cells were seeded per well in a 6-well cell culture plate (Corning, USA) using the complete medium overnight. Once the cells reached 100% confluence, wounds were created through the monolayers by using a sterile pipette tip. The cells were then rinsed multiple times with phosphate buffered saline to clear away debris and cultured in a medium containing 2% FBS. Healing was observed via microscopy (ZEISS) and quantified using the ImageJ software.
Transwell Migration and Invasion Assay
Cell migration and invasion assays were conducted using a 24-well cell culture plate with transwell chambers (3422, Corning, USA). For the invasion assay, Matrigel (#356234, Corning, USA) was applied to the upper chamber to mimic the extracellular matrix. After resuspending the cells in DMEM medium, 1 × 106 cells were added to the upper chamber, while the lower chamber contained complete culture medium. After 24 hours of incubation, cells were fixed with 4% paraformaldehyde and subsequently stained with crystal violet. Five random fields were captured via microscopy (ZEISS) and quantified using the ImageJ software.
GSEA Enrichment Analysis
The Gene Set Enrichment Analysis (GSEA) was employed to investigate the pathways potentially associated with the varying expression levels of MERTK and SERPINF1 on cervical cancer phenotypes. Pathways were deemed significantly enriched when |NES| > 1, P < 0.05, and FDR < 0.25.
Drug Sensitivity Analysis
RNAactDrug (http://bio-bigdata.hrbmu.edu.cn/RNAactDrug) is a comprehensive database of RNAs associated with drug sensitivity derived from multi-omics data, such as GDSC, CellMiner, and CCLE datasets.31 The RNAactDrug database was utilized to conduct a comprehensive analysis for screening drugs that are sensitive to MERTK and SERPINF1. Depending on the characteristics of the data, either Pearson or Spearman correlation analysis was selected for evaluation.
Prognostic Analysis
Clinical information on cervical cancer and mRNA expression data were collected for MERTK and SERPINF1 from the TCGA dataset (https://portal.gdc.com). To ensure optimal fit with minimal error, lasso regression was employed as a filtering method to select genes associated with clinical prognosis in cervical cancer. Diagnostic performance analysis was conducted for the co-expressed genes through fitting modeling. Overfitting was prevented by incorporating a penalty term. Using the R package “survival”, a univariate Cox regression model was constructed. The prognostic value of MERTK and SERPINF1 in cervical cancer was assessed through three clinical outcomes: overall survival (OS), disease-specific survival (DSS), and progression-free interval (PFI).
Statistical Analysis
All data were analyzed using GraphPad Prism 8.0 (GraphPad Software) and presented as “mean ± standard deviation (SD)”. Comparisons between the two groups were performed using the t-test, while multiple comparisons between groups were analyzed using Two-way ANOVA. A P-value of less than 0.05 was considered statistically significant.
Results
Datasets Normalization and DEGs Identification
Through a comparative search of the GEO datasets, three gene expression microarrays were included in our study: GSE122697, GSE127265, and GSE166466 (Table 1), which comprised a total of 15 control samples (5, 3, and 7 samples, respectively) and 34 disease samples (14, 7, and 13 samples, respectively). Using R version 4.2.2, we normalized and merged the expression values of each gene across the datasets and then employed PCA to mitigate batch effects. Before bath correction, there exists significant variation between the three datasets along both first principal component (PC1) and second principal component (PC2) (Supplementary Figure 1A). After correction, the samples from each dataset cluster closely along the PC1 and the PC2, indicating that all samples within the datasets achieved acceptable homogeneity (Supplementary Figure 1B).
 |
Table 1 Characteristic of the Three GEO Datasets |
The differential expression analysis revealed that a smaller p-value indicates higher reliability in gene ranking to identify DEGs. Ultimately, we identified 2,801 upregulated DEGs and 1,646 downregulated DEGs between the control and disease groups (Supplementary Table 1). The volcano plot exhibited a near-symmetric distribution around the center, suggesting a comparable predominance of these two gene categories in cervical cancer (Supplementary Figure 1C). The heatmap represented the detailed information regarding top 50 upregulated and top 50 downregulated DEGs across the samples (Supplementary Figure 1D).
MR Analysis
To investigate the causal relationship between DEGs and cervical cancer, we conducted a MR analysis. According to the screening criteria, we identified 26,152 SNPs associated with cervical cancer (Supplementary Table 2). Given that the F-statistics for all these SNPs exceeded the threshold of 10, they met our pre-defined selection criteria and showed no signs of linkage disequilibrium (r2 < 0.001). As a result, these SNPs were not regarded as weak instrumental factors. Subsequently, employing the two-sample MR analysis based on the above chosen SNPs, we identified 212 cervical cancer-associated genes (Supplementary Table 3). Through cross-comparative analysis, we discovered 21 co-expressed genes, comprising 14 upregulated genes (Figure 1A, left) and 7 downregulated genes (Figure 1A, right).
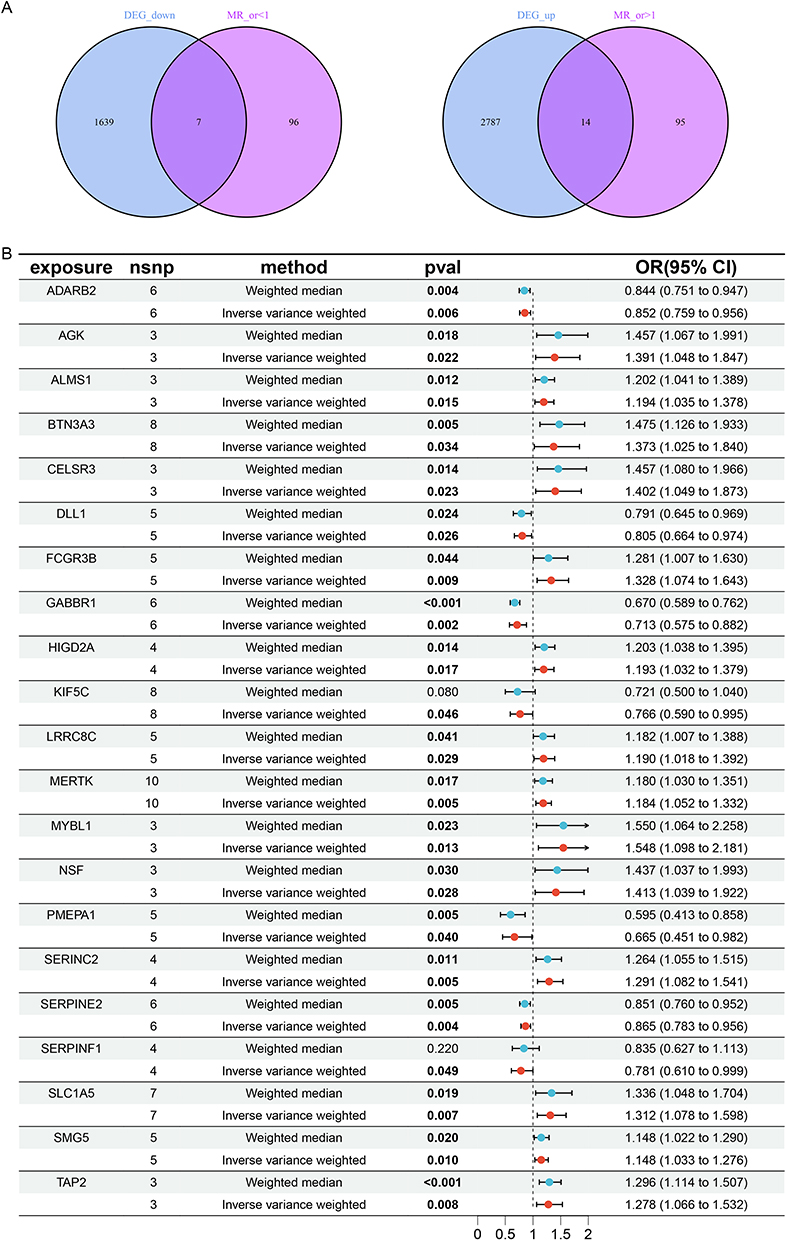 |
Figure 1 Co-expressed genes identified through cross-comparison. (A) Upregulated and downregulated genes generated through cross-comparison. (B) MR of the 21 intersecting genes. (The bold text represent statistically significant differences (p< 0.05). |
The primary MR analysis using the IVW approach demonstrated that all 14 upregulated co-expressed genes have a significant positive causal association with cervical cancer (Figure 1B). Specifically, the odds ratios (ORs) with their corresponding 95% confidence intervals (CIs) were as follows: AGK (OR = 1.391, 95% CI: [1.048–1.847], P = 0.022), ALMS1 (OR = 1.194, 95% CI: [1.035–1.378], P = 0.015), BTN3A3 (OR = 1.373, 95% CI: [1.025–1.840], P = 0.034), CELSR3 (OR = 1.402, 95% CI: [1.049–1.873], P = 0.023), FCGR3B (OR = 1.328, 95% CI: [1.074–1.643], P = 0.009), HIGD2A (OR = 1.193, 95% CI: [1.032–1.379], P = 0.017), LRRC8C (OR = 1.190, 95% CI: [1.018–1.392], P = 0.029), MERTK (OR = 1.184, 95% CI: [1.052–1.332], P = 0.005), MYBL1 (OR = 1.548, 95% CI: [1.098–2.181], P = 0.013), NSF (OR = 1.413, 95% CI: [1.039–1.922], P = 0.028), SERINC2 (OR = 1.291, 95% CI: [1.082–1.541], P = 0.005), SLC1A5 (OR = 1.312, 95% CI: [1.078–1.598], P = 0.007), SMG5 (OR = 1.148, 95% CI: [1.033–1.276], P = 0.022), TAP2 (OR = 1.278, 95% CI: [1.066–1.532], P = 0.008). Thus, these 14 upregulated genes exhibited as risk factors for cervical cancer. Meanwhile, 7 downregulated genes represented protective effects against cervical cancer (Figure 1B): ADARB2 (OR = 0.852, 95% CI: [0.759–0.956], P = 0.006), DLL1 (OR = 0.805, 95% CI: [0.664–0.974], P = 0.026), GABBR1 (OR = 0.713, 95% CI: [0.575–0.882], P = 0.002), KIF5C (OR = 0.766, 95% CI: [0.590–0.995], P = 0.046), PMEPA1 (OR = 0.665, 95% CI: [0.451–0.982], P = 0.040), SERPINE2 (OR = 0.865, 95% CI: [0.783–0.956], P = 0.004), SERPINF1 (OR = 0.781, 95% CI: [0.610–0.999], P = 0.049). These findings implicated that the 7 downregulated genes might play a crucial role in impeding cervical cancer development.
In addition, MR-Egger, simple mode, weighted median, and weighted mode were also used to estimate causal effects for further validation. All methods consistently indicated an increased risk of cervical cancer (OR > 1) for the 14 upregulated genes, and a decreased risk of cervical cancer (OR < 1) for the 7 downregulated genes (Figure 1B). Plots of the leave-one-out analysis (Supplementary Figure 2) indicated that there were no potentially influential SNP driving the causal link. Heterogeneity assessments were performed using Cochran’s Q statistic and MR-PRESSO test, revealing no statistical significance for these co-expressed genes (Supplementary Figure 2). The funnel plots were consistent with heterogeneity analysis (Supplementary Figure 2). Furthermore, none of these co-expressed genes showed significant directional horizontal pleiotropy according to the MR-Egger regression intercept analysis (Supplementary Figure 2). The tests for heterogeneity and pleiotropy for these genes all yielded p-values greater than 0.05, indicating that there were no potentially influential SNP driving the causal link and the results were robust.
GO/KEGG Enrichment Analysis
To further elucidate the chromosomal distribution of the aforementioned genes, we performed a gene localization analysis, visualized using a circus diagram (Figure 2A). Subsequently, we conducted GO and KEGG analyses to explore the functions and pathways related to the co-expressed genes. The GO analysis indicated that the 21 intersecting genes are primarily enriched in the BP among the three major functional categories (MF, BP, and CC) (Figure 2B). The bubble plot reveals that the co-expressed genes were mainly involved in dicarboxylic acid transport, amino acid transport, retina development in the camera-type eye, and neuronal cell body. The KEGG enrichment analysis identified that the co-expressed genes predominantly affected GABAergic synapse and phagosome (Figure 2C). Detailed data regarding these analyses can be found in Supplementary Table 4.
 |
Figure 2 Genomic location, enrichment analysis, and immune infiltration of the co-expressed genes. (A) Circos plot showing the genomic distribution of the co-expressed genes. (B and C) GO and KEGG enrichment analysis of the intersecting genes. (D) Bar plot of immune cell infiltration. (E) Differential analysis of immune cells. (F) Correlation analysis of the shared genes with immune features. (*p< 0.05, **p< 0.01). |
Evaluation of Immune Cell Infiltration in Cervical Cancer
The GO enrichment analysis delineated that the intersecting genes were also involved in IgG binding, suggesting a potential contribution of immune system in cervical cancer. Immune infiltration assessment were conducted to unveil the immune cell infiltration and its correlation with co-expressed genes in cervical cancer. The bar graph provided the proportions of 22 different immune cell types across all individuals (Figure 2D). The violin plot further revealed that compared to healthy individuals, Macrophages M0 exhibited higher infiltration levels in individuals with cervical cancer (P < 0.05), while Mast cells resting were lower (P < 0.01) (Figure 2E). Furthermore, the correlation analysis showed that TAP2 was positively correlated with T follicular helper cells (Tfhs), macrophages M1, and dendritic cells activated, while negatively with regulatory T cells (Tregs); CELSR3 was positively associated with NK cells activated, mast cells activated, and Macrophages M0, whereas negatively with plasma cells, T cells gamma delta, and mast cells resting; SLC1A5 was positively related to Tfhs, macrophages M1, dendritic cells activated, and neutrophils, while negatively with monocytes; SMG5 was positively correlated with Tfhs cells and dendritic cells activated, while negatively with monocytes; NSF exhibited a negative correlation with Tregs and NK cells resting; HIGD2A was positively related to T cells gamma delta, macrophages M2, and dendritic cells activated, while negatively with Tregs and NK cells resting; BTN3A3 was positively associated with T cells gamma delta and macrophages M2, while negatively with monocytes; AGK was positively related with T cells gamma delta, memory CD4 T cells resting, and mast cells resting, but negatively with mast cells activated; ALMS1 was positively correlated with CD4 T cells naive; LRRC8C showed a positive correlation with plasma cells; MERTK was positively associated with NK cells activated and Macrophages M0; FCGR3B was positively correlated with macrophages M1, while negatively with NK cells resting and monocytes; SERINC2 was positively associated with macrophages M1, mast cells activated, and neutrophils, but negatively with monocytes; KIF5C showed a positive correlation with NK cells resting, while negative with macrophages M0 and mast cells activated; SERPINE2 was positively correlated with monocytes, while negatively with macrophages M0; PMEPA1 was positively related to dendritic cells activated and negatively with Tregs; and SERPINF1 showed a negative correlation with eosinophils (Figure 2F). Besides, MYBL1, DLL1, GABBR1, and ADARB2 exhibited no significant correlation with various immune cells.
Importantly, consistent with immune infiltration analysis, macrophage M0 was positively correlated with CELSR3 and LRRC8C, exhibited as risk factors for cervical cancer, while negatively associated with KIF5C and SERPINE2, displayed protective effect against cervical cancer. In addition, mast cells resting was negatively associated with CELSR3, risk factor for cervical cancer, also supporting the findings of immune infiltration analysis. Yet, the mast cells resting was positively related to AGK, as a risk factor for cervical cancer. In conclusion, except MYBL1, DLL1, GABBR1, and ADARB2, other 17 co-expressed can affect the occurrence and progression of cervical cancer through regulating the immune responses, especially macrophages M0 and mast cells resting. It should be noted that given the complexity of cervical cancer pathogenesis, immune infiltration is merely one of the mechanisms involved, the co-expressed genes may adopt other significant mechanisms to modulate this process.
Validation of the TCGA Database
To validate the results of our MR analysis, the TCGA database was used to assess the differential expression of 21 co-expressed genes between the cervical cancer and adjacent non-cancerous tissue samples. CELSR3, MYBL1, SERINC2, SLC1A5, SMG5, and TAP2 were found to be upregulated in cervical cancer compared to controls, while GABBR1 and SERPINF1 were downregulated (P < 0.05) (Figure 3A–H). The other genes showed no significant differences in their expression between the cervical cancer and adjacent non-cancerous tissue samples (Supplementary Figure 3). These findings are consistent with our MR results, thereby confirming the reliability of our MR analysis through external data validation.
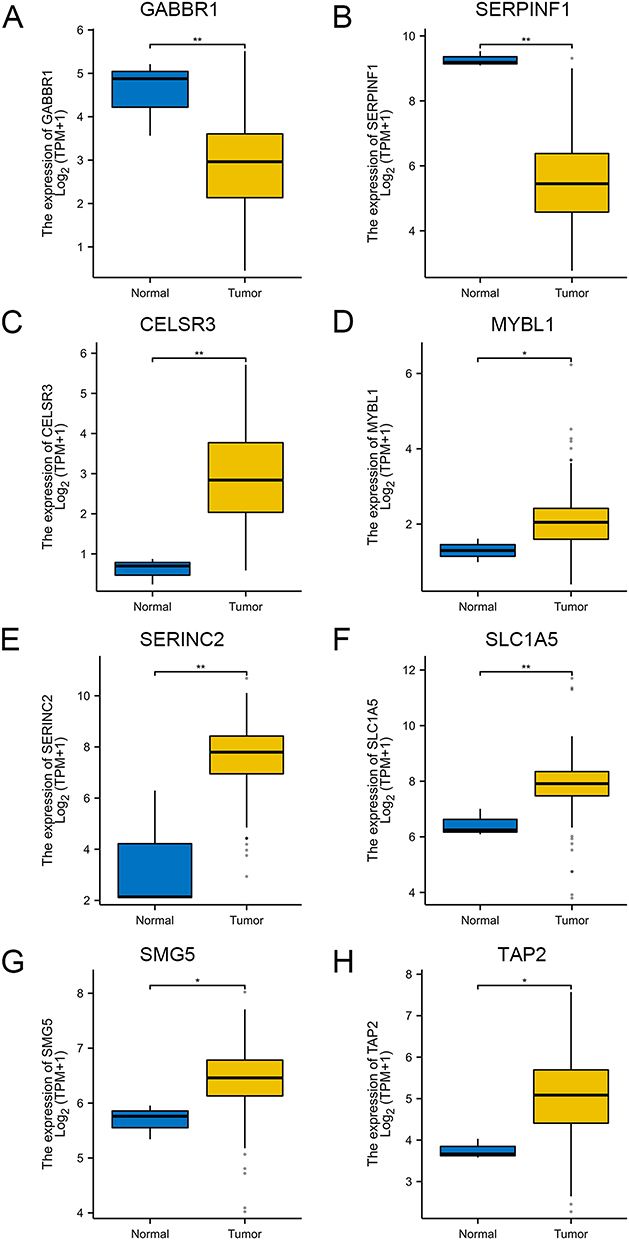 |
Figure 3 Validation of the expression levels of eight co-expressed genes in the TCGA database. (A) Box plot of differential gene expression of GABBR1 between normal group and cervical cancer group. (B) Box plot of differential gene expression of SERPINF1 between normal group and cervical cancer group. (C) Box plot of differential gene expression of CELSR3 between normal group and cervical cancer group. (D) Box plot of differential gene expression of MYBL1 between normal group and cervical cancer group. (E) Box plot of differential gene expression of SERINC2 between normal group and cervical cancer group. (F) Box plot of differential gene expression of SLC1A5 between normal group and cervical cancer group. (G) Box plot of differential gene expression of SMG5 between normal group and cervical cancer group. (H) Box plot of differential gene expression of TAP2 between normal group and cervical cancer group. (*p< 0.05, **p< 0.01). |
Construction of PPI Networks and Nomogram Model
To elucidate the crosstalk among co-expressed gene proteins, STRING was utilized to construct a PPI network of 21 co-expressed genes (Figure 4A) and GeneMANIA to develop another PPI network, including 21 co-expressed genes and additional 20 potential interacting genes (Figure 4B). According to STRING analysis, we observed that AGK interacted with ADARB2; GABBR1 interacted with KIF5C, NSF, and CELSR3; and TAP2, BTN3A3, FCGR3B, LRRC8C, DLL1, MERTK, ALMS1, SERPINF1, and SERPINE2 exhibited various interactions among themselves, while the remaining 6 co-expressed genes did not show any interactions, thus excluded from diagram (Figure 4A). Among them, MERTK exhibited as the most prominent node, interacting with four intersecting genes to generate ten connections and encompassing three distinct functions. The GeneMANIA report revealed that TAP2, TAP1 and TIMM22 were highlighted as being associated with peptide transmembrane transporter activity, aligning with the findings of the GO enrichment analysis (Figure 4B).
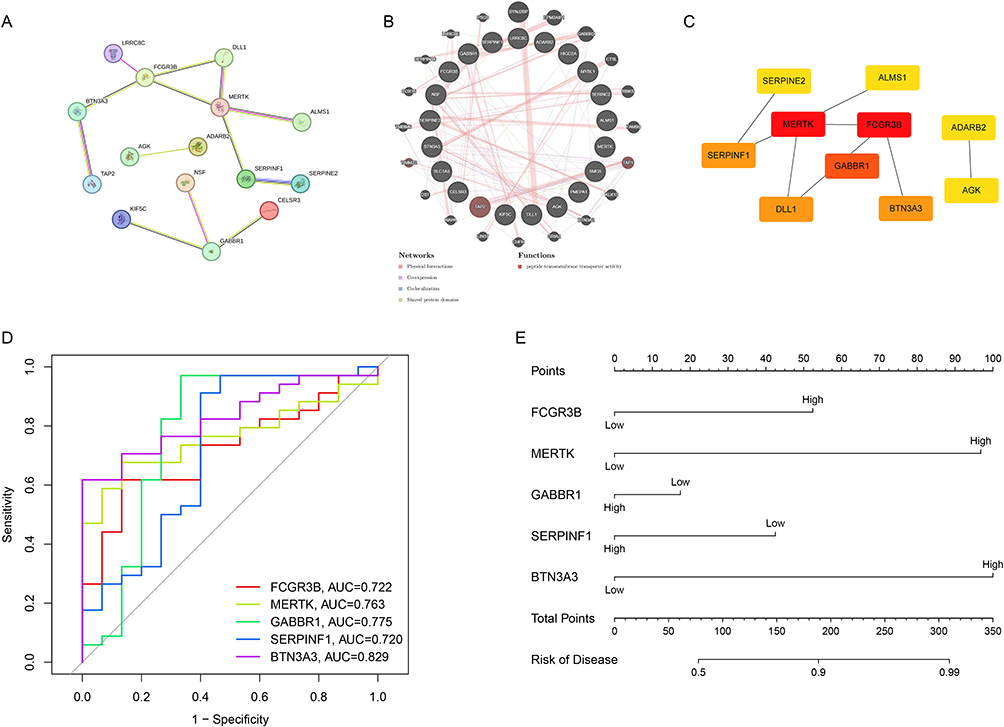 |
Figure 4 PPI analysis and clinical significance of co-expressed genes. (A) Interactions among the shared genes. (B) PPI network of the intersecting genes and other proteins. (C) Hub genes within the PPI network. (D) ROC curve illustrating the diagnostic value of the intersecting genes for cervical cancer. (E) Nomogram for predicting the risk of cervical cancer based on co-expressed genes. |
Based on the PPI analysis of the intersecting genes, the Cytoscape tool was utilized to extract 10 core genes (Figure 4C). The genes FCGR3B, MERTK, GABBR1, SERPINF1, BTN3A3, and DLL1 were specifically identified and highlighted. The color intensity corresponded to the magnitude of the scores, with darker hues indicating higher values. We then developed an R-based clinical prediction model to elucidate the clinical relevance of the core genes. We calculated the area under the curve (AUC) of ROC curves for the top 5 hub genes to assess their diagnostic performance. The AUC values were as follows: BTN3A3 (AUC = 0.829), GABBR1 (AUC = 0.775), MERTK (AUC = 0.763), FCGR3B (AUC = 0.722), and SERPINF1 (AUC = 0.720) (Figure 4D). Finally, we developed a nomogram to predict the risk of cervical cancer occurrence associated with these five central genes (Figures 4E and S4). Our results showed that the nomogram model, incorporated these 5 core genes, exhibited a satisfactory capability in predicting the risk of cervical cancer occurrence. Notably, there existed a certain degree of prediction bias: at low-risk levels, the risk of cervical cancer occurrence was overestimated, while at high-risk levels, the risk was underestimated (Supplementary Figure 4). Additionally, MERTK and BTN3A3 emerged as strong predictors for cervical cancer occurrence, with their expression levels positively correlated with the risk of disease development (Figure 4E). This suggested their potential as valuable biomarkers for the diagnosis of cervical cancer.
In vitro Functional Investigation into the Contribution of MERTK and SERPINF1 in Cervical Cancer
The cellular functional experiments were then conducted to further investigate the effects of the top five hub genes on cervical cancer. Given the experimental challenges associated with FCGR3B, GABBR1, and BTN3A3, which are all cell membrane proteins involved in the interactions between a variety of components within the tumor microenvironment, we focused on the functions of MERTK and SERPINF1 on cervical cancer cells. Currently, the role of MERTK on cervical cancer remains largely elusive. SERPINF1 is known to function primarily through its transcript, pigment epithelium-derived factor (PEDF). PEDF has been shown to indirectly inhibit cervical cancer through its anti-angiogenic properties, but its direct inhibitory effects have not been thoroughly explored.32,33 CC and AC represent the most prevalent histological subtypes of cervical cancer, with the majority being HPV-positive.6–8 The SiHa and HeLa cell lines, derived from HPV-positive CC and AC, respectively, serve as representative models for the predominant forms of cervical cancer. Thus, functional studies were performed to assess the phenotypic effects of siRNA-mediated knockdown of MERTK and SERPINF1 in cervical cancer cell lines SiHa and HeLa.
After transfecting the two cervical cancer cell lines with siRNAs targeting MERTK and SERPINF1, the knockdown efficiency was verified by measuring the RNA expression levels (Figures 5A and 6A). MTS assays revealed that compared to the control, knockdown of MERTK remarkably reduced the proliferation of HeLa cells (P < 0.05) but did not affect SiHa cells (P > 0.05) (Figure 5B), while SERPINF1 downregulation had no significant effects on their proliferative activity (P > 0.05) (Figure 6B), suggesting the promotive role of MERTK in proliferation of HeLa cells. In the scratch assay, the downregulation of MERTK slowed the migration of both cervical cancer cell lines compared to the control group (P < 0.05) (Figure 5C), whereas silencing SERPINF1 obviously facilitated the wound healing (P < 0.05) (Figure 6C), implicating the promotive and inhibitory roles of MERTK and SERPINF1 in the migration of cervical cancer cells, respectively. Transwell experiments further evaluated the migration ability of SiHa and HeLa cells, revealing that MERTK knockdown reduced, while SERPINF1 silencing enhanced the migration and invasion capabilities of both cell lines (P < 0.05) (Figures 5D and 6D), consistent with the scratch assay results. It was indicated by our findings that MERTK was a risk factor for cervical cancer through the promotion of proliferation, migration and invasion of cervical cancer cells, while SERPINF1 was exhibited as a protective factor against migration and invasion, which was consistent with our MR predictions.
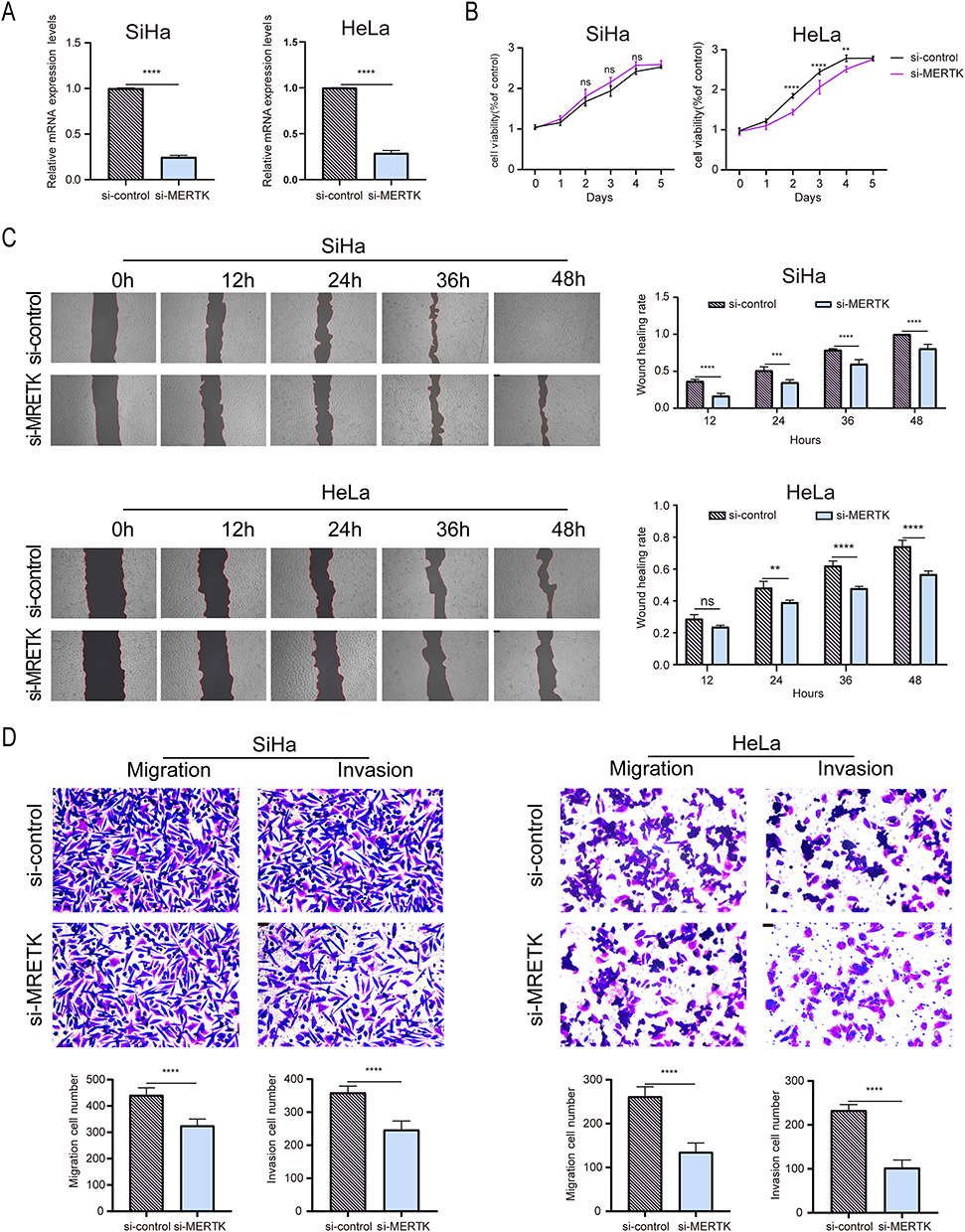 |
Figure 5 Functional assays of MERTK in cervical cancer cells. (A) mRNA levels following MERTK knockdown with siRNA(n=3). (B) Assessment of the impact of MERTK knockdown on the proliferation capacity of cervical cancer cells using the MTS assay(n=3). (C) Evaluation of the migratory ability of SiHa and HeLa cells following low expression of MERTK through a scratch assay(n=3). (D) Assessment of the effects of MERTK silencing on the migratory and invasive capabilities of cervical cancer cells using transwell migration and invasion assays(n=3). Data are presented as mean±SD. (**p< 0.01, ***p< 0.001, ****p< 0.0001). Abbreviation: ns, not significant. |
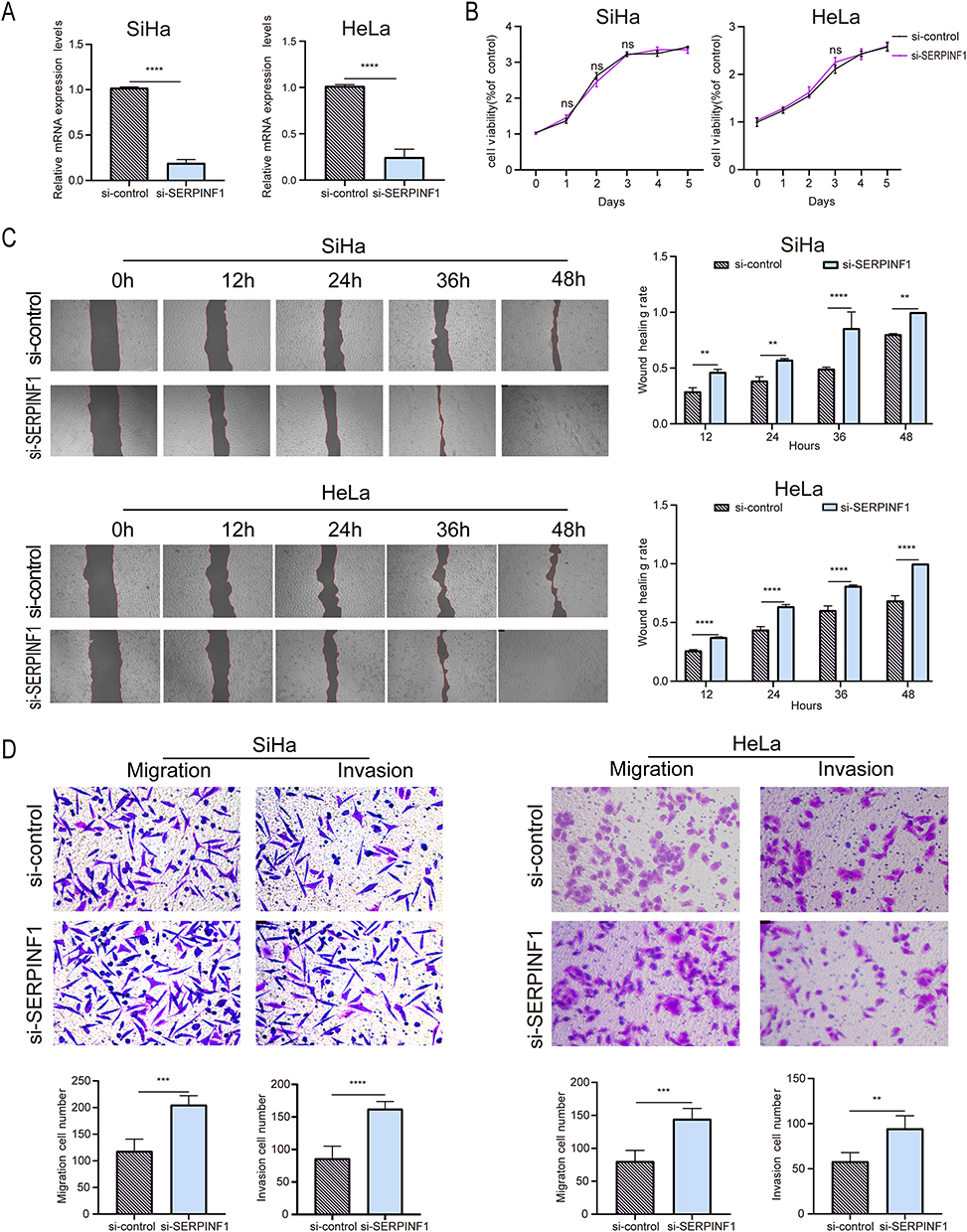 |
Figure 6 Functional assays of SERPINF1 in cervical cancer cells. (A) Validation of SERPINF1 mRNA levels following siRNA-mediated knockdown(n=3). (B) Evaluation of the changes in proliferation capacity of SiHa and HeLa cells after SERPINF1 knockdown using the MTS assay(n=3). (C) Assessment of the impact of low SERPINF1 expression on the migratory ability of SiHa and HeLa cells through a scratch assay(n=3). (D) Transwell migration and invasion assays of SiHa and HeLa cells with SERPINF1 silencing (n=3). Data are presented as mean±SD. (**p< 0.01, ***p< 0.001, ****p< 0.0001). Abbreviation: ns, not significant. |
GSEA Enrichment Analysis
GSEA enrichment analysis was performed to further explore which functions or pathways associated with MERTK and SERPINF1 were enriched at the top or bottom of the ranking, indicating trends of upregulation or downregulation, respectively. The high expression of SERPINF1 was primarily correlated with innate immune responses, such as cell adhesion molecules, chemokine signaling pathways, cytokines, intestinal immune networks, and neutrophil chemotaxis and migration (Figure 7A and C). Conversely, the low expression of SERPINF1 was related to gene activity and cell cycle-related processes, including homologous recombination, DNA replication, nucleotide excision repair, mismatch repair, and chromosome segregation (Figure 7B and D). Additionally, high expression of MERTK was associated with immune responses, including exogenous antigen presentation, B-cell receptor signaling pathway, antigen receptor-mediated signaling pathway, regulation of immune response by receptor binding, MHC protein complex binding, and allograft rejection, Leishmania infection, and systemic lupus erythematosus (Figure 7E and G). In contrast, low expression of MERTK was primarily associated with keratinocyte differentiation in the skin (Figure 7F). These insights provided a novel perspective on the roles of these genes in physiological and pathological processes in cervical cancer.
 |
Figure 7 GSEA enrichment analysis of MERTK and SERPINF1 expression levels. (A) GO enrichment analysis in high SERPINF1 group. (B) GO enrichment analysis in low SERPINF1 group. (C) KEGG enrichment analysis in high SERPINF1 group. (D) KEGG enrichment analysis in low SERPINF1 group. (E) GO enrichment analysis in high MERTK group. (F) GO enrichment analysis in low MERTK group. (G) KEGG enrichment analysis in high MERTK group. |
Drug Sensitivity Analysis of MERTK and SERPINF1
Furthermore, drug sensitivity analyses were conducted to explore the relationship between commonly used clinical anticancer drugs and the expression levels of MERTK and SERPINF1. MERTK overexpression was associated with increased sensitivity to a greater number of drugs (Supplementary Figure 5A), while the expression level of SERPINF1 was inversely correlated with the half-maximal inhibitory concentrations (IC50) of several drugs (Supplementary Figure 5B). This indicated that these anticancer drugs could effectively target the upregulated MERTK and downregulated SERPINF1 observed in cervical cancer, as determined by MR analysis. Supplementary Tables 5 and 6 listed the drugs to which MERTK and SERPINF1 exhibited sensitive, respectively. Specifically, the group with high MERTK expression exhibited increased sensitivity to irinotecan, temsirolimus, and topotecan (Figure 8A–C); conversely, the SERPINF1 low expression group showed greater sensitivity to 5-Fluorouracil, paclitaxel, and piperlongumine (Figure 8D–G). Importantly, the irinotecan, temsirolimus, topotecan, 5-Fluorouracil, paclitaxel, and piperlongumine were commonly utilized in clinical practice for cervical cancer treatment. These findings held substantial significance for guiding clinical medication practices to optimize treatment plans and enhance therapeutic efficacy.
 |
Figure 8 Sensitivity analysis of MERTK and SERPINF1 to commonly used anti-cervical cancer drugs. (A) Sensitivity correlation analysis of MERTK and Irinotecan. (B) Sensitivity correlation analysis of MERTK and Temsirolimus. (C) Sensitivity correlation analysis of MERTK and Topotecan. (D) Sensitivity correlation analysis of SERPINF1 and 5-Fluorouracil. (E) Sensitivity correlation analysis of SERPINF1 and Paclitaxel. (F) Sensitivity correlation analysis of SERPINF1 and Piperlongumine. (G) Sensitivity correlation analysis of SERPINF1 and Vinblastine. |
Effect of MERTK and SERPINF1 on the Prognosis of Cervical Cancer
To further elucidate the potential clinical utility of MERTK and SERPINF1 as biomarkers, we evaluated their association with OS, DSS, and PFI. The LASSO regression fitting analysis revealed that when the deviance reached its minimum (indicating optimal model fit), 14 genes including SERPINF1 can be selected for subsequent LASSO regression analysis (Supplementary Figure 5C). Detailed parameters are provided in Supplementary Table 7A. At the point of best model fit, LASSO regression identified SERPINF1 as having a significant impact on cervical cancer (coefficient = −0.33) (Supplementary Figure 5D). Coefficients for all genes are presented in Supplementary Table 7B. SERPINF1 expression levels showed a significant association with clinical outcomes (Figure 9A–C). The HR (95% CI) for OS, DSS, and PFI in the high SERPINF1 expression group compared to the low expression group were 0.48 (0.30–0.77), 0.46 (0.27–0.79), and 0.47 (0.30–0.76), respectively. The p-values for these comparisons were 0.002, 0.005, and 0.002, respectively, suggesting that high SERPINF1 expression was significantly associated with improved OS, DSS, and PFI. Thus, high SERPINF1 expression was associated with significantly better survival outcomes in individuals with cervical cancer. The upregulated SERPINF1 emerged as a promising biomarker for predicting OS, DSS, and PFI in cervical cancer.
 |
Figure 9 Clinical prognosis analysis of SERPINF1. (A) Correlation analysis between SERPINF1 expression and overall survival in cervical cancer. (B) Correlation analysis between SERPINF1 expression and disease specific survival in cervical cancer. (C) Correlation analysis between SERPINF1 expression and progress free interval in cervical cancer. |
Discussion
In this study, we employed bioinformatics analysis to identify potential therapeutic targets for cervical cancer from a genetic perspective. MR analysis revealed 21 co-expressed genes significantly associated with cervical cancer. These genes are predominantly involved in amino acid and dicarboxylic acid transport as well as retinal development, underscoring their potential role in cancer progression. Importantly, the external validation through the TCGA database confirmed differential expression of these co-expressed genes in cervical cancer tissues, further supporting the reliability of our MR results. PPI analysis further revealed five most significantly associated genes (FCGR3B, MERTK, GABBR1, SERPINF1, and BTN3A3), which may play a crucial role in the occurrence and development of cervical cancer. Except GABBR1 and MERTK, the involvement of other hub genes has been reported in cervical cancer. The current findings indicate that FCGR3B is a risk factor for cervical cancer, associated with immune infiltration.34,35 BTN3A3 gene is demethylated and activated by HPV pathogenic genes in cervical cancer.36 SERPINF1 inhibits the progression of cervical cancer through its transcript PEDF.37,38
Our integrated analysis demonstrates that MERTK, a TAM family receptor tyrosine kinase, promotes cervical cancer progression through both direct tumorigenic effects and immune modulation, as evidenced by cellular experiments showing that MERTK knockdown significantly enhances cancer cell proliferation, migration and invasion. Numerous studies have shown that MERTK is known to facilitate cancer progression through mechanisms like exocytosis and immune modulation.39–42 It has been associated with poorer prognosis in various cancers. Immune profiling revealed MERTK expression correlates with activated NK cells and M0 macrophages – the latter being substantially elevated in individuals with cervical cancer and previously established as key mediators of an unfavorable immune microenvironment43–53 through associations with hypoxia-inducible factor 1-alpha (HIF-1α),47 diagnostic potential,45 and prognostic value.51 Mechanistically, MERTK is upregulated in M0 macrophages by neutrophil gelatinase-associated lipocalin (NGAL) and transforming growth factor-beta (TGF-β),54–56 driving their differentiation into immunosuppressive M2 phenotypes, while in NK cells, tumor-derived the growth arrest-specific 6(GAS6) activates MERTK to suppress antitumor immunity through Cbl-b-dependent ubiquitination.57,58 Importantly, pharmacological MERTK inhibition reverses these immunosuppressive effects, restoring NK cell cytotoxicity and reducing metastasis,57,58 supporting the therapeutic potential of emerging MERTK-targeted agents including small molecule inhibitors, degraders, and biological traps39 for cervical cancer immunotherapy.
SERPINF1 dysfunction is implicated in over 20 human diseases, with cancer being the most common manifestation, where the SERPINF1-PEDF axis exerts broad anti-tumor effects through multiple mechanisms including inhibition of proliferation, migration and invasion, induction of apoptosis, and promotion of cancer cell differentiation across various malignancies.59–63 While SERPINF1 primarily mediates its anti-cervical cancer effects indirectly through PEDF,37,38 our experimental validation in HeLa and SiHa cells confirmed its direct inhibitory role in migration and invasion, consistent with MR analysis and TCGA data, and demonstrated significant associations with improved OS, DSS, and PFI, supporting its prognostic biomarker potential. Notably, both our findings and previous research33 showed no effect of SERPINF1/PEDF modulation on cervical cancer cell proliferation or apoptosis, suggesting potential divergence in their mechanisms of action. GSEA analysis further revealed that SERPINF1’s anti-tumor activity in cervical cancer may involve base excision repair, mismatch repair, cell cycle regulation, DNA replication, and homologous recombination pathways, mediated through key transcription factors including methyl-CpG binding domain protein 4 (MBD4), high mobility group box 1 (HMGB1), TP53, and E1A Binding Protein p300 (EP300).
Immune cells play a pivotal role in tumor progression through their involvement in anti-inflammatory responses and immune microenvironment modulation.64 Beyond Macrophages M0, our study revealed a downregulation of resting mast cells in cervical cancer, though their exact role remains controversial. While most research-including our findings-suggests an inverse correlation between resting mast cells and cervical cancer malignancy,47,48,65–67 as well as its risk factors (eg, HIF-1α and transmembrane protein 33 (TMEM33)),47,65 with a positive association to OS,67 some studies paradoxically identify them as risk factors.44,45,49 This discrepancy underscores the need for further investigation into their functional impact. Nonetheless, our results highlight the potential of immune cells, including resting mast cells, as inflammatory biomarkers for cervical cancer prevention and therapeutic strategies.
While this study advances our understanding of MERTK and SERPINF1 in cervical cancer, several limitations must be addressed: (1) The modest sample size (15 normal vs 34 tumor samples) in initial screening may yield false-positive associations, necessitating validation in larger cohorts; (2) Technical constraints precluded experimental validation of bioinformatics-predicted roles for Macrophages M0 and resting mast cells in tumor progression; (3) While SiHa/HeLa cell assays provided mechanistic insights, the absence of in vivo models and clinical tissue validation limits translational relevance; (4) Focus on HPV18+ CC and HPV16+ AC subtypes overlooks potential heterogeneity in other subtypes;6–8 and (5) The exact mechanisms of MERTK/SERPINF1 action—particularly whether SERPINF1 operates via PEDF—remain unclear. Future studies addressing these gaps will strengthen the clinical applicability of these findings.
In conclusion, the molecular mechanisms of cervical cancer have been further elucidated through this study, with evidence being provided for the potential utility of MERTK and SERPINF1 as both therapeutic targets and prognostic indicators. The exploration of their roles and mechanisms, along with the identification of drugs sensitive to these genes provide a promising direction for the development of precision medicine in cervical cancer. Despite these advancements, our findings remain preliminary and require further validation through additional experiments and clinical trials to confirm the mechanisms and therapeutic potential of these genes.
Abbreviations
AC, adenocarcinoma; ALDH1, aldehyde dehydrogenase 1; ASIR, age-standardized incidence rate; ASMR, age-standardized mortality rate; ASR, age-standardized rate; AUC, area under the curve; BP, biological processes; C4B, complement component 4B; CC, cellular components; CCND2, Cyclin D2; ceRNA, endogenous RNA; CIN2+, cervical intraepithelial neoplasia grade 2 or worse; circRNA, circular RNA; DEGs, differentially expressed genes; DSS, disease-specific survival; EP300, E1A Binding Protein p300; eQTL, expression quantitative trait loci; FGFRs, fibroblast growth factor receptors; GAS6, growth arrest-specific 6; GEO, Gene Expression Omnibus; GO, Gene Ontology; GWAS, genome-wide association studies; HDR, homologous recombination-directed repair; HIF-1α, hypoxia-inducible factor 1-alpha; HLA-A, human leukocyte antigen-A; HMGB1, high mobility group box 1; HPV, human papillomavirus; HR, hazard ratios; IVW, Inverse Variance Weighted; KEGG, Kyoto Encyclopedia of Genes and Genomes; LEPR, leptin receptor; lncRNA, long non-coding RNA; LogFC, logFoldChange; MBD4, methyl-CpG binding domain protein 4; MCL1, myeloid cell leukemia sequence 1; MF, molecular function; MMR, mismatch repair; MR, Mendelian Randomization; NGAL, neutrophil gelatinase-associated lipocalin; OS, overall survival; PCA, Principal Component Analysis; PD-1, programmed cell death protein 1; PEDF, pigment epithelium-derived factor; PFI, progression-free interval; PPI, protein–protein interaction; qRT-PCR, Quantitative real-time polymerase chain reaction; ROC, receiver operator characteristic; SCC, squamous cell carcinoma; SD, standard deviation; SHKBP1, SH3KBP1-binding protein 1; SNP, polymorphisms; TGFBR2, Transforming growth factor-beta receptor II; TGF-β, transforming growth factor-beta; TMEM33, transmembrane protein 33; UCP2, uncoupling protein 2; VEGF, vascular endothelial growth factor; YTHDF2, YTH Domain-Containing Family Protein 2.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Acknowledgment
Rong Zhang and Shengjun Chai are co-first authors for this study. This work was supported by the Qinghai University Research Ability Enhancement Project (2025KTSQ01).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The Qinghai University Research Ability Enhancement Project (2025KTSQ01).
Disclosure
The authors declare no competing interests in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–263. doi:10.3322/caac.21834
2. Filho AM, Laversanne M, Ferlay J, et al. The GLOBOCAN 2022 cancer estimates: data sources, methods, and a snapshot of the cancer burden worldwide. Int J Cancer. 2025;156:1336–1346. doi:10.1002/ijc.35278
3. Li Y, Song W, Gao P, et al. Global, regional, and national burden of breast, cervical, uterine, and ovarian cancer and their risk factors among women from 1990 to 2021, and projections to 2050: findings from the global burden of disease study 2021. BMC Cancer. 2025;25:330. doi:10.1186/s12885-025-13741-9
4. Singh D, Vignat J, Lorenzoni V, et al. Global estimates of incidence and mortality of cervical cancer in 2020: a baseline analysis of the WHO global cervical cancer elimination initiative. Lancet Glob Health. 2023;11:e197–e206. doi:10.1016/S2214-109X(22)00501-0
5. Drolet M, Benard E, Perez N, Brisson M; Group, H.P.V.V.I.S. Population-level impact and herd effects following the introduction of human papillomavirus vaccination programmes: updated systematic review and meta-analysis. Lancet. 2019;394:497–509. doi:10.1016/S0140-6736(19)30298-3
6. Small W, Bacon MA, Bajaj A, et al. Cervical cancer: a global health crisis. Cancer. 2017;123:2404–2412. doi:10.1002/cncr.30667
7. Arezzo F, Cormio G, Loizzi V, et al. HPV-negative cervical cancer: a narrative review. Diagnostics. 2021;11.
8. Shao N. Research progress on human papillomavirus-negative cervical cancer: a review. Medicine. 2024;103:e39957. doi:10.1097/MD.0000000000039957
9. Hu K, Wang W, Liu X, Meng Q, Zhang F. Comparison of treatment outcomes between squamous cell carcinoma and adenocarcinoma of cervix after definitive radiotherapy or concurrent chemoradiotherapy. Radiat Oncol. 2018;13:249. doi:10.1186/s13014-018-1197-5
10. Yokoi E, Mabuchi S, Takahashi R, et al. Impact of histological subtype on survival in patients with locally advanced cervical cancer that were treated with definitive radiotherapy: adenocarcinoma/adenosquamous carcinoma versus squamous cell carcinoma. J Gynecol Oncol. 2017;28:e19. doi:10.3802/jgo.2017.28.e19
11. Manjili DA, Babaei FN, Younesirad T, et al. Dysregulated circular RNA and long non-coding RNA-mediated regulatory competing endogenous RNA networks (ceRNETs) in ovarian and cervical cancers: a non-coding RNA-mediated mechanism of chemotherapeutic resistance with new emerging clinical capacities. Arch Biochem Biophys. 2025;768:110389. doi:10.1016/j.abb.2025.110389
12. Shirani N, Abdi N, Chehelgerdi M, Yaghoobi H, Chehelgerdi M. Investigating the role of exosomal long non-coding RNAs in drug resistance within female reproductive system cancers. Front Cell Dev Biol. 2025;13:1485422. doi:10.3389/fcell.2025.1485422
13. Xia Z, Zhang L, Zhou H, Ran W, Tu J. LncRNA-FGD5-AS1 promotes 5-Fu resistance of cervical cancer cells through modulating the miR-130a-3p-YTHDF2 axis. J Chemother. 2024;1–13. doi:10.1080/1120009X.2024.2436803
14. Qu N, Li Z, Wei J, et al. Bevacizumab increases cisplatin efficacy by inhibiting epithelial-mesenchymal transition via ALDH1 in cervical carcinoma. Int Immunopharmacol. 2025;145:113736. doi:10.1016/j.intimp.2024.113736
15. Bou Antoun N, Afshan Mahmood H-T-N, Walker AJ, et al. Development and characterization of three novel FGFR inhibitor resistant cervical cancer cell lines to help drive cervical cancer research. Int J Mol Sci. 2025;26:1799. doi:10.3390/ijms26051799
16. Meyer M, Fourie C, van der Merwe H, Botha H, Engelbrecht AM. Targeting treatment resistance in cervical cancer: a new avenue for senolytic therapies. Adv Med Sci. 2024;70:33–43. doi:10.1016/j.advms.2024.11.001
17. Mate S. [Medical therapy of cervical cancer]. Magy Onkol. 2022;66:315–323. Czech
18. Gopu P, Antony F, Cyriac S, Karakasis K, Oza AM. Updates on systemic therapy for cervical cancer. Indian J Med Res. 2021;154:293–302. doi:10.4103/ijmr.IJMR_4454_20
19. Hyeon DY, Nam D, Shin H-J, et al. Proteogenomic characterization of molecular and cellular targets for treatment-resistant subtypes in locally advanced cervical cancers. Mol Cancer. 2025;24:77. doi:10.1186/s12943-025-02256-3
20. Kokemuller L, Ramachandran D, Schürmann P, et al. Germline variants of homology-directed repair or mismatch repair genes in cervical cancer. Int J Cancer. 2025;156:700–710. doi:10.1002/ijc.35221
21. Yao Y, Yan Z, Li C, et al. Association of HLA class I and II genes with cervical cancer susceptibility in a Han Chinese population. HLA. 2024;103:e15340. doi:10.1111/tan.15340
22. Beckhaus T, Kachuri L, Nakase T, et al. Genome-Wide Association Analyses of HPV16 and HPV18 Seropositivity Identify Susceptibility Loci for Cervical Cancer. J Med Virol. 2025;97:e70195. doi:10.1002/jmv.70195
23. Eisenblatter R, Seifert F, Schürmann P, et al. Validation and functional follow-up of cervical cancer risk variants at the HLA locus. HLA. 2024;104:e15597. doi:10.1111/tan.15597
24. Burk RD, Chen Z, Saller C, et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378–384. doi:10.1038/nature21386
25. Fang HY, Ji LM, Hong CH. An innovative glutamine metabolism-related gene signature for predicting prognosis and immune landscape in cervical cancer. Discov Oncol. 2025;16:368. doi:10.1007/s12672-025-02109-x
26. Tang R, Yao Q, Zhang K, et al. Revolutionizing prognosis: introducing cell death index (CDI) as a powerful prognostic tool for CSCC patients. Environ Toxicol Int J. 2025;40:481–492. doi:10.1002/tox.24265
27. Li S, Gao K, Yao D. Comprehensive analysis of autophagy associated genes and immune infiltrates in cervical cancer. Iran J Basic Med Sci. 2024;27:813–824. doi:10.22038/IJBMS.2024.74431.16168
28. Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. doi:10.1038/ng.2756
29. Dudbridge F. Polygenic Mendelian randomization. Cold Spring Harb Perspect Med. 2021;11:a039586. doi:10.1101/cshperspect.a039586
30. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318:1925–1926. doi:10.1001/jama.2017.17219
31. Dong Q, Li F, Xu Y, et al. RNAactDrug: a comprehensive database of RNAs associated with drug sensitivity from multi-omics data. Brief Bioinform. 2020;21:2167–2174. doi:10.1093/bib/bbz142
32. Xu B, Li J, Liu X, Li C, Chang X. TXNDC5 is a cervical tumor susceptibility gene that stimulates cell migration, vasculogenic mimicry and angiogenesis by down-regulating SERPINF1 and TRAF1 expression. Oncotarget. 2017;8:91009–91024. doi:10.18632/oncotarget.18857
33. Yang J, Chen S, Huang X, et al. Growth suppression of cervical carcinoma by pigment epithelium-derived factor via anti-angiogenesis. Cancer Biol Ther. 2010;9:967–974. doi:10.4161/cbt.9.12.11635
34. Zhao W, Li Q, Wen S, et al. Novel biomarkers of inflammation-associated immunity in cervical cancer. Front Oncol. 2024;14:1351736. doi:10.3389/fonc.2024.1351736
35. Le TM, Nguyen HDT, Lee E, et al. Transcriptomic immune profiles can represent the tumor immune microenvironment related to the tumor budding histology in uterine cervical cancer. Genes. 2022;13:1405. doi:10.3390/genes13081405
36. Milutin Gasperov N, Farkas SA, Nilsson TK, Grce M. Epigenetic activation of immune genes in cervical cancer. Immunol Lett. 2014;162:256–257. doi:10.1016/j.imlet.2014.09.019
37. Francis MK, Appel S, Meyer C, et al. Loss of EPC-1/PEDF expression during skin aging in vivo. J Invest Dermatol. 2004;122:1096–1105. doi:10.1111/j.0022-202X.2004.22510.x
38. Lanza RP, Cibelli JB, Blackwell C, et al. Extension of cell life-span and telomere length in animals cloned from senescent somatic cells. Science. 2000;288:665–669. doi:10.1126/science.288.5466.665
39. Lahey KC, Gadiyar V, Hill A, et al. Mertk: an emerging target in cancer biology and immuno-oncology. Int Rev Cell Mol Biol. 2022;368:35–59.
40. Aehnlich P, Powell RM, Peeters MJW, Rahbech A, Thor Straten P. TAM receptor inhibition-implications for cancer and the immune system. Cancers. 2021;13:1195. doi:10.3390/cancers13061195
41. Davra V, Kumar S, Geng K, et al. Axl and Mertk receptors cooperate to promote breast cancer progression by combined oncogenic signaling and evasion of host antitumor immunity. Cancer Res. 2021;81:698–712. doi:10.1158/0008-5472.CAN-20-2066
42. Du W, Zhu J, Zeng Y, et al. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Differ. 2021;28:1284–1300. doi:10.1038/s41418-020-00651-5
43. Chen H, Ma R, Zhou B, et al. Integrated immunological analysis of single-cell and bulky tissue transcriptomes reveals the role of interactions between M0 macrophages and naive CD4(+) T cells in the immunosuppressive microenvironment of cervical cancer. Comput Biol Med. 2023;163:107151. doi:10.1016/j.compbiomed.2023.107151
44. Mo X, Wang N, He Z, et al. The sub-molecular characterization identification for cervical cancer. Heliyon. 2023;9:e16873. doi:10.1016/j.heliyon.2023.e16873
45. Liu QF, Feng Z-Y, Jiang -L-L, et al. Immune cell infiltration as signatures for the diagnosis and prognosis of malignant gynecological tumors. Front Cell Dev Biol. 2021;9:702451. doi:10.3389/fcell.2021.702451
46. Wu Z, Lin Q, Sheng L, et al. A novel immune-related risk-scoring system associated with the prognosis and response of individuals with cervical cancer treated with radiation therapy. Front Mol Biosci. 2023;10:1297774. doi:10.3389/fmolb.2023.1297774
47. Li Z, Wei R, Yao S, Meng F, Kong L. HIF-1A as a prognostic biomarker related to invasion, migration and immunosuppression of cervical cancer. Heliyon. 2024;10:e24664. doi:10.1016/j.heliyon.2024.e24664
48. Wang X, Tang W, Lu Y, et al. Prognostic significance of alternative splicing genes in cervical squamous cell carcinoma and endocervical adenocarcinoma. Int J Gen Med. 2021;14:7933–7949. doi:10.2147/IJGM.S335475
49. Yao H, Jiang X, Fu H, et al. Exploration of the immune-related long noncoding RNA prognostic signature and inflammatory microenvironment for cervical cancer. Front Pharmacol. 2022;13:870221. doi:10.3389/fphar.2022.870221
50. Petrillo M, Zannoni GF, Martinelli E, et al. Polarisation of tumor-associated macrophages toward M2 phenotype correlates with poor response to chemoradiation and reduced survival in patients with locally advanced cervical cancer. PLoS One. 2015;10:e0136654. doi:10.1371/journal.pone.0136654
51. Yin R, Zhai X, Han H, et al. Characterizing the landscape of cervical squamous cell carcinoma immune microenvironment by integrating the single-cell transcriptomics and RNA-Seq. Immun Inflamm Dis. 2022;10:e608. doi:10.1002/iid3.608
52. Qiu Q, Zhou Q, Luo A, et al. Integrated analysis of virus and host transcriptomes in cervical cancer in Asian and Western populations. Genomics. 2021;113:1554–1564. doi:10.1016/j.ygeno.2021.03.029
53. Liu J, Yang J, Gao F, et al. A microRNA-messenger RNA regulatory network and its prognostic value in cervical cancer. DNA Cell Biol. 2020;39:1328–1346. doi:10.1089/dna.2020.5590
54. Healy LM, Perron G, Won S-Y, et al. MerTK is a functional regulator of myelin phagocytosis by human myeloid cells. J Immunol. 2016;196:3375–3384. doi:10.4049/jimmunol.1502562
55. Shen D, Chen J. Cardiac macrophages promote polarization of macrophages toward M2 phenotype to improve myocardial remodeling via NGAL after myocardial infarction. Cell Biochem Biophys. 2025. doi:10.1007/s12013-025-01726-1
56. Zizzo G, Cohen PL. The PPAR-gamma antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas6 axis: a key role for PPAR-gamma in human macrophage polarization. J Inflamm. 2015;12(36). doi:10.1186/s12950-015-0081-4
57. Jung SH, Park -S-S, Lim J-Y, et al. Single-cell analysis of multiple myelomas refines the molecular features of bortezomib treatment responsiveness. Exp Mol Med. 2022;54:1967–1978. doi:10.1038/s12276-022-00884-z
58. Paolino M, Choidas A, Wallner S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507:508–512. doi:10.1038/nature12998
59. Becerra SP, Notario V. The effects of PEDF on cancer biology: mechanisms of action and therapeutic potential. Nat Rev Cancer. 2013;13:258–271. doi:10.1038/nrc3484
60. Tan ML, Choong PF, Dass CR. Anti-chondrosarcoma effects of PEDF mediated via molecules important to apoptosis, cell cycling, adhesion and invasion. Biochem Biophys Res Commun. 2010;398:613–618. doi:10.1016/j.bbrc.2010.05.098
61. Yang H, Grossniklaus HE. Constitutive overexpression of pigment epithelium-derived factor inhibition of ocular melanoma growth and metastasis. Invest Ophthalmol Vis Sci. 2010;51:28–34. doi:10.1167/iovs.09-4138
62. Feng Y, Bao W, Luo Y, et al. Phosphomimetic mutants of pigment epithelium-derived factor with enhanced anti-choroidal melanoma cell activity in vitro and in vivo. Invest Ophthalmol Vis Sci. 2012;53:6793–6802. doi:10.1167/iovs.12-10326
63. Orgaz JL, Ladhani O, Hoek KS, et al. Loss of pigment epithelium-derived factor enables migration, invasion and metastatic spread of human melanoma. Oncogene. 2009;28:4147–4161. doi:10.1038/onc.2009.284
64. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79:4557–4566. doi:10.1158/0008-5472.CAN-18-3962
65. Zhang H, Wang J, Yang J, et al. TMEM33 as a prognostic biomarker of cervical cancer and its correlation with immune infiltration. Mediators Inflamm. 2023;2023:5542181. doi:10.1155/2023/5542181
66. Huang J, Luo F, Shi M, et al. Construction and validation of a metabolic gene-associated prognostic model for cervical carcinoma and the role on tumor microenvironment and immunity. Aging. 2021;13:25072–25088. doi:10.18632/aging.203723
67. Yang S, Wu Y, Deng Y, et al. Identification of a prognostic immune signature for cervical cancer to predict survival and response to immune checkpoint inhibitors. Oncoimmunology. 2019;8:e1659094. doi:10.1080/2162402X.2019.1659094
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.