Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
The retinoblastoma protein: a master tumor suppressor acts as a link between cell cycle and cell adhesion
Authors Engel B, Cress W D, Santiago-Cardona P
Received 20 September 2014
Accepted for publication 4 November 2014
Published 18 December 2014 Volume 2015:7 Pages 1—10
DOI https://doi.org/10.2147/CHC.S28079
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Denis Wirtz
Brienne E Engel,1 W Douglas Cress,1 Pedro G Santiago-Cardona2
1Molecular Oncology Program, H Lee Moffitt Cancer Center and Research Institute, Tampa, FL, USA; 2Department of Biochemistry, Ponce School of Medicine, Ponce, Puerto Rico, USA
Abstract: RB1 was the first tumor suppressor gene discovered. Over 4 decades of work have revealed that the Rb protein (Rb) is a master regulator of biological pathways influencing virtually every aspect of intrinsic cell fate including cell growth, cell-cycle checkpoints, differentiation, senescence, self-renewal, replication, genomic stability, and apoptosis. While these many processes may account for a significant portion of RB1's potency as a tumor suppressor, a small but growing stream of evidence suggests that RB1 also significantly influences how a cell interacts with its environment, including cell-to-cell and cell-to-extracellular matrix interactions. This review will highlight Rb’s role in the control of cell adhesion and how alterations in the adhesive properties of tumor cells may drive the deadly process of metastasis.
Keywords: cadherin, integrin, Rb, cancer, aggressiveness, metastasis
Introduction
Cancer cells are known to have many hallmarks that distinguish them from their normal counterparts.1 Two prominent hallmarks of cancer cells are the ability to survive and proliferate under inappropriate circumstances, and the ability to migrate from their original tissue location to distant places and there continue to proliferate. This cellular migration is thought to be dependent upon the cancer cell’s ability to change its cell-to-cell and cell-to-matrix adhesive properties and to survive during the process. Interestingly, accumulating evidence suggests that the RB1 tumor suppressor gene may regulate both of these key cancer hallmarks, explaining its prominence as a tumor suppressor.
The retinoblastoma protein (Rb): the classic paradigm
Existence of the RB1 gene was predicted in 1971 from epidemiological evidence from retinoblastoma families,2 and the RB1 gene was identified over 15 years later.3 The initial characterization of Rb function was guided by studies of DNA tumor viruses4,5 which pointed to Rb’s role as a regulator of the G1/S transition.6 It is now known that both the G1/S and G2/M phases of the mammalian cell cycle are controlled by a complex and redundant molecular pathway (highlighted schematically in Figure 1) that involves members of the E2 promoter binding factor (E2F),7 dimerization partner,8 Rb,4 cyclin-dependent kinase (CDKs), cyclin,9 and CDK inhibitor (CDKN)10 families. This pathway is disrupted in most, if not all, solid tumors.11
 | Figure 1 The canonical Rb/E2F pathway in cancer. |
While initial work on the function of Rb in the cell cycle highlighted its role in the G1/S transition, work over the last 3 decades demonstrates that Rb controls most cellular processes related to cell fate and DNA metabolism, including cell-cycle checkpoints, tissue differentiation and morphogenesis, senescence, self-renewal, replication, tissue-specific gene expression, mitotic fidelity, genomic stability, and apoptosis.12–14 In this review we will refer to these various intrinsic cell processes collectively as the cell cycle. There are a number of outstanding articles15–18 that review Rb’s role in the cell cycle processes and they will not be repeated in detail here.
Rb’s activity is regulated by posttranslational modifications, phosphorylation being the most predominant.19 Rb phosphorylation by CDK4–cyclin D and CDK2–cyclin E induces S-phase entry.20 Rb is phosphorylated on at least 13 different serine/threonine residues suggesting that specific patterns of Rb phosphorylation may represent a “Rb code” in which different Rb conformational variants mediate distinct protein–protein interactions. In noncancerous cells, antiproliferative signals activate Rb by promoting its dephosphorylation by serine and threonine type I phosphoprotein phosphatases and by inhibiting the cyclin–CDK complexes that phosphorylate Rb.21,22 This activation allows Rb to block progression to S-phase, promoting entry to G0 instead. A recent review has addressed the complexity of these Rb kinases.19
Rb’s strong tumor-suppressive nature is evident in the fact that Rb function is lost in most human cancers,21,23,24 and also by the fact that oncogenic insults, such as Ras activation, trigger a strong antioncogenic senescence program that depends on Rb.12,25 Every component of the Rb pathway that represses the cell cycle is subject to mutational inactivation in some human cancers and every component that induces the cell cycle is subject to oncogenic upregulation, providing genetic evidence that the pathway as a whole is essential in tumor development. CDKN proteins are subjected to inactivating mutations and epigenetic silencing. Cyclins, CDKs, and, rarely, E2Fs themselves are upregulated by translocations and gene amplifications.26–29 The rate of RB1 gene mutation varies significantly among different tumor types, but is highest in retinoblastoma, osteosarcoma, and small-cell lung cancer (SCLC).23,30 Mutations targeting the RB1 gene directly affect Rb function by either completely abrogating its expression or by producing a nonfunctional protein.31 Other cancer types bearing wild-type RB1 alleles still have impaired Rb function due to alterations in genes coding for upstream Rb regulators. These alterations range from inactivating mutations, deletions, or epigenetic silencing of the p16INK4A locus (a CDKN family member), to alterations leading to cyclin D or CDK4 overexpression.27 The latter scenario results in Rb inactivation by chronic hyperphosphorylation. Therefore, oncogenesis usually entails either a complete loss of Rb expression or its inactivation by hyperphosphorylation.
Deregulation of adhesion proteins in cancer
Figure 2 highlights the role that cell adhesion is thought to play in cancer spread and metastasis. Cadherins are calcium-dependent cell adhesion proteins that mediate cell-to-cell adhesion. They are named for the tissue that they were first identified in (eg, E-cadherin, N-cadherin, and OB-cadherin were discovered in epithelial, neural, and osteoblast tissues, respectively), but are not restricted exclusively to those tissues. Cadherins, together with catenins, are the main components of adherens junctions, which are membrane protein complexes that are stabilized by association with actin filaments densely packed under the cell membrane.32 Their disruption is part of the epithelial–mesenchymal transition (EMT) during oncogenic progression and contributes to metastasis by facilitating detachment of cancer cells from the primary tumor.33 This disruption consists of a “cadherin switch” whereby expression of E-cadherin is repressed and N-cadherin is upregulated.
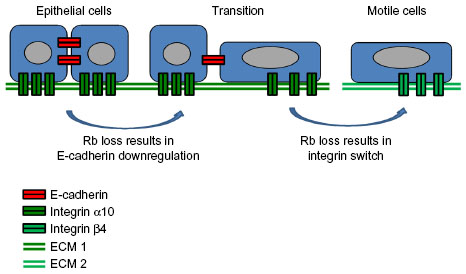 | Figure 2 Rb’s role in cell adhesion in cancer. |
Alterations in integrin expression have also been noted in cancers. Similar to the “cadherin switch” occurring during EMT, an “integrin switch” has been observed in multiple solid tumors consisting of overexpression of integrin β4, and underexpression of integrins α7, α8, and α10.34 Integrins are a family of 26 cell-to-extracellular matrix (ECM) adhesion receptor subunits. Each functional integrin heterodimer consists of two type-1 (single membrane-spanning domain with the C-terminus located cytoplasmically) transmembrane subunits: one α- and one β-subunit. Integrins bind to multiadhesive ECM components, organizing the cytoskeleton and activating intracellular signaling pathways. They have been shown to affect cell shape, polarization, cytoskeletal organization, cell motility, proliferation, survival, and differentiation. Integrins are unusual among transmembrane receptors in that they signal bidirectionally, carrying both mechanical and chemical signals. “Inside-out” signaling, known as “priming”, is responsible for a conformational change in the integrin heterodimer which extends outward and induces adhesiveness to the ECM. Adhesion is further strengthened by the lateral reorganization of integrins into clusters, which may progress to dot-like focal complexes that mature into larger focal adhesions and finally into streak-like fibrillar adhesions.35 The “integrin adhesome” is comprised of 156 signaling, structural, and adaptor molecules that contribute to cytoskeletal reorganization and catalytic activity as integrin tails have no catalytic activity of their own.35 Integrin signaling, and the associated cross talk with adjacent receptor tyrosine kinases, has been linked to many pathways important in human cancer including the Ras/Raf/MEK/MAPK, PI3K/PIP3/AKT, NF-κB, and Rb/E2F pathways.
New roles for Rb in cell adhesion: regulation of cadherin- and integrin-mediated adhesion
Cancer types showing high frequencies of mutational inactivation of the RB1 gene are very aggressive relative to tumors with chronically hyperphosphorylated wild-type Rb. An example is osteosarcoma, which at the time of diagnosis was consistently high grade and poorly differentiated. These patients have a poor prognosis, with 20% of diagnosed cases already having detectable metastases36 and only 10% achieving long-term disease-free intervals.37 Osteosarcoma incidence is increased 1,000-fold in patients who inherit RB1 mutations relative to the general population,38,39 implicating Rb loss in osteosarcoma formation. Rb loss occurs in over 70% of sporadic osteosarcomas, and loss of RB1 heterozygosity is present in 60%–70% of osteosarcomas and is indicative of a poor prognosis.40
Like osteosarcoma, SCLC is characterized by a high rate (~90%) of mutational inactivation of the RB1 locus.41 Patients with SCLC have a 5-year survival rate of only 6%,41,I which can increase to 54% if detected at a localized stage. Unfortunately, only 15% of SCLCs are detected at the localized stage. This extreme aggressiveness is in stark contrast to the 5-year survival rates of tumors with lower rates of RB1 mutations such as breast, prostate, and colorectal cancers when detected at a localized stage, which are 99%, 100%, and 90%, respectively, according to the American Cancer Society Cancer Facts and Figures, 2014. Interestingly, lung cancer survival rate triples to 18% in the non-SCLC (NSCLC) subtype, which usually bears wild-type RB1 alleles but instead exhibits preferential loss of the p16INK4A locus with consequent Rb hyperphosphorylation.42 The differences in survival rates between SCLC and NSCLC suggest that increased aggressiveness may be associated more with direct alterations of the RB1 locus than with chronic Rb hyperphosphorylation resulting from alterations in other loci.
This RB1 effect is also observed in epithelial cancers. Although RB1 mutations are rare in prostate cancer, a recent analysis of 50 castration-resistant prostate cancer patients43 demonstrated that patients with inactivating RB1 mutations have a 35-month reduction in median overall survival relative to patients with WT RB1 (P=0.025). Specifically, the 16 patients with RB1 mutations had a median overall survival of 70 months, versus 105 months in 34 patients with WT RB1.
The data discussed above highlight the association between direct mutational targeting of the RB1 gene and high mortality as demonstrated by the low 5-year survival rates of Rb-null cancers. Given that metastases indicate aggressiveness and cause over 90% of cancer deaths,44 we propose that Rb deficiency leads to a proclivity for early metastasis, that is, for early detachment of tumor cells from the primary tumor and invasion of adjacent and distal tissues. If so, blocking cellular events associated with metastasis (eg, loss of cell polarity, EMT, loss of cell adhesion) may be part of Rb’s tumor-suppressive arsenal. The first hints of a relationship between Rb and metastasis came from studies published over a decade ago implicating Rb in the stabilization of adherens junctions. Disruption of these structures is part of EMT and contributes to metastases by facilitating detachment of cancer cells from the primary tumor mass.33 Early studies showed that retinoblastomas, osteosarcomas, and SCLC, known for their high frequencies of RB1 mutations, are composed of cells that lack stable adherens junctions. In retinoblastoma, adherens junctions fail to anchor to the cortical actin cytoskeleton.45 In osteosarcoma and SCLC, adherens junction proteins are downregulated and aberrantly localized in the cytoplasm rather than at the cell membrane.46,47 Furthermore, a strong correlation was found in retinoblastoma and osteosarcoma between abnormal adherens junctions and invasive capacity,45,46 underscoring the notion that disruption of these structures is related to invasion, and for the first time, implicating Rb loss in invasive behavior.
These early studies linking Rb to adherens junction integrity remained largely ignored for years following their publication, possibly since they were mostly correlative and did not establish a causal relationship between Rb loss and cell adhesion perturbations. An exception was a study showing that Rb inactivation by SV40 large T antigen in MDCK epithelial cells resulted in a mesenchymal conversion associated with invasiveness that could be reversed by Rb reactivation.48 This study also offered the first mechanistic explanation of Rb’s involvement in cell adhesion by showing that Rb, together with the AP-2 transcription factor, activated transcription of the E-cadherin promoter in epithelial cells.48 It took approximately a decade for the next reports confirming the link between Rb and cell adhesion. These studies showed that Rb depletion disrupted cellular adhesion and induced a mesenchymal-like phenotype. They further established that transcriptional regulation of E-cadherin expression by Rb is a molecular link between Rb and cell adhesion.49,50 Further implicating Rb loss in EMT, Rb depletion results in upregulation of several EMT-related transcriptional factors including Slug and ZEB-1, which are known E-cadherin transcriptional repressors.49,51
The studies described above strongly implicate Rb loss as a promoter of metastasis of carcinomas or epithelial tumors, specifically via the loss of epithelial markers such as E-cadherin and the acquisition of mesenchymal and migratory phenotypes (see Figure 2). Additionally, it has been shown that OB-cadherin, the predominant osteoblast cadherin, is also transcriptionally regulated by Rb,51 implicating Rb loss in the molecular etiology of non-epithelial tumors such as osteosarcomas. Conditional deletion of Rb in osteoblasts produces a “cadherin switch” in which OB-cadherin is replaced by N-cadherin,51 suggesting that Rb promotes the expression of adhesion molecules characteristic of the fully differentiated state, regardless of cell type, while repressing the expression of cell adhesion genes related to an undifferentiated phenotype. The global nature of Rb’s influence on cell adhesion was revealed by microarray analyses comparing Rb-proficient versus Rb-deficient osteoblasts, which found that Rb affects the expression of a variety of cell adhesion genes beyond cadherins, and that cellular processes related to cell adhesion are strongly affected by Rb.51 Integrins were also found among the cell adhesion genes whose expression is strongly affected by Rb, and cellular pathways involved in integrin-mediated cell-to-ECM adhesion were also found to be under Rb control.51 In a follow-up study, it was shown that Rb induces transcription of integrin α10 in osteoblasts, regardless of Rb phosphorylation status, with a corresponding increase in osteoblast binding to a collagen substrate.34 This suggests that Rb mediates not only cadherin-dependent cell-to-cell adhesion, but also promotes integrin-dependent cell-to-ECM adhesion. Taken together, the studies summarized above indicate a strong influence by Rb on cell adhesion that can either be activating or repressive depending on the genes involved, upregulating adhesion genes in differentiated cells (eg, integrin α10) while downregulating expression of adhesion genes associated with invasiveness and metastasis (eg, N-cadherin).
Rb appears to regulate the formation of functional cell adhesive structures beyond transcriptional regulation of cell adhesion genes, including facilitating the assembly of cell adhesion gene products at the cell membrane. In the absence of Rb, the Rho GTPase Rac1 and its effector, the p21-activated protein kinase (Pak1), become upregulated with consequent phosphorylation of the Merlin tumor suppressor at Serine 518 by Pak1, which in turn causes Merlin to detach from the cell membrane.51 Therefore, Rb seems to promote adherens junction assembly at the cell membrane by blocking the inactivating phosphorylation of Merlin by Pak1. Merlin is a membrane-bound tumor suppressor and cytoskeleton adapter that stabilizes adherens junctions by anchoring them to the cortical actin cytoskeleton under the plasma membrane.52,53 Merlin loss, which is frequent in the human cancer syndrome neurofibromatosis type 2, results in adherens junction disruption with consequent inactivation of contact-dependent growth arrest.52 In summary, studies demonstrate that in the absence of functional Rb, transcription of adherens junction components as well as their assembly at the cell membrane are both compromised. This explains the observation that in Rb-deficient tumors, such as retinoblastoma and osteosarcoma, not only do adherens junction proteins show diminished expression, but they also fail to anchor to the cell membrane, instead showing aberrant cytoplasmic localization.45–47
Invasiveness and metastases arise from a combination of: loss of cell adhesion, onset of migration facilitated by cytoskeletal reorganization and loss of cell polarity, and the capacity to degrade basal laminae in order to escape the primary tumor site and penetrate adjacent tissues. The data summarized above link Rb loss predominantly to perturbations in cell adhesion, but Rb loss could exacerbate invasiveness by affecting other aspects of metastasis. For example, Rb loss has been linked to increased expression of the matrix metalloproteinases that remodel the ECM during cell invasion and metastasis.54,55 Matrix metalloproteinase genes such as MMP9, MMP14, and MMP15, which are usually overexpressed in NSCLC, have been shown to be regulated by Rb.55 Rb reactivation was sufficient to inhibit MMP transcription, to reduce the invasion and migration of cancer cells in vitro, and to reduce metastatic foci development in a tail vein lung metastasis model in mice.55 Rb depletion also exacerbates the invasiveness of erbB2-positive breast cancer, suggesting that Rb loss may play a predominant role in the progression of in situ breast ductal cell carcinoma to the invasive stages of the disease.56
The data implicating Rb control of cell adhesion in cultured cells are abundant and provide mechanistic insights that were lacking in early correlative studies. There are also data providing insights into the consequences of Rb loss for in vivo tissue morphogenesis. When a mouse model of osteosarcoma was generated by conditionally knocking out RB1 in osteoblasts,51 structural defects indicative of impaired osteoblast adhesion were observed in the calvaria of Rb knockout mice. Specifically, Rb knockout mice lacked properly organized osteoblast layers, and showed osteoblasts that had migrated away from their proper position in the calvaria and invaded the adjacent cartilage.51 Rb-deficient osteoblasts also expressed elevated levels of ezrin, a membrane-cytoskeleton linker and osteosarcoma metastasis marker.51,57,58 Other mouse models of osteosarcoma based on abrogation of Rb function have resulted in mice that develop fully penetrant, highly metastatic early onset osteosarcomas.59 Given the importance of cell-to-cell adhesion for osteoblast differentiation, Rb loss can be predicted to alter osteoblast differentiation and lead to the formation of osteosarcoma. Osteoblasts originate from pluripotent mesenchymal stem cells that differentiate into stroma, adipocytes, myoblasts, chondroblasts, fibroblasts, or osteoblasts.60,61 Stem cells committed to osteoblastic differentiation are sorted from the rest of the mesenchymal precursors and align with, and adhere to, each other. Homotypic, cadherin-based cell-to-cell interactions play a major role in sorting the pluripotent stem cells into distinct lineages. Consistently, osteoprogenitor cells express a spatio-temporally regulated repertoire of cadherins that provide cues for their alignment into a distinct subpopulation within the bone marrow that will later differentiate into mature osteoblasts.62,63 Adherens junction loss in Rb-null osteoblasts is accompanied by abnormal expression patterns of the predominant osteoblast-specific cadherins OB- and N-cadherins, suggesting that the timing of cadherin expression during osteoblast differentiation can be altered by Rb loss.51 This in turn suggests that Rb is required to ensure that expression of specific cadherins proceeds with the right timing during differentiation, and that Rb loss could hamper proper homotypical intercellular contacts, resulting in defective osteoblast differentiation with consequent disruption of bone integrity and/or formation of bone tumors. Based on in vivo observations, it is plausible that Rb is instrumental in the orchestration of cell proliferation and cell adhesion as part of differentiation and bone morphogenesis – disruption of which may be central to the molecular etiology of osteosarcomas, which are characterized by poor differentiation and high frequencies of RB1 mutations.
A link between cell cycle control and cell adhesion
In metastatic cancer cells, adhesion is aberrantly regulated by a variety of pathways, resulting in loss of cell-to-cell and cell-to-ECM contact and dissemination of cancer cells throughout the body. While more work is needed to elucidate those pathways, in many instances this loss of adhesion has been tied to cell cycle regulators, including members of the Rb–E2F pathway.
Signaling from integrins through their downstream pathways occurs cooperatively through cross talk with growth factor receptors and has been linked to a variety of pathways involved with cell cycle progression. Integrin-mediated cell-to-ECM adhesion acts as a checkpoint for cell cycle entry. For example, in early work using Rb-positive LNCaP and Rb-negative RU145 prostate epithelial cell lines, loss of β1 integrin contact to ECM inhibited G1 CDK activity leading to an accumulation of hypophosphorylated Rb and subsequent Bcl-2-mediated apoptosis.64 More recently, Wang et al65 found that overexpression of integrin α5 and knockdown of integrin α6 decreased pulmonary metastasis of the highly invasive breast cancer cell line 4T1 by inhibiting entry to S-phase through p27 upregulation, resulting in downregulation of cyclin E/CDK2 complexes. They also found that this modulation of integrin expression upregulated E2F, which may then induce expression of Chk1 to regulate cdc25A/Cyclin E/CDK2/Rb in a feedback loop. These findings implicate integrin α5 as a metastasis suppressor and α6 as a metastasis promoter in breast cancer. For a comprehensive review of how integrins control downstream entry to cell cycle progression, see the review by Moreno-Layseca and Streuli.66
Expression of E2Fs 1–3 was shown to indirectly increase integrin β4 mRNA, protein, and cell surface expression.67 These E2Fs were found to be downstream of active H-Ras in SUM-159 breast carcinoma cells. Integrin α6β4 has been previously shown to enhance carcinoma invasion, so the mechanism proposed by Yoon et al67 links active H-Ras, active E2Fs, and integrin α6β4 in a single pathway to promote invasion.
Long-term treatment of three NSCLC cell lines with recombinant cysteine-rich 61 (CCN1), a secreted matrix-associated molecule, led to permanent cell cycle arrest in G1. Addition of CCN1 increased abundance of hypophosphorylated Rb and increased levels of p53 and p21 accumulation. A CCN1 mutant defective for binding integrin α6β1 and coreceptor heparan sulfate proteoglycans was incapable of inducing senescence.68
The finding that Rb’s effect on integrin expression is unaltered by its phosphorylation state34 is particularly informative of the mechanisms linking Rb to cell adhesion and of the coupling between cell cycle and cell adhesion. As discussed above, phosphorylation is a mechanism of regulation of Rb function that abrogates Rb’s capacity to bind and block E2F transcription factors. The integrin α10 findings34 suggest that regulation of cell cycle progression and cell adhesion by Rb may be mechanistically uncoupled since, while Rb hyperphosphorylation abrogates Rb’s capacity to bind E2F and repress the cell cycle, it leaves intact the capacity to induce integrin-mediated cell-to-ECM adhesion. This could shed some light onto the aggressive behavior of Rb-deficient tumors. The tendency of Rb-deficient tumors to metastasize early in their development could be explained by the loss of both cell cycle control and cell adhesion resulting from Rb loss. The residual Rb activity retained by tumors with chronically hyperphosphorylated Rb, while not enough to halt initial tumor growth, may result in a less aggressive tumor and in deterring metastasis by helping to anchor the tumor structure.
The data discussed above expand the paradigm of Rb function beyond cell cycle to include roles in cell adhesion, and therefore implicate Rb loss in later stages of tumor metastasis. Figure 3 shows a model depicting how Rb can integrate cell cycle control and cell adhesion. These dual roles of Rb mechanistically explain how impairment of Rb function contributes to the aggressive nature of some tumor types, expands Rb’s arsenal of tumor suppressive abilities, and explains the potency of this preeminent tumor suppressor more adequately than the notion that Rb acts predominantly as a cell cycle repressor.
 | Figure 3 Rb links cell cycle and cell adhesion. |
Targeting Rb loss and aberrant adhesion
The activity of the Rb kinases, the CDKs, is central to the Rb pathway. For this reason, small molecule CDK inhibitors are being developed and examined in clinical trials for a number of malignancies.69
Aberrant adhesion has been determinant of potential treatment options in several cancers. For example, in erlotinib-resistant lung cancer cells harboring activating EGFR mutations, there was increased expression of Src, integrins β1, α2, and α5 along with increased adhesion. Silencing of integrin β1 restored erlotinib sensitivity. There was also increased expression of integrins β1, α2, and/or α5 in refractory tumor samples from patients treated with erlotinib and/or gefitinib.70
Furanodiene, a natural terpenoid derived from Curcuma Wenyujin, was found to have antiproliferative activity in 95-D human lung cancer cells when combined with paclitaxel. These effects included downregulation of protein levels of cyclins D1 and B1, CDK6, and c-Myc, as well as downregulation of expression of integrin β4, focal adhesion kinase, and paxillin.71 Previous studies had shown that combining furanodiene and paclitaxel had synergistic antiproliferative effects in NCI-H1299 and 95-D human lung cancer cell lines,72 and that furanodiene decreased integrin β1 expression in breast cancer cells in a concentration-dependent manner.73
Unsurprisingly, aberrant integrin signaling has been implicated in several human cancers and specific therapies are being developed to target the integrin pathway in a number of diseases including development of anti-integrin α4 antibodies (eg, Natalizumab74), integrin-linked kinase (ILK),75 and arginylglycylaspartic acid peptides such as eptifibatide.76 Perhaps most relevant to cancer, focal adhesion kinase (FAK) inhibitors such as GSK2256098 are in multiple clinical trials in solid tumors.77–79 Unfortunately, integrins are also known mediators of cell adhesion-mediated drug resistance. Specifically, melanoma cells expressing α4β1 and α5β1 integrins are resistant to doxorubicin and melphalan once bound to their fibronectin ligands.80 This resistance is the result of cell cycle arrest in G1 and is associated with increased levels of the CDKN p27 and its inhibition of cyclins A and E.81
Ligand-induced adhesion, an integrin-mediated Rap-1-independent pathway that allows unstimulated leukocytes to adhere to and migrate through exposed endothelial matrix or high-density ligand, is CDK4-mediated, but Rb-independent. CDKNs were able to block this leukocyte adhesion and migration.82
The treatment options listed above are all designed to target either the Rb pathway or the process of adhesion. With the new work being pioneered on the link between these two pathways, it is our hope that, by either combining these drug classes or developing new drugs to specifically target this newly discovered link, treatment options will be more tailored to individual cancers and will be increasingly effective in the future.
Conclusion: implications for Rb as a metastasis suppressor
Recent work linking Rb to cell adhesion should reinvigorate the Rb field by challenging the classic paradigm of Rb acting predominantly as a cell cycle regulator. New information about Rb, as well as other oncogenes and tumor suppressors discovered decades ago, continues to uncover novel effects and potentialities beyond cell cycle control.
In the currently accepted model of tumor evolution, a stepwise accumulation of mutations results in the progressive acquisition of aberrant cellular behaviors, with each behavior elicited by a particular mutation or sets of mutations. Mutations that inactivate tumor suppressors like RB1 or that activate proto-oncogenes like KRAS would contribute to early stages of tumor evolution by conferring a proliferative advantage to incipient tumor cells. These mutations target cell cycle control by rendering cells capable of bypassing proliferative arrest, contributing to unchecked tumor growth. However, a paradigm in which Rb acts predominantly as a cell cycle repressor does not explain how Rb inactivation in early tumorigenesis would contribute to later stages of metastasis, particularly to the detachment of tumor cells from their original site and dispersion to distant tissues. The current model mandates the acquisition of additional secondary mutations that confer metastasis potential at later stages of tumor evolution. This multistep model has been challenged, however, and deemed conceptually inconsistent since the additional genetic hits that confer metastatic capabilities at later stages of tumorigenesis do not necessarily exacerbate the proliferative advantage conferred by the initial hits that disrupt cell cycle control.83 In a tumor history that is essentially a microevolutionary process, if secondary metastasis-related mutations do not further enhance the previously acquired replicative advantage, the cells that acquired them will remain rare within the tumor mass, outcompeted by more proliferative counterparts. Rather, it has been suggested that mutations acquired by incipient tumor cells early in tumorigenesis confer not only the replicative advantage that allows the initial tumor growth, but also later in tumorigenesis the proclivity to metastasize.83 Thus, the tendency to metastasize could be determined by mutant alleles acquired early in tumor history.83 This revised model predicts that fewer mutations are required for a full-blown malignant phenotype if they target multifunctional genes such as RB1. Several lines of evidence support this. First, primary human breast cancers can shed malignant cells into the bone marrow even when tumors are small and well-localized; second, DNA microarray analyses reveal that metastatic tumor cells show gene expression profiles remarkably similar to the cells contained in the primary tumor from which they were derived; third, certain early gene expression profiles in primary breast cancer tumors strongly predict metastasis and can be detected before metastasis actually occurs.84–88 A dual role for Rb in cell cycle and cell adhesion is fully consistent with a model of metastases arising from fewer mutations. Thus, Rb inactivation enhances proliferative capacity and growth of tumor mass during early carcinogenesis, and also contributes to later stages of metastasis by promoting cell detachment from the primary tumor. Further characterization of Rb’s role in cell adhesion could contribute to what has been described as
the hope to achieve an understanding of the complex process of neoplastic transformation at the cellular level in terms of a small number of genetic changes.89
Disclosure
The authors report no conflicts of interest in this work.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. | |
Fung YK, Murphree AL, T’Ang A, Qian J, Hinrichs SH, Benedict WF. Structural evidence for the authenticity of the human retinoblastoma gene. Science. 1987;236(4809):1657–1661. | |
Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65(6):1053–1061. | |
Cress WD, Nevins JR. Use of the E2F transcription factor by DNA tumor virus regulatory proteins. Curr Top Microbiol Immunol. 1996;208:63–78. | |
Sellers WR, Kaelin WG Jr. Role of the retinoblastoma protein in the pathogenesis of human cancer. J Clin Oncol. 1997;15(11):3301–3312. | |
Kovesdi I, Reichel R, Nevins JR. Role of an adenovirus E2 promoter binding factor in E1A-mediated coordinate gene control. Proc Natl Acad Sci U S A. 1987;84(8):2180–2184. | |
La Thangue NB. DP and E2F proteins: components of a heterodimeric transcription factor implicated in cell cycle control. Curr Opin Cell Biol. 1994;6(3):443–450. | |
Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993;73(3):487–497. | |
Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512. | |
Kaelin WG Jr. Functions of the retinoblastoma protein. Bioessays. 1999;21(11):950–958. | |
Thomas DM, Yang HS, Alexander K, Hinds PW. Role of the retinoblastoma protein in differentiation and senescence. Cancer Biol Ther. 2003;2(2):124–130. | |
Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA. Rb regulates fate choice and lineage commitment in vivo. Nature. 2010;466(7310):1110–1114. | |
Manning AL, Dyson NJ. pRB, a tumor suppressor with a stabilizing presence. Trends Cell Biol. 2011;21(8):433–441. | |
Attardi LD, Sage J. RB goes mitochondrial. Genes Dev. 2013;27(9):975–979. | |
Viatour P, Sage J. Newly identified aspects of tumor suppression by RB. Dis Model Mech. 2011;4(5):581–585. | |
Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14(5):297–306. | |
Manning AL, Dyson NJ. RB: mitotic implications of a tumour suppressor. Nat Rev Cancer. 2012;12(3):220–226. | |
Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38(1):12–19. | |
Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18(2):753–761. | |
Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81(3):323–330. | |
Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther. 2002;1(3):226–231. | |
Thomas DM, Carty SA, Piscopo DM, et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001;8(2):303–316. | |
Knudsen ES, Knudsen KE. Retinoblastoma tumor suppressor: where cancer meets the cell cycle. Exp Biol Med (Maywood). 2006;231(7):1271–1281. | |
Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37(5):961–976. | |
Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9(11):785–797. | |
Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–112. | |
Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. | |
Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. | |
Tiemann F, Musunuru K, Hinds P. The retinoblastoma tumour suppressor protein and cancer. In: DM Swallow, Edwards YH, editors. Protein Dysfunction and Human Genetic Disease. Oxford: BIOS Scientific Publishers; 1997:163–185. | |
Shew JY, Lin BT, Chen PL, Tseng BY, Yang-Feng TL, Lee WH. C-terminal truncation of the retinoblastoma gene product leads to functional inactivation. Proc Natl Acad Sci U S A. 1990;87(1):6–10. | |
Tsukita S, Tsukita S, Nagafuchi A, Yonemura S. Molecular linkage between cadherins and actin filaments in cell-cell adherens junctions. Curr Opin Cell Biol. 1992;4(5):834–839. | |
Lozano E, Betson M, Braga VM. Tumor progression: Small GTPases and loss of cell-cell adhesion. Bioessays. 2003;25(5):452–463. | |
Engel BE, Welsh E, Emmons MF, Santiago-Cardona PG, Cress WD. Expression of integrin alpha 10 is transcriptionally activated by pRb in mouse osteoblasts and is downregulated in multiple solid tumors. Cell Death Dis. 2013;4:e938. | |
Legate KR, Wickström SA, Fässler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23(4):397–418. | |
Dahlin DC. Pathology of osteosarcoma. Clin Orthop Relat Res. 1975(111):23–32. | |
Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: osteosarcoma and related tumors. Cancer Genet Cytogenet. 2003;145(1):1–30. | |
Lueder GT, Judisch F, O’Gorman TW. Second nonocular tumors in survivors of heritable retinoblastoma. Arch Ophthalmol. 1986;104(3):372–373. | |
MacCarthy A, Bayne AM, Brownbill PA, et al. Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951–2004. Br J Cancer. 2013;108(12):2455–2463. | |
Araki N, Uchida A, Kimura T, et al. Involvement of the retinoblastoma gene in primary osteosarcomas and other bone and soft-tissue tumors. Clin Orthop Relat Res. 1991(270):271–277. | |
Kaye F, Kubo A. Molecular Genetics of Cancer. In: JK C, editor. Oxford: Bios Scientific Publishers; 2001:387–413. | |
Kaye FJ. RB and cyclin dependent kinase pathways: defining a distinction between RB and p16 loss in lung cancer. Oncogene. 2002;21(45):6908–6914. | |
Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. | |
Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6(6):449–458. | |
Van Aken EH, Papeleu P, De Potter P, et al. Structure and function of the N-cadherin/catenin complex in retinoblastoma. Invest Ophthalmol Vis Sci. 2002;43(3):595–602. | |
Kashima T, Kawaguchi J, Takeshita S, et al. Anomalous cadherin expression in osteosarcoma. Possible relationships to metastasis and morphogenesis. Am J Pathol. 1999;155(5):1549–1555. | |
Rodríguez-Salas N1, Palacios J, de Castro J, Moreno G, González-Barón M, Gamallo C. Beta-catenin expression pattern in small cell lung cancer: correlation with clinical and evolutive features. Histol Histopathol. 2001;16(2):353–358. | |
Batsché E, Muchardt C, Behrens J, Hurst HC, Crémisi C. RB and c-Myc activate expression of the E-cadherin gene in epithelial cells through interaction with transcription factor AP-2. Mol Cell Biol. 1998;18(7):3647–3658. | |
Arima Y, Inoue Y, Shibata T, et al. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res. 2008;68(13):5104–5112. | |
Arima Y, Hayashi H, Sasaki M, et al. Induction of ZEB proteins by inactivation of RB protein is key determinant of mesenchymal phenotype of breast cancer. J Biol Chem. 2012;287(11):7896–7906. | |
Sosa-García B, Gunduz V, Vázquez-Rivera V, et al. A Role for the Retinoblastoma Protein As a Regulator of Mouse Osteoblast Cell Adhesion: Implications for Osteogenesis and Osteosarcoma Formation. PloS One. 2010;5(11):e13954. | |
Lallemand D, Curto M, Saotome I, Giovannini M, McClatchey AI. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 2003;17(9):1090–1100. | |
McClatchey AI, Giovannini M. Membrane organization and tumorigenesis – the NF2 tumor suppressor, Merlin. Genes Dev. 2005;19(19):2265–2277. | |
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. | |
Johnson JL, Pillai S, Pernazza D, Sebti SM, Lawrence NJ, Chellappan SP. Regulation of matrix metalloproteinase genes by E2F transcription factors: Rb–Raf-1 interaction as a novel target for metastatic disease. Cancer Res. 2012;72(2):516–526. | |
Witkiewicz AK, Cox DW, Rivadeneira D, et al. The retinoblastoma tumor suppressor pathway modulates the invasiveness of ErbB2-positive breast cancer. Oncogene. 2014;33(30):3980–3991. | |
Khanna C, Wan X, Bose S, et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004;10(2):182–186. | |
Curto M, McClatchey AI. Ezrin … a metastatic detERMinant? Cancer Cell. 2004;5(2):113–114. | |
Berman SD, Calo E, Landman AS, et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci U S A. 2008;105(33):11851–11856. | |
Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276(5309):71–74. | |
Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98(5):1076–1084. | |
Hynes RO, Lander AD. Contact and adhesive specificities in the associations, migrations, and targeting of cells and axons. Cell. 1992;68(2):303–322. | |
Cheng SL, Shin CS, Towler DA, Civitelli R. A dominant negative cadherin inhibits osteoblast differentiation. J Bone Miner Res. 2000;15(12):2362–2370. | |
Day ML, Foster RG, Day KC, et al. Cell anchorage regulates apoptosis through the retinoblastoma tumor suppressor/E2F pathway. J Biol Chem. 1997;272(13):8125–8128. | |
Wang Y, Shenouda S, Baranwal S, et al. Integrin subunits alpha5 and alpha6 regulate cell cycle by modulating the chk1 and Rb/E2F pathways to affect breast cancer metastasis. Mol Cancer. 2011;10:84. | |
Moreno-Layseca P, Streuli CH. Signalling pathways linking integrins with cell cycle progression. Matrix Biol. 2014;34:144–153. | |
Yoon SO, Shin S, Mercurio AM. Ras stimulation of E2F activity and a consequent E2F regulation of integrin alpha6beta4 promote the invasion of breast carcinoma cells. Cancer Res. 2006;66(12):6288–6295. | |
Jim Leu SJ, Sung JS, Chen MY, et al. The matricellular protein CCN1 suppresses lung cancer cell growth by inducing senescence via the p53/p21 pathway. J Cell Biochem. 2013;114(9):2082–2093. | |
Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin Cancer Res. 2014;20(13):3379–3383. | |
Kanda R, Kawahara A, Watari K, et al. Erlotinib resistance in lung cancer cells mediated by integrin β1/Src/Akt-driven bypass signaling. Cancer Res. 2013;73(20):6243–6253. | |
Xu WS, Dang YY, Chen XP, Lu JJ, Wang YT. Furanodiene presents synergistic anti-proliferative activity with paclitaxel via altering cell cycle and integrin signaling in 95-D lung cancer cells. Phytother Res. 2014;28(2):296–299. | |
Xu WS, Dang YY, Guo JJ, et al. Furanodiene induces endoplasmic reticulum stress and presents antiproliferative activities in lung cancer cells. Evid Based Complement Alternat Med. 2012;2012:426521. | |
Zhong Z, Dang Y, Yuan X, et al. Furanodiene, a natural product, inhibits breast cancer growth both in vitro and in vivo. Cell Physiol Biochem. 2012;30(3):778–790. | |
Rudick R, Polman C, Clifford D, Miller D, Steinman L. Natalizumab: bench to bedside and beyond. JAMA neurology. 2013;70(2):172–182. | |
Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. Sep 2014;14(9):632–641. | |
Kononczuk J, Surazynski A, Czyzewska U, et al. AlphaIIbbeta3-Integrin Ligands: Abciximab and Eptifibatide as Proapoptotic Factors in MCF-7 Human Breast Cancer Cells. Curr Drug Targets. 2014. Epub 2014 Aug 4. | |
GlaxoSmithKline. A Dose Escalation Study to Assess Safety of GSK2256098 (FAK Inhibitor) in Combination With Trametinib (MEK Inhibitor) in Subjects With Advanced Solid Tumors. Available from: http://clinicaltrials.gov/ct2/show/NCT01938443. NLM identifier: NCT01938443. Accessed November 21, 2014. | |
GlaxoSmithKline. A Phase I, Randomized, Single-Blind, Placebo-Controlled Dose-Escalation Study to Evaluate the Safety, Pharmacokinetics, Pharmacodynamics and Preliminary Food Effect Following Single Oral Doses of the Focal Adhesion Kinase Inhibitor, GSK2256098, in Healthy Subjects. 2014; Available from: http://clinicaltrials.gov/ct2/show/NCT00996671. NLM identifier NCT00996671. Accessed Nov 21, 2014. | |
GlaxoSmithKline. Study of a Focal Adhesion Kinase Inhibitor in Subjects With Solid Tumors. 2014; Available from: https://clinicaltrials.gov/ct2/show/NCT01138033. NLM identifier: NCT01138033. Accessed November 21, 2014. | |
Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93(5):1658–1667. | |
Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene. 2000;19(38):4319–4327. | |
Liu L, Schwartz B, Tsubota Y, et al. Cyclin-dependent kinase inhibitors block leukocyte adhesion and migration. J Immunol. 2008;180(3):1808–1817. | |
Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418(6900):823. | |
Heitzer E, Auer M, Gasch C, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013;73(10):2965–2975. | |
Vignot S, Frampton GM, Soria JC, et al. Next-generation sequencing reveals high concordance of recurrent somatic alterations between primary tumor and metastases from patients with non-small-cell lung cancer. J Clin Oncol. 2013;31(17):2167–2172. | |
Pozzatti R, Muschel R, Williams J, et al. Primary rat embryo cells transformed by one or two oncogenes show different metastatic potentials. Science. 1986;232(4747):223–227. | |
Yokota J. Tumor progression and metastasis. Carcinogenesis. 2000;21(3):497–503. | |
van ‘t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. | |
Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347(20):1593–1603. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.