Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
The potential of CAR T therapy for relapsed or refractory pediatric and young adult B-cell ALL
Authors Forsberg MH, Das A, Saha K ![]() , Capitini CM
, Capitini CM ![]()
Received 1 June 2018
Accepted for publication 9 July 2018
Published 3 September 2018 Volume 2018:14 Pages 1573—1584
DOI https://doi.org/10.2147/TCRM.S146309
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Video abstract presented by Matthew H Forsberg.
Views: 1899
Matthew H Forsberg,1 Amritava Das,2,3 Krishanu Saha,2,4,5 Christian M Capitini,1,6
1Department of Pediatrics, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA; 2Wisconsin Institute for Discovery, University of Wisconsin, Madison, WI, USA; 3Morgridge Institute for Research, University of Wisconsin, Madison, WI, USA; 4Department of Medical History & Bioethics, University of Wisconsin, Madison, WI, USA; 5Department of Biomedical Engineering, University of Wisconsin, Madison, WI, USA; 6Carbone Comprehensive Cancer Center, University of Wisconsin, Madison, WI, USA
Abstract: Recent advancements in immuno-oncology have resulted in the generation of novel therapies such as chimeric antigen receptor (CAR) T cells, which have revolutionized the treatment of pediatric patients with relapsed or refractory B-cell acute lymphoblastic leukemia. The journey of tisagenlecleucel (formerly CTL019) from early preclinical success to the US Food and Drug Administration approval is summarized in this review. Strategies that are currently being investigated to improve the efficacy and safety profile of CAR T-cells are also explored, as well as the factors contributing to the present state of patient access to CAR T therapy.
Keywords: CAR T-cells, tisagenlecleucel, acute lymphoblastic leukemia, CD19, cancer immunotherapy, chimeric antigen receptor
Introduction
Treatment strategies for children and adolescents with acute lymphoblastic leukemia (ALL) developed over the past few decades have managed to cure up to 90% of cases.1,2 While these advancements are encouraging, the most common cause of treatment failure stems from patients who are either refractory to chemotherapy or experience one or more relapse of disease, which occurs in up to 20% of children3 with overall poor outcomes. Success rates of anywhere from 30% to 50% (as defined by 5-year disease-free survival) can be achieved using current intensive chemotherapy treatments in concordance with allogeneic hematopoietic stem cell transplantation (HSCT).4 The variance in outcome is dependent on a multitude of factors, with the two most important being length of first complete remission (CR) and site of relapse.5 However, children who experience two or more relapses of disease have an even more dismal prognosis, with 5-year disease-free survival rates of 27% and 15% in children who achieve a second and third CR, respectively.6
Prior to the approval of tisagenlecleucel, the US Food and Drug Administration (FDA) approved two drugs specifically for children with relapsed or refractory ALL. Clofarabine, a nucleoside analog, showed a 30% response rate for a median CR duration of 6 weeks, in a Phase II study involving 61 pediatric patients.7 It was FDA approved in 2004. Blinatumomab, a CD3/CD19 bispecific antibody, demonstrated in a Phase I/II study consisting of 70 children that a CR was achieved in 39% of patients.8 While the success of blinatumomab compared favorably with other treatment strategies for similar relapsed patients9 leading to its FDA approval in 2017, remission rates were still below 50%. In addition, durability was difficult to determine given that patients were allowed to withdraw from threatment, after two cycles, for consolidation chemotherapy or allogeneic HSCT. Fortunately, chimeric antigen receptor (CAR) T-cell therapy can provide high remission rates with durable survival benefits, leading to its FDA approval in 2017 for the treatment of relapsed/refractory pediatric and young adult ALL.
CAR T-cell overview
CAR T-cells are a powerful tool that allows for the specific targeting of tumor cells with engineered T cells.10 These T cells express synthetic receptors on their cell membrane designed to recognize specific antigens overexpressed on tumor cells in a major histocompatibility complex independent manner.11 This is achieved through the synthesis of a single-chain variable fragment (scFv) consisting of the variable regions of heavy and light chains from a monoclonal antibody specific for a given tumor antigen.12 The scFv is cloned into a genetic construct that includes a hinge domain, a transmembrane segment, and the CD3ζ chain in the construction of first-generation CARs.13 More recent CARs incorporate one (second-generation) or two (third-generation) intracellular signaling domains from costimulatory molecules such as CD28 or 4-1BB.14–18 After inserting the CAR construct into a viral vector,19 autologous or allogeneic T cells are transduced with the viral vector to express the CAR on their cell surface.20 These CAR-expressing T cells are then expanded and infused directly into the patient, where they specifically target tumor cells expressing the antigen recognized by the scFv. CAR T-cell therapy is not without side effects, one of which being cytokine release syndrome (CRS). CRS is characterized by increased levels of inflammatory cytokines such as IL-6 and interferon γ (IFNγ) in the patient, which can be fatal if untreated.21,22 Other side effects include tumor lysis syndrome,23 neurotoxicity,24 and on-target, off-tumor effects,25 a condition where CAR T-cells recognize their antigen on normal cells, resulting in destruction of healthy tissue. In the context of treatment for B-ALL, the destruction of healthy tissue results in low numbers or absence of B cells (B-cell aplasia), leading to hypogammaglobulinemia.
Development of CTL019
Initial second-generation CARs were constructed with the addition of the CD28 signaling domain into existing first-generation CAR constructs. The introduction of the CD28 domain enhanced IFNγ and IL-2 secretion as well as the proliferative capacity of CAR T-cells in vitro.14,15 The in vivo antitumor capabilities of these CARs were also enhanced in xenogeneic and syngeneic colon carcinoma mouse models.26 A subsequent study demonstrated that costimulatory domains of the tumor necrosis factor receptor family, which includes CD137 (4-1BB), could also be utilized in combination with CD3ζ demonstrating similar effects to those seen in CD28 CARs.27 CD137 signaling sustains cytotoxic T-cell activity,28 favors CD8-positive T-cell expansion,29 and has been shown to be vital in the antitumor response in animal models and in humans.30,31 From here, both CD28 and CD137 signaling domains were utilized in the development of CARs against B-ALL.30,32–34 CD19 was chosen as a target for B-ALL due to its high expression on most malignant B cells, while at the same time lacking expression on hematopoietic stem cells, limiting the risk of aplastic anemia.35–38 Secondary B-cell aplasia could be managed with immunoglobulin infusions similar to children with agammaglobulinemia. The first direct comparison of CD19-specific second-generation CARs containing a CD137 or CD28 signaling domain was performed in preclinical models.34 In this study, NOD-SCID-γc−/− (NSG) mice were engrafted with a human pre-B-cell ALL and then treated with CD137 or CD28 expressing CD19 CAR T-cells.34 The results demonstrated not only that a significantly higher percentage of NSG mice treated with CD19-CD137 CAR T-cells remained leukemia free but also that CD19-CD137 CAR T-cells could be detected in the spleen 6 months after transfer.34 The superior antileukemic activity and prolonged persistence of CD19-CD137 CAR T-cells, specifically CTL019 (Figure 1), opened the door for their use in initial human trials.
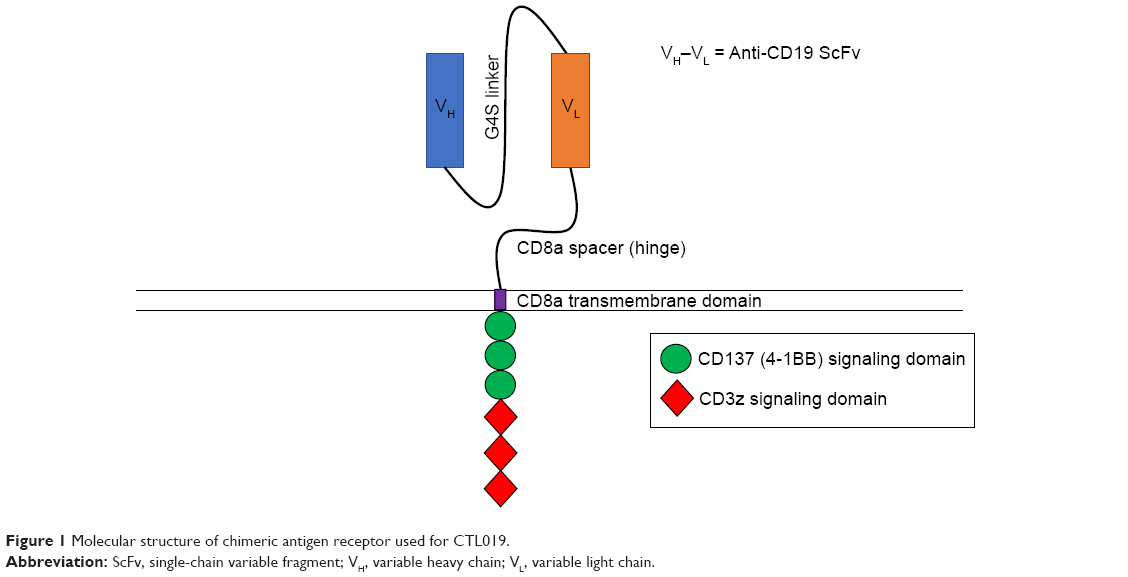 | Figure 1 Molecular structure of chimeric antigen receptor used for CTL019. |
Early successes of CTL019
Early applications of CTL019 in humans provided very encouraging results. Three adult patients ranging in age from 64 to 77 years with chemotherapy-resistant chronic lymphocytic leukemia (CLL) were given CTL019 following extensive pretreatment with various biological and chemotherapy regimens. Two of the three patients achieved a CR to CTL019 therapy with the other patient showing a partial response.39,40 Importantly, no detectable CLL was present in the bone marrow or circulating blood 6 months after treatment in the two patients with a CR, with remissions extending for more than 10 months.39,40 Additionally, functional CTL019 cells were present for at least 6 months, with each cell on average expanding greater than 1,000-fold and eradicating at least 1,000 CLL cells.
Following the initial success of CTL019 in the treatment of adult CLL, a pilot trial investigating the efficacy of CTL019 in children with refractory and relapsed B-ALL was initiated.41 Two children aged 7 (patient 1) and 10 (patient 2) years were treated with CTL019 after both patients had experienced a second relapse of B-ALL.41 Robust expansion was observed in both patients, with CTL019 T cells making up to 72% and 34% of total circulating T cells in patient 1 and patient 2, respectively.41 As seen in the adult CLL patients, the infused CAR T-cells persisted for at least 6 months and expanded to a level that was more than 1,000 times the original amount.41 Most importantly, both patients achieved CR, with one ongoing at the time of publication, 11 months after initial treatment.41 The other patient experienced a CD19− relapse, indicating that it was not due to nonfunctional CTL019 T cells.41 The initial success of CTL019 T-cell therapy in the treatment of pediatric relapsed/refractory B-ALL led to clinical trials with larger cohorts, producing even more noteworthy results.
Clinical trials of CTL019
In a single institution pediatric trial, 30 children and adults with relapsed/refractory B-ALL (of which 25 were 5–22 years of age) were given CTL019.23 A CR was achieved in 90% of patients, with 6-month disease-free survival of 67% and overall survival of 78%. Of note was the 73% probability of a patient who had achieved remission to experience relapse-free B-cell aplasia over the same 6-month time period. A clinical trial (KTE-C19) investigating the efficacy of a second-generation CAR T-cell containing a CD28 signaling domain in the treatment of pediatric B-ALL also demonstrated impressive results, with few differences to CTL019. For instance, only 70% of patients who were treated with the second-generation CD28 CAR T-cells achieved a CR according to the report on the first 21 patients,42 a percentage that fell to 61% when the total cohort of 38 patients were analyzed.43 Furthermore, B-cell recovery was observed at 28 days post-treatment in 13 of the 14 responding patients, demonstrating the superior persistence of CD137-expressing CTL019 compared with those with a CD28 signaling domain.
A subsequent trial by the same group treated an additional 59 pediatric patients, aged 20 months to 24 years, with CTL019.44 CR rates reached 93% one-month postinfusion in this larger cohort. Disease-free survival numbers were also improved at 6 months with 76%, and overall survival was 79% at 12 months. B-cell aplasia was observed in 24 of the 34 (70.6%) patients with ongoing CR at the time of last assessment.
Based on the success of the previous two single-center clinical trials, a global study of CTL019 therapy was initiated at 25 sites in 11 countries across North America, Europe, Asia, and Australia.45 In this multicenter study, 75 pediatric and young adult patients underwent CTL019 infusion. CR rates at 3 months postinfusion remained high (81%), while disease-free survival was 73% at 6 months and 50% at 12 months. The median duration of remission was not reached, which compares favorably with the median remission rate of 17.7 months seen in the KTE-C19 trial. With overall survival numbers of 90% at 6 months and 76% at 12 months, the reproducibility and feasibility of CTL019 therapy were conclusively demonstrated, leading to FDA approval of CTL019 as tisagenlecleucel on August 30, 2017.
Comparing overall remission rates across separate studies can be misleading, as researchers choose to report different statistics. For example, the KTE-C19 study reported CR and overall survival rates based on an intent-to-treat analysis, while some of the CTL019 studies only reported rates based on patients who received a CAR T-cell infusion. If intent-to-treat analysis is compared between the CTL019 and KTE-C19 studies, initial CR rates are very similar. Future studies might consider reporting rates of event-free survival, relapse-free survival, and overall survival on an intent-to-treat basis, as all these parameters are necessary and informative in determining the efficacy of CAR T-cell therapies in the real world. The percentages for all clinical trials discussed above are reported in Table 1.
 | Table 1 Summary of results reported from CD19 CAR T-cell pediatric clinical trials |
Challenges with CTL019 therapy
There are substantial risks associated with CAR T-cell therapy, primary among them being CRS. In the clinical trials of CTL019 mentioned earlier, anywhere from 77% to 100% of patients experienced an episode of CRS, with 27%–37% presenting with severe CRS requiring treatment with the anti-IL6 receptor agent tocilizumab, and in many cases, corticosteroids. The development of CRS was followed in the first 39 patients treated with CTL019 to characterize the timing and severity of CRS.46 This retrospective cohort study found that 46% of patients followed developed grade 3 or 4 CRS, with prolonged fever and organ dysfunction. Institutional preparation for the management of CRS, as well as for the rest of CTL019 treatment, from leukapheresis to infusion, is reviewed elsewhere.47
Neurotoxicity, or CAR T-related encephalopathy syndrome (CRES), can occur before, in association with, or after CRS. Around 40% of patients who received a CTL019 infusion experienced some sort of neurologic event, the most common being encephalopathy, confusion, delirium, tremors, agitation, somnolence, and hallucinations. The exact causes of neurologic events are currently unknown, and almost all cases resolve themselves without intervention beyond standard supportive care. Due to the potential severity of CRS and CRES, the FDA limited administration of tisagenlecleucel to just over 40 certified treatment centers under a risk evaluation and mitigation strategy (REMS) program with elements to assure safe use (ETASU). The REMS/ETASU program ensures that high-grade CRS and CRES will be monitored post-FDA approval.
One limitation of CAR T therapy is insufficient T-cell collection during apheresis. Many children are heavily pretreated from prior chemotherapy and have subsequent lymphopenia that preclude obtaining an adequate leukapheresis product to generate CAR T-cells. In an analysis of apheresis efficiency conducted on three pediatric/young adult CAR T-cell trials, it was found that the target of 2×109 CD3+ cells was achieved in 55 of 71 (77%) patients, while 69 of 71 (97%) patients reached the minimum number of 0.6×109 CD3+ cells.48 Of note, 16 patients with yields below the target had significantly lower percentages and overall numbers of CD3+ cells compared with patients who achieved the target.48 Efforts have been made to construct a universal CAR T-cell, one that could be used as an “off-the-shelf” therapy for patients whose apheresis may not be feasible due to severe lymphopenia. This universal CAR T-cell, termed UCART19, has been genetically engineered to not only express the CD19 targeting CAR construct but also knock out the endogenous TRAC locus and CD52 using transcription activator-like effector nucleases.49 Removal of the endogenous TRAC locus aids in the prevention of UCART19 exhaustion, while the lack of CD52 expression renders these cells resistant to the anti-CD52 monoclonal antibody alemtuzumab. Endogenous mature T and B cells are depleted with the administration of alemtuzumab, which can be given either before or concurrently with UCART19 infusion, enhancing its engraftment. However, boosting UCART19 engraftment with alemtuzumab is not without risk, as its use has been linked to significant increases in opportunistic infections such as cytomegalovirus reactivation.50 Finally, UCART19 also includes an RQR8 “safety switch” that allows for targeted elimination of RQR8+ cells by the anti-CD20 antibody rituximab. Initial uses of UCART19 in infants unable to generate enough T cells for autologous CAR T-cell therapy have been promising, with both patients achieving CR at Day 28 postinfusion.49 There are currently two ongoing clinical trials investigating the efficacy of UCART19 therapy, one in adults (NCT02746952) and the other in pediatric patients (NCT02808442) with relapsed or refractory B-ALL. Universal allogeneic CAR T-cells have the potential to elicit host immune responses, a problem that may be addressed by knocking out human leukocyte antigen (HLA) class I. However, such HLA class I-deficient cells would remain susceptible to deletion through the “missing-self recognition” ability of natural killer (NK) cells. Researchers have addressed this problem in human pluripotent stem cells, where HLA class I genes A, B, and C have been knocked out, with an HLA-E single-chain dimer fused to beta-2 microglobulin being knocked in.51 These cells are not recognized as allogeneic by host T cells and are resistant to NK-mediated lysis. The application of this strategy to CAR T-cells may facilitate the generation of a safer universal product.
In an attempt to limit the exclusion of research participant enrollment, a clinical trial was initiated by Gardner et al with a defined formulation of CAR T-cell therapy.52 In this trial, a CD19 CAR product with a 1:1 CD4:CD8 T-cell ratio, uniform CAR expression, and limited effector differentiation was manufactured for 93% of the pediatric and young adult patients enrolled. Furthermore, CR was achieved in 89% of the intent-to-treat population, an important advancement compared with prior trials, where up to 24% of patients were excluded based on a predicted failure to produce a CAR T-cell product.53 The estimated 12-month disease-free survival was 50.8%, with a 12-month overall survival of 69.5%. CD19− relapse was seen in seven patients, a type of relapse that was also seen in the CTL019 clinical trials.
Additional sources of variation between clinical trials of CD19 CAR T-cell therapies are the drugs/dosages used for lymphodepletion, as well as the number of CAR T-cells infused into the patient (Table 2). The most common lymphodepletion drug combination was fludarabine and cyclophosphamide, but doses of cyclophosphamide varied from 900 mg/m2 to 1,000 mg/m2 per course when combined with fludarabine. Some patients received alternate lymphodepletion regimens including high-dose cyclophosphamide (up to 4 g/m2), fludarabine with or without cytarabine, and cyclophosphamide with etoposide. While potentially many regimens are adequate enough to generate lymphodepletion, it is not clear which is most optimal and if a target absolute lymphocyte count is needed. The maximum tolerated dose of CAR T-cells was determined to be 1×106/kg in both the Gardner et al and KTE-C19 Phase I trials,42,52 and 20.6×106/kg in the CTL019 Phase I trial.23
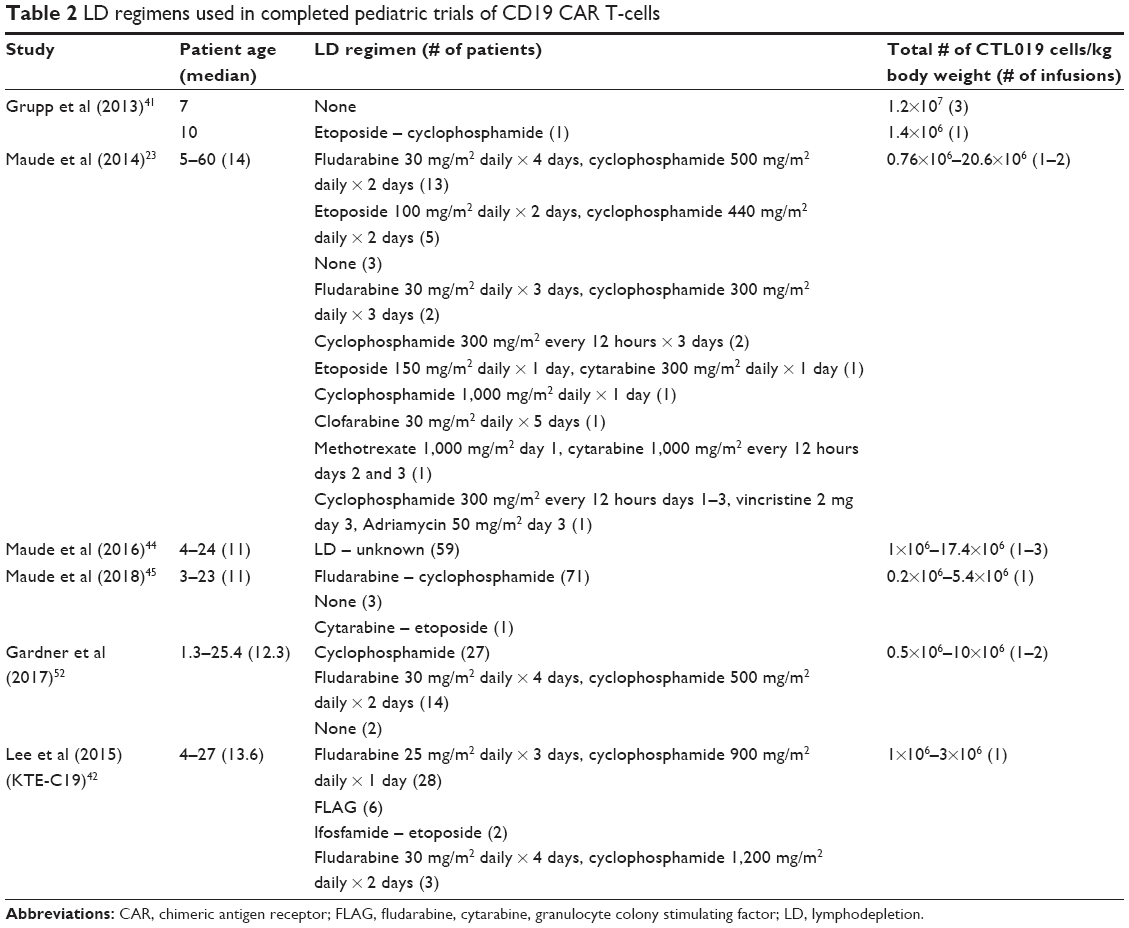 | Table 2 LD regimens used in completed pediatric trials of CD19 CAR T-cells |
Relapses after CD19 CAR T-cell therapy can be CD19+ or CD19−. It is unknown if CD19+ relapses can be salvaged with additional infusions of CAR T-cells. In an effort to treat CD19− relapse patients, other B-cell-specific antigens have been investigated for selection as targets for novel CAR T-cell generation (Figure 2). Promising new single antigen CARs have been developed for the treatment of B-cell malignancies that target ROR1,54 CD20,55 CD22,56 and thymic stromal lymphopoietin receptor.57 Worldwide ongoing clinical trials investigating CAR T-cell therapy for pediatric B-ALL are available (Table 3). Of note, a bispecific CAR targeting the B-cell antigens CD19 and CD20 simultaneously has shown encouraging results,58,59 while a separate bispecific CAR targeting CD19 and CD22 is currently being tested in an ongoing clinical trial (NCT03241940). A successful clinical trial has been conducted that treated relapsed B-ALL patients who had previously undergone CTL019 infusion, but then experienced a CD19− relapse, with CD22 CAR T-cells.60 Seventy-three percent of patients who received 1×106 CD22 CAR T-cells per kilogram body weight achieved a CR, including 5/5 patients with CD19− B-ALL. Remissions were not durable as nine patients relapsed, seven of which were associated with diminished CD22 surface expression. Finally, the efficacy of a sequential infusion of anti-CD19 and anti-CD20 CAR T-cells as a single treatment is also currently being investigated (NCT03207178).
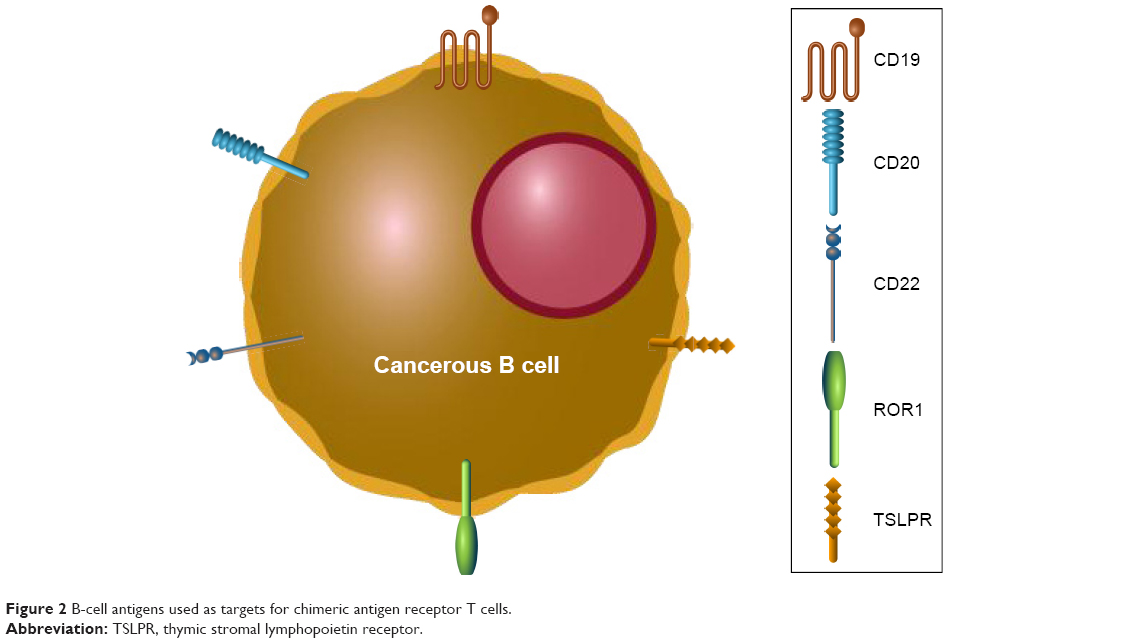 | Figure 2 B-cell antigens used as targets for chimeric antigen receptor T cells. |
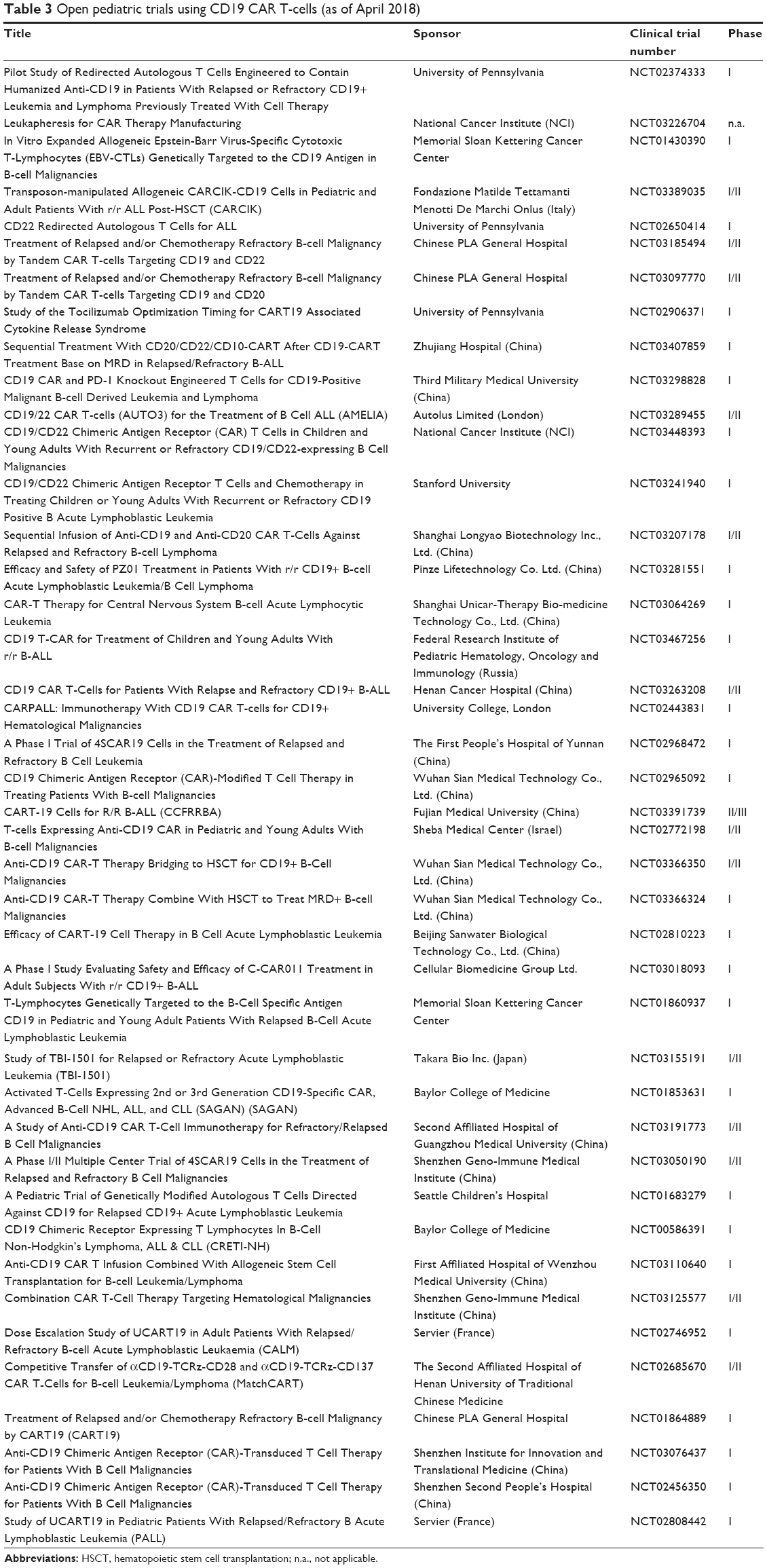 | Table 3 Open pediatric trials using CD19 CAR T-cells (as of April 2018) |
Potential future directions of CD19 CAR T therapy
Ongoing preclinical research in CAR T-cell therapy could potentially be used to enhance the efficacy and safety profile of CD19-directed CARs. One method to reduce on-target off-tumor toxicity is through the development of a tunable CAR T-cell. This can be achieved with an “on” or an “off” switch whose activation is dependent on the administration of a small molecule. In the case of “on” switch CARs, research has demonstrated that the CD19 binding scFv domain can be separated from the intracellular signaling domain, with their fusion being dependent on the small molecule rapalog.61 A different type of “on” switch has also been developed, where the expression of the CD19 CAR construct is dependent on the administration of doxycycline.62 CAR T-cell activity could also be controlled through the activation of an “off” switch. In this scenario, the dimerization of caspase-9, engineered to be part of the cytoplasmic domain, is made inducible through the administration of a specific small molecule. A recent study has shown that >90% of CARs can be induced to undergo apoptosis using this approach, with the percentage of apoptotic cells dependent on the amount of caspase-9 binding small molecules administered.63 Both “on” and “off” switch CAR T-cells would allow physicians to precisely control the timing and dosage of CAR T-cell activation.
A major factor contributing to CD19 CAR T-cell exhaustion is the constitutively active cytoplasmic signaling domains. Incorporation of the CAR construct by lentiviral or retroviral transduction can result in overexpression and contribute to low-level constitutive (tonic) signaling in an antigen-independent manner.64 With the recent advancement in genome editing technology, it is now possible to directly target the genomic integration of the CAR transgene. A recent study used CRISPR/Cas9 to direct genomic integration of a CAR construct to the T-cell receptor locus (TRAC).65 Results demonstrated not only robust CAR expression but also enhanced T-cell potency, a reduction of inhibitory receptor expression characteristic of exhaustion (such as PD-1) and prevention of tonic signaling. CRISPR/Cas9 has also been used to generate PD-1 knockout CD19 CAR T-cells, which was demonstrated to enhance CAR T-cell-mediated killing of tumor cells in vitro and increased clearance of PD-L1+ tumor xenografts in vivo.66
In a recent study, researchers reported on the unique case of a 78-year-old patient who had undergone CTL019 therapy for relapsed/refractory CLL.67 At peak response in this patient, it was discovered that 94% of CD19 CAR T-cells originated from a single clone in which CTL019 transgene integration occurred in the TET2 gene, disrupting its function.67 Further analysis revealed that TET2-disrupted CART T cells had a greater proliferative capacity, increased levels of degranulation, and exhibited a central memory phenotype. This patient remained relapse-free 5 years postinfusion.67 This surprising discovery, when combined with CRISPR genome editing tools, suggests that disruption of TET2 locus may increase the potency of CTL019 cells.
Besides enhancing CAR T function through innovative bioengineering, one could imagine using tisagenlecleucel either for first relapse or upfront for patients with high risk or very high-risk disease. Because chemotherapy and allogeneic HSCT are associated with long-term, end-organ toxicities, usage of tisagenlecleucel has appeal since most of the toxicity appears in the first 30–60 days after infusion. Other than secondary B-cell aplasia, long-term toxicities have yet to be reported. Using CAR T earlier in treatment, such as when there is little to no blast burden after induction chemotherapy, may reduce the risk of severe CRS because bone marrow blast counts >50% correlate with severe CRS.23 In addition, it is not yet known if tisagenlecleucel is best used as a bridge to allogeneic HSCT or as a standalone curative therapy.
Access to CAR T-cell therapy
An emerging bioethical concern regarding CAR T-cell therapy is access – primarily overcoming geographical barriers and financial barriers. Providing access to CAR T-cell therapy globally involves a broader improvement in medical facilities and the development of subsidized oncology diagnosis and treatment facilities. Within high-income countries, the primary barriers currently are cost and reimbursement strategies. Manufacturers of CAR T-cell therapies are working with governments to streamline reimbursements and implement new pricing models and have justified the high one-time cost of CAR T-cell therapies,68,69 while patient advocates and government representatives question the high sticker price of these therapies.70 It is hoped that further improvements in technology and wider adoption of CAR T-cell therapy will ultimately reduce its cost and increase its availability to a larger segment of the population.
For geographical barriers, there is limited access to standard infrastructure globally for cancer care including access to radiation (>50% of the world population lack access)71 and publicly funded pathology services (only 26% of low-income countries have access).72 CAR T-cell therapy delivery requires facilities and expertise exceeding those required for bone marrow transplantation. Currently, 57% of cancer cases occur in low- and middle-income countries, and 65% of cancer deaths occur in those countries as well.71 It is also likely that many cancers that are diagnosed through regular screening and accessibility to pathology services are underreported in those numbers. Given these broad trends, methods for increasing global access to oncology and specifically to CAR T-cell therapy need to be developed.
CAR T-cell therapy costs more than other leading cancer therapies – tisagenlecleucel has been made available at a price of $475,000 in the USA, while comparable new orally administered cancer medicines cost at least $135,000 per year as of 2014.73 This high cost places it out of reach for most patients paying out-of-pocket and increases the burden on private and public insurers.74 Novartis has proposed an outcome-based agreement, or a “money-back guarantee,” for tisagenlecleucel: the price of the drug is refunded in case of failed treatment within the first month.73 However, it should be noted that a significant number of patients who are in CR at 1 month will later relapse with disease. In addition, the exact terms of the outcome-based agreement are not publicly available. This uncertainty prompted a letter from members of Congress to Novartis asking for clarification on a number of issues including who the responsible party is for reimbursement, and what the specific criteria is for determining a successful 1-month response, among others.70
Tisagenlecleucel has been approved for refractory diffuse large B-cell lymphoma recently,75 which is the primary indication for the other approved CAR T-cell product axicabtagene ciloleucel. Announcements from Novartis have indicated that the price of tisagenlecleucel will be based on indication, meaning that the price of the drug will vary depending on the clinical condition it is being used for.76 One analysis argued that indication-based pricing can help increase access to oncology medications.76 Such an approach lowered the price of tisagenlecleucel to $373,000 for use against large B-cell lymphomas, pricing it competitively with axicabtagene ciloleucel.77
Pricing of CAR T-cell therapy has remained contentious since their approval in August 2017.78 While the Centers for Medicare and Medicaid Services (CMS) have issued statements stating their commitment toward the “development of innovative pricing systems that reflect value delivered to patients,”79 there have not been any definitive statements outlining these systems. The CMS decides on reimbursement of medical care for patients receiving public subsidies for health care within the USA; collectively, they are responsible for payment of health care expenses for 72 million American citizens.80 The ambiguity in CMS pricing has been questioned by a select group of members of the US House of Representatives70 and one of the largest private insurers in the USA.81
An analysis of the effectiveness and value of CAR T-cell therapies for B-cell cancers by the Institute for Clinical and Economic Review, an independent nonprofit organization, found that, “these therapies seem to be priced in alignment with clinical benefits over a lifetime time horizon.”82 They found that tisagenlecleucel provided at least seven more quality-adjusted life years (QALY) compared with clofarabine. The cost difference between tisagenlecleucel and clofarabine indicated that tisagenlecleucel is cost-effective if a QALY is valued >$45,871. Current estimations indicate that the minimum value of a QALY is around $50,000 and should be higher.83 Analyses of cost-effectiveness performed by Novartis funded researchers have reached similar conclusions within the US context68 and within the UK.69 These political and ethical pressures to increase access to CAR T-cell therapies are likely to shape the standards by how CAR T-cell therapies are deployed for the next decade.
Conclusion
Prior advancements in the treatment of relapsed or refractory pediatric B-ALL have been limited by low 5-year survival, with early bone marrow relapses far too common.84 CTL019, now known as tisagenlecleucel, is the first gene therapy and CAR T-cell therapy to be FDA approved, and its early success provides a new hope for the thousands of children and young adults who relapse with B-ALL each year. The efficacy of CAR T-cell therapy will likely be improved due to the optimization of its delivery, as well as new advancements in CAR T-cell engineering. As we scratch the surface of personalized medicine through adoptive cell therapy with CAR T-cells, the future of relapsed or refractory pediatric B-ALL treatment looks bright.
Acknowledgments
This work was supported by grants from Stand Up To Cancer St Baldrick’s Pediatric Dream Team Translational Research Grant SU2C-AACR-DT1113 (CMC), the NCI/NIH K08 CA174750 (CMC), NIH/NCI P30 CA014520 to the University of Wisconsin Carbone Cancer Center (KS and CMC), NSF CBET-1350178 (KS), and NSF CBET-1645123 (KS and CMC). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. The contents of this article neither necessarily reflect the views or policies of the Department of Health and Human Services nor do mention trade names, commercial products, or organizations imply endorsement by the US Government.
Disclosure
CMC received honorarium for serving on a one-time advisory board for Novartis. The authors report no other conflicts of interest in this work.
References
Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012;30(14):1663–1669. | ||
Möricke A, Zimmermann M, Reiter A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010;24(2):265–284. | ||
Annesley CE, Brown P. Novel agents for the treatment of childhood acute leukemia. Ther Adv Hematol. 2015;6(2):61–79. | ||
Locatelli F, Schrappe M, Bernardo ME, Rutella S. How I treat relapsed childhood acute lymphoblastic leukemia. Blood. 2012;120(14):2807–2816. | ||
Cooper SL, Brown PA. Treatment of pediatric acute lymphoblastic leukemia. Pediatr Clin North Am. 2015;62(1):61–73. | ||
Ko RH, Ji L, Barnette P, et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol. 2010;28(4):648–654. | ||
Jeha S, Gaynon PS, Razzouk BI, et al. Phase II study of clofarabine in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. J Clin Oncol. 2006;24(12):1917–1923. | ||
von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381–4389. | ||
von Stackelberg A, Völzke E, Kühl JS, et al. Outcome of children and adolescents with relapsed acute lymphoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of the ALL-REZ BFM Study Group. Eur J Cancer. 2011;47(1):90–97. | ||
Eshhar Z, Bach N, Fitzer-Attas CJ, et al. The T-body approach: potential for cancer immunotherapy. Springer Semin Immunopathol. 1996;18(2):199–209. | ||
Chmielewski M, Hombach AA, Abken H. Antigen-specific T-cell activation independently of the MHC: chimeric antigen receptor-redirected T cells. Front Immunol. 2013;4:371. | ||
Huston JS, Levinson D, Mudgett-Hunter M, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci U S A. 1988;85(16):5879–5883. | ||
Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, Eshhar Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J Immunol. 1993;151(11):6577–6582. | ||
Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161(6):2791–2797. | ||
Hombach A, Wieczarkowiecz A, Marquardt T, et al. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol. 2001;167(11):6123–6131. | ||
Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. | ||
Hombach AA, Chmielewski M, Rappl G, Abken H. Adoptive immunotherapy with redirected T cells produces CCR7- cells that are trapped in the periphery and benefit from combined CD28-OX40 costimulation. Hum Gene Ther. 2013;24(3):259–269. | ||
Tammana S, Huang X, Wong M, et al. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. 2010;21(1):75–86. | ||
Hu WS, Pathak VK. Design of retroviral vectors and helper cells for gene therapy. Pharmacol Rev. 2000;52(4):493–511. | ||
Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92–101. | ||
Hay KA, Hanafi LA, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130(21):2295–2306. | ||
Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6(6):664–679. | ||
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. | ||
Brentjens RJ, Rivière I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. | ||
Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012;14(6):405–415. | ||
Haynes NM, Trapani JA, Teng MW, et al. Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol. 2002;169(10):5780–5786. | ||
Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172(1):104–113. | ||
Hurtado JC, Kim YJ, Kwon BS. Signals through 4-1BB are costimulatory to previously activated splenic T cells and inhibit activation-induced cell death. J Immunol. 1997;158(6):2600–2609. | ||
May KF, Chen L, Zheng P, Liu Y. Anti-4-1BB monoclonal antibody enhances rejection of large tumor burden by promoting survival but not clonal expansion of tumor-specific CD8+ T cells. Cancer Res. 2002;62(12):3459–3465. | ||
Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. | ||
Mogi S, Sakurai J, Kohsaka T, et al. Tumour rejection by gene transfer of 4-1BB ligand into a CD80(+) murine squamous cell carcinoma and the requirements of co-stimulatory molecules on tumour and host cells. Immunology. 2000;101(4):541–547. | ||
Brentjens RJ, Latouche JB, Santos E, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9(3):279–286. | ||
Brentjens RJ, Santos E, Nikhamin Y, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13(18 Pt 1):5426–5435. | ||
Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. | ||
Müschen M, Lee S, Zhou G, et al. Molecular portraits of B cell lineage commitment. Proc Natl Acad Sci U S A. 2002;99(15):10014–10019. | ||
Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma. 1995;18(5–6):385–397. | ||
Schwonzen M, Pohl C, Steinmetz T, et al. Immunophenotyping of low-grade B-cell lymphoma in blood and bone marrow: poor correlation between immunophenotype and cytological/histological classification. Br J Haematol. 1993;83(2):232–239. | ||
Stamenkovic I, Seed B. CD19, the earliest differentiation antigen of the B cell lineage, bears three extracellular immunoglobulin-like domains and an Epstein-Barr virus-related cytoplasmic tail. J Exp Med. 1988;168(3):1205–1210. | ||
Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;395(95):ra73. | ||
Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. | ||
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. | ||
Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. | ||
Lee DW, Stetler-Stevenson M, Yuan CM, et al. Safety and response of incorporating CD19 chimeric antigen receptor T cell therapy in typical salvage regimens for children and young adults with acute lymphoblastic leukemia. Blood. 2015;126(23):684. | ||
Maude SL, Teachey DT, Rheingold SR, et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. J Clin Oncol. 2016;34(15_suppl):3011. | ||
Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. | ||
Fitzgerald JC, Weiss SL, Maude SL, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017;45(2):e124–e131. | ||
Mcguirk J, Waller EK, Qayed M, et al. Building blocks for institutional preparation of CTL019 delivery. Cytotherapy. 2017;19(9):1015–1024. | ||
Allen ES, Stroncek DF, Ren J, et al. Autologous lymphapheresis for the production of chimeric antigen receptor T cells. Transfusion. 2017;57(5):1133–1141. | ||
Qasim W, Zhan H, Samarasinghe S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9(374):eaaj2013. | ||
Laurenti L, Piccioni P, Cattani P, et al. Cytomegalovirus reactivation during alemtuzumab therapy for chronic lymphocytic leukemia: incidence and treatment with oral ganciclovir. Haematologica. 2004;89(10):1248–1252. | ||
Gornalusse GG, Hirata RK, Funk SE, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. 2017;35(8):765–772. | ||
Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–3331. | ||
Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. 2016;8(320):320ra3. | ||
Hudecek M, Schmitt TM, Baskar S, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116(22):4532–4541. | ||
Zhang WY, Wang Y, Guo YL, et al. Treatment of CD20-directed chimeric antigen receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal Transduct Target Ther. 2016;1:16002. | ||
Qin H, Zhang L, Orentas RJ, Fry TJ. CD22-targeted chimeric antigen receptor (CAR) T cells containing the 4-1BB costimulatory domain demonstrate enhanced persistence and superior efficacy against B-cell precursor acute lymphoblastic leukemia (ALL) compared to those containing CD28. Blood. 2013;122(21):1431. | ||
Qin H, Cho M, Haso W, et al. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood. 2015;126(5):629–639. | ||
Martyniszyn A, Krahl AC, André MC, Hombach AA, Abken H. CD20-CD19 bispecific CAR T cells for the treatment of B-cell malignancies. Hum Gene Ther. 2017;28(12):1147–1157. | ||
Schneider D, Xiong Y, Wu D, et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer. 2017;5(1):42. | ||
Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. | ||
Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077. | ||
Sakemura R, Terakura S, Watanabe K, et al. A tet-on inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol Res. 2016;4(8):658–668. | ||
Diaconu I, Ballard B, Zhang M, et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther. 2017;25(3):580–592. | ||
Frigault MJ, Lee J, Basil MC, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3(4):356–367. | ||
Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. | ||
Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. | ||
Fraietta JA, Nobles CL, Sammons MA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558(7709):307–312. | ||
Hao Y, Eldjerou LK, Yang H, Qi C, Globe D. Cost-Effectiveness Analysis of CTL019 for the Treatment of Pediatric and Young Adult Patients with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia in the United States. Blood. 2017;130 (Suppl 1):609. | ||
Batt JS, Michelle B, Yanni H. The Economic Value of CTL019 Therapy for Pediatric Patients with Relapsed and Refractory Acute Lymphoblastic Leukemia in the United Kingdom. Blood. 2017;130 (Suppl 1):1330. | ||
Rep. Doggett Calls for Transparency in New Cancer Drug Pricing Agreement; 2017. Available from: https://doggett.house.gov/media-center/press-releases/rep-doggett-calls-transparency-new-cancer-drug-pricing-agreement. Accessed July 26, 2018. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Cancer; 2018. Available from: http://www.who.int/news-room/fact-sheets/detail/cancer. Accessed May 31, 2018. | ||
Dolgin E. Bringing down the cost of cancer treatment. Nature. 2018;555(7695):S26–S29. | ||
Barlas S. Are specialty drug prices destroying insurers and hurting consumers? A number of efforts are under way to reduce price pressure. P T. 2014;39(8):563–566. | ||
Kymriah® (tisagenlecleucel)first-in-class CAR-T therapy from Novartisreceives second FDA approval to treat appropriate r/r patients with large B-cell lymphoma Novartis; 2018. Available from: https://www.novartis.com/news/media-releases/kymriahr-tisagenlecleucel-first-class-car-t-therapy-from-novartis-receives-second-fda-approval-treat-appropriate-rr-patients-large-b-cell-lymphoma. Accessed July 26, 2018. | ||
Chandra A, Garthwaite C. The Economics of Indication-Based Drug Pricing; 2017. N Engl J Med. 2017;377(2):103–106. | ||
Tisagenlecleucel (Kymriah) Approved to Treat Some Lymphomas – National Cancer Institute; 2018. Available from: https://www.cancer.gov/news-events/cancer-currents-blog/2018/tisagenlecleucel-fda-lymphoma. Accessed May 31 2018. | ||
Novartis receives first ever FDA approval for a CAR-T cell therapy, Kymriah (TM) (CTL019). For children and young adults with B-cell ALL that is refractory or has relapsed at least twice|Novartis. 2018. Available from: https://www.novartis.com/news/media-releases/novartis-receives-first-ever-fda-approval-car-t-cell-therapy-kymriahtm-ctl019. Accessed May 31, 2018. | ||
CMS: Innovative treatments call for innovative payment models and arrangements; 2018. Available from: https://www.cms.gov/Newsroom/MediaReleaseDatabase/Press-releases/2017-Press-releases-items/2017-08-30-2.html. Accessed May 31, 2018. | ||
CMS Fast Facts; 2018. Available from: https://www.cms.gov/fastfacts/. Accessed July 26, 2018. | ||
Castillo E. Formal Request for National Coverage Determination for Chimeric Antigen Receptor T-Cell Therapies; 2018. Available from: https://www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/id291.pdf. | ||
Chimeric Antigen Receptor T-Cell Therapy for B-Cell Cancers: Effectiveness and Value. Institute for Clinical and Economic Review; 2018. | ||
Neumann PJ, Cohen JT, Weinstein MC. Updating Cost-Effectiveness – The Curious Resilience of the $50,000-per-QALY Threshold. N Engl J Med. 2014;371(9):796–797. | ||
Bailey LC, Lange BJ, Rheingold SR, Bunin NJ. Bone-marrow relapse in paediatric acute lymphoblastic leukaemia. Lancet Oncol. 2008;9(9):873–883. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.