Back to Journals » International Journal of Nanomedicine » Volume 11
The potential applications of fibrin-coated electrospun polylactide nanofibers in skin tissue engineering
Authors Bacakova M, Musilkova J, Riedel T ![]() , Stranska D, Brynda E, Zaloudkova M, Bacakova L
, Stranska D, Brynda E, Zaloudkova M, Bacakova L ![]()
Received 28 October 2015
Accepted for publication 7 January 2016
Published 25 February 2016 Volume 2016:11 Pages 771—789
DOI https://doi.org/10.2147/IJN.S99317
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Marketa Bacakova,1,2 Jana Musilkova,1 Tomas Riedel,3 Denisa Stranska,4 Eduard Brynda,3 Margit Zaloudkova,5 Lucie Bacakova1
1Department of Biomaterials and Tissue Engineering, Institute of Physiology, Czech Academy of Sciences, 2Second Faculty of Medicine, Charles University in Prague, 3Institute of Macromolecular Chemistry, Czech Academy of Sciences, Prague, 4InStar Technologies, Liberec, 5Institute of Rock Structure and Mechanics, Czech Academy of Sciences, Prague, Czech Republic
Abstract: Fibrin plays an important role during wound healing and skin regeneration. It is often applied in clinical practice for treatment of skin injuries or as a component of skin substitutes. We prepared electrospun nanofibrous membranes made from poly(L-lactide) modified with a thin fibrin nanocoating. Fibrin surrounded the individual fibers in the membrane and also formed a thin fibrous mesh on several places on the membrane surface. The cell-free fibrin nanocoating remained stable in the cell culture medium for 14 days and did not change its morphology. On membranes populated with human dermal fibroblasts, the rate of fibrin degradation correlated with the degree of cell proliferation. The cell spreading, mitochondrial activity, and cell population density were significantly higher on membranes coated with fibrin than on nonmodified membranes, and this cell performance was further improved by the addition of ascorbic acid in the cell culture medium. Similarly, fibrin stimulated the expression and synthesis of collagen I in human dermal fibroblasts, and this effect was further enhanced by ascorbic acid. The expression of beta1-integrins was also improved by fibrin, and on pure polylactide membranes, it was slightly enhanced by ascorbic acid. In addition, ascorbic acid promoted deposition of collagen I in the form of a fibrous extracellular matrix. Thus, the combination of nanofibrous membranes with a fibrin nanocoating and ascorbic acid seems to be particularly advantageous for skin tissue engineering.
Keywords: electrospun nanofibers, nanocoating, skin tissue engineering, fibroblasts, fibrin, ascorbic acid, nanotechnology, nanomedicine, collagen I synthesis, beta1-integrins
Introduction
Nanostructured materials are advantageously applied for constructing a tissue replacement, because they are more attractive for cell adhesion, proliferation, and differentiation than flat or microstructured surfaces. The nanostructure of the material mimics the nanoarchitecture of natural extracellular matrix (ECM), eg, its nanofibrous component, and it enhances the cell–material interaction. Nanostructured materials are capable of adsorbing cell adhesion-mediating ECM molecules from the body fluid or from the cell culture medium in an appropriate spatial conformation. This conformation is important for good accessibility of specific oligopeptidic ligands of ECM molecules to cell adhesion receptors called integrins.1,2
Nanostructured materials can promote skin regeneration by improving the adhesion and proliferation of skin cells and the neovascularization of a tissue-engineered skin implant. They enable cells to be supplied with oxygen and nutrition and prevent fluid accumulation at the wound site.3
Most models of skin substitutes are based on a nonresorbable artificial material and allogenic cells. These substitutes cannot provide permanent coverage because they will finally be rejected.4 A promising approach could be to fabricate nanofibrous biodegradable scaffolds colonized with autologous fibroblasts and keratinocytes. The degradable scaffolds are slowly resorbed in the organism and are finally replaced by a newly formed tissue.
The most widespread technique for preparing nanofibrous scaffold is electrospinning. Due to increasing requirements of the electrospun nanofibrous scaffold for biomedical purposes, several innovative methods of electrospinning have been developed. One approach is the needleless electrospinning enabling the production of nanofibers on a large scale.5 Another method is the creation of structural nanofibers using multiple-fluid electrospinning such as coaxial,6,7 triaxial,8 or side-by-side9 process. The multiple-fluid electrospinning enables encapsulation of drugs or other biological agents into polymeric nanofibrous scaffold and can be potentially applied for drug delivery. In addition, the electrospinning can be combined with other traditional methods or after-treatment of electrospun nanofibers. Our structural nanofibers were fabricated by coating fibrin on the electrospun polylactide (PLA) nanofibers that were created using a needleless electrospinning technique.
Promising materials for developing nanofibrous degradable scaffolds are synthetic degradable polymers, namely PLAs,10–12 and their copolymers with glycolides13 or polyethylene glycol.14 Other examples include poly (epsilon-caprolactone)15 or segmented copolymers of polyethylene oxide terephthalate and polybutylene terephthalate.16
However, synthetic polymers used for skin tissue engineering often do not favor cellular adhesion, growth, and matrix deposition in their pristine unmodified state.11 In our earlier study, PLA nanofibrous membranes were modified by plasma treatment in order to enhance the adhesion and growth of human keratinocytes on these carriers.12 Other modifications included etching of the polymers in NaOH in order to increase their wettability and their electrical charge,10 copolymerization or blending of the polymers with other more hydrophilic synthetic polymers,17 and particularly combining synthetic polymers with molecules physiologically present in the skin, such as collagen,10 hyaluronan,18 and fibronectin.17 Another approach is through modification by fibrin.19,20 Fibrin is a temporary matrix molecule that forms during wound healing. It offers the advantage that fibrinogen, a precursor of fibrin, can be isolated in a considerable amount from the patient’s blood in autologous form to prevent unwanted immune reactions.21,22
Fibrin plays an important role in skin regeneration. It is a biopolymer resulting from enzymatic reactions of the coagulation cascade. Activation of this cascade leads to the conversion of soluble fibrinogen by thrombin into a network of insoluble fibrin fibers.23 The fibrin network stabilizes the platelet plug by binding platelets to the fibrin. The platelets secrete growth factors, namely platelet-derived growth factor, which stimulates fibroblasts to proliferate, migrate into the wound site, and produce collagen I and III, glycosaminoglycans, and proteoglycans. Moreover, the fibrin network binds other cell types, such as smooth muscle cells and endothelial cells, through integrin adhesion receptors. In addition, fibrin binds cell adhesion-mediating ECM proteins, fibronectin, and vitronectin, which support the adhesion of fibroblasts.24
During the wound healing process, dermal fibroblasts secrete ECM proteins, mainly collagen of types I and III, which are the major component of ECM in the skin dermis.23 The synthesis of collagen is enhanced by water-soluble ascorbic acid (vitamin C). Ascorbic acid plays a role as a cofactor for enzymes, prolyl, and lysyl hydroxylases, catalyzing the synthesis of hydroxyproline and hydroxylysine in collagen. In addition, ascorbic acid also stimulates collagen biosynthesis by increasing the steady-state level of procollagen mRNA.25,26 A previous study has shown that the fibroblasts cultivated in a three-dimensional system consisting of ECM molecules not only secreted collagen into the cell culture medium but also deposited it on the cultivation substrate as collagen fibrils.27 Similarly, human dermal fibroblasts cultured on composite polymeric nanofibrous scaffolds blended with ascorbic acid synthesized more collagen than that synthesized on nonblended scaffolds.3
For skin tissue engineering and wound healing, fibrin has been used mostly in the form of self-supporting structures, such as gels,18,28,29 a glue,30,31 and microbeads.30,32 It has been relatively rarely used for surface modification of scaffolds, with the exception of collagen–chitosan porous membranes,31 or on electrospun poly(lactide-glycolide-caprolactone) nanofibers.33 To the best of our knowledge, a combination of fibrin and ascorbic acid has not previously been used for constructing potential skin replacements, although this combination has been applied in tissue engineering of tendon and ligament,34 cartilage,35 bone,36 smooth muscle,37 and heart valves.38 In these applications, ascorbic acid promoted appropriate differentiation and phenotypic maturation of cells and increased the collagen production in these cells.
The aim of this study is therefore to evaluate the adhesion and growth of human dermal fibroblasts on PLA nanofibrous membranes modified with a cell-degradable fibrin nanocoating. We further investigated the effect of a combination of nanofibrous membranes, fibrin nanocoating, and ascorbic acid on the synthesis of collagen by fibroblasts, mainly in the form of collagen fibrils deposited on the membrane surface as ECM.
Materials and methods
Preparation of nanofibrous membranes
Poly(L-lactide) (PLA; Ingeo™ Biopolymer 4032D) was purchased from NatureWorks, Minnetonka, MN, USA. The molecular parameters of the polymer material, determined by size exclusion chromatography, were Mw=124,000 g/mol and Mn=48,000 g/mol.39 A 7 wt% solution of PLA in a mixture of chloroform, dichloroethane, and ethyl acetate in the volume ratio of 61:29:10 was used for the electrospinning process. The polymer was diluted only in chloroform, and the other two solvents were added after the dilution. The polymer solution was made electrically conductive with the use of tetraethylammonium bromide, which was first dissolved in dimethylformamide at a concentration of 3 wt%, and then 3 g of this solution was added to 100 g of the PLA solution.40
Nanofibrous membranes were prepared using the novel Nanospider needleless electrospinning technology (NS Lab 500; Elmarco Ltd., Liberec, Czech Republic). The process conditions were: electrode distance: 180 mm; voltage: 60–10 kV; the spinning electrode rotated at 4 rpm; relative humidity: 25%–30%, and room temperature. The fiber density, ie, the area weight of the prepared nanofibers, was from 12 to 15 g/m2.
Preparation of a fibrin nanocoating on PLA membranes
The fibrin nanocoating on a nanofibrous membrane was created by activation of human fibrinogen (EMD Millipore, Billerica, MA, USA) with human thrombin (Sigma-Aldrich Co., St Louis, MO, USA), as published previously.41 Human fibrinogen and human thrombin are commercially available, and in our study both chemicals were purchased from international well-known companies and therefore no ethical approval was required.
A total of 10 μg/mL of fibrinogen in Tris buffer (consisting of 50 mM Tris–HCl, 100 nM NaCl, and 2.5 mM CaCl2) was adsorbed on the surface of the membrane for 1 hour. The adsorbed fibrinogen was rinsed with Tris buffer and was activated with thrombin (2.5 U/mL in Tris buffer) for 15 minutes. After thrombin activation, the membranes were rinsed with Tris Buffer. Finally, a solution of 200 μg/mL of fibrinogen and 0.5 U/mL of antithrombin III (Chromogenix, Milano, Italy) was added to the membranes for 1 hour. A fibrin network was formed by a catalytic reaction of the surface-attached thrombin with the ambient fibrinogen solution. Antithrombin III blocked unreacted thrombin to form a two-dimensional fibrin layer.
Scanning electron microscopy
The morphology of the PLA membrane in pristine state (nonmodified membrane) was studied by scanning electron microscopy. The membranes were sputter-coated with gold and were evaluated by a Quanta 450 scanning electron microscope (FEI, Hillsboro, OR, USA) in high vacuum mode. The images were taken using ETD – the Everhart–Thornley detector in secondary electrons mode at high voltage of 20 kV and magnification of 2,000× and 10,000×. The diameter of the fibers was measured on the scanning electron microscopy images using Atlas software (Tescan Ltd., Brno, Czech Republic).
Cell culture
The nanofibrous membranes were fixed in Cell Crown inserts (Scaffdex Ltd., Tampere, Finland) and were inserted into polystyrene 24-well or six-well cell culture plates (TPP, Trasadingen, Switzerland; well diameter: 1.56 or 3.38 cm). The samples were seeded with neonatal human dermal fibroblasts, purchased from Lonza (Basel, Switzerland), in passage 5 at a density of 10,000 cells/cm2. Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich Co.) supplemented with 10% of fetal bovine serum (FBS; Sebak GmbH, Aidenbach, Germany) and 40 μg/mL of gentamicin (LEK, Ljubljana, Slovenia) was used for cultivating the cells on the samples. The volume of cell culture medium was 1.5 mL/sample for the 24-well cell culture plate and 6 mL/sample for the six-well cell culture plate. The cells were cultivated for 1, 3, 5, 7, 10, and 14 days in a cell incubator at 37°C and in a humidified atmosphere with 5% of CO2 in the air. The medium was changed after 1 week of cell cultivation and subsequently every 3 days. Polystyrene culture dishes (24-well or six-well plates) or glass coverslips were used as control materials. On day 1 (for β1-integrin evaluation) or on day 3 (for other experiments) after cell seeding, 50 μg/mL 2-phospho-L-ascorbic acid trisodium salt (AA) was added into the cell culture medium of one half of the samples to stimulate the cells to produce collagen.
Cell mitochondrial activity
Cell mitochondrial activity was used as an indirect marker of cell proliferation activity, because it usually increases with increasing cell number. This parameter was measured in six intervals of cell cultivation (on days 1, 3, 5, 7, 10, and 14 after cell seeding) by CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS; Promega Corporation, Madison, WI, USA) on samples of nanofibrous membranes incubated in a 24-well cell culture plate. The principle of this assay is based on the cleavage of yellow tetrazolium salt MTS and on the formation of water-soluble brown formazan salt by the activity of mitochondrial enzymes in cells. The formazan dye is then quantified by measuring the absorbance using a spectrophotometer (ELISA reader).
Four independent samples for each experimental group and time interval were used. One sample without cells for each experimental group and time interval was used as a control to set the background of the measured absorbance. A 24-well polystyrene culture dish was used as a control material for cell proliferation. The samples of nanofibrous membranes were moved into fresh 24-well plates to avoid the influence of the cells adhered to the bottom of the wells. The assay was performed in accordance with the manufacturer’s protocol. The absorbance was measured using the VersaMax ELISA Microplate Reader (Molecular Devices LLC, Sunnyvale, CA, USA) in Nunc-Immuno MicroWell 96-well cell culture plates (Sigma-Aldrich Co., St Louis, MO, USA) with wavelength at 490 nm.
Cell morphology and spreading
The spreading and morphology of the cells on PLA nanofibrous membranes with or without fibrin nanocoating were evaluated in six culture intervals (on days 1, 3, 5, 7, 10, and 14 after cell seeding). A 24-well plate polystyrene culture dish was used as a control material. The cells were rinsed with phosphate-buffered saline (PBS), fixed with −20°C cold ethanol for 10 minutes, and then stained for 1 hour at room temperature with a combination of fluorescent dyes diluted in PBS. The cell cytoplasm was stained with Texas Red C2-maleimide (Molecular Probes, eugene, OR, USA; Thermo Fisher Scientific, Waltham, MA, USA, 20 ng/mL). F-actin, an important molecule of the cell cytoskeleton, was stained with phalloidin conjugated with TRITC (Sigma-Aldrich Co., 5 μg/mL), and cell nucleus was stained with Hoechst #33258 (Sigma-Aldrich Co., 5 μg/mL). Images of the cell morphology were taken using an epifluorescence microscope (IX51, Olympus Corporation, Tokyo, Japan, objective [obj] 10×) equipped with a digital camera (DP 70, Olympus Corporation, Tokyo, Japan) or scanned using a Leica TCS SPE DM2500 upright confocal microscope (Leica Microsystems, Wetzlar, Germany), obj 40×/1.15 numerical aperture (NA) oil.
Real-time polymerase chain reaction
Real-time polymerase chain reaction (real-time PCR) was used for measuring relative mRNA expression of β1-integrin (important molecule marker of cell adhesion) and collagen I (main component of ECM produced by fibroblasts) on days 7, 10, and 14 after cell seeding. The first evaluated interval was chosen after 1 week of cultivation on the basis of our previous experiments, when fibroblasts started to form collagen fibers after they reached confluence. Moreover, higher cell population density was needed for RNA isolation. Four independent samples of PLA membranes fixed into a six-well cell culture plate for each experimental group and time interval were used. Each membrane sample was pooled in two independent experiments. The polystyrene culture dishes were used as a control material for cell proliferation.
Total RNA was isolated from the samples using the Total RNA Purification Micro Kit (Norgen Biotek Corp., Thorold, ON, Canada), according to the manufacturer’s protocol. The cells were rinsed with PBS. The PLA membranes were transferred into 1.5 mL tubes, and RNA was isolated using the lysis solution enriched with mercaptoethanol (1%). The cells on the control polystyrene dish were directly lysed in the plate.
Reverse transcription was performed using the Omniscript Reverse Transcription Kit (Qiagen NV, Venlo, the Netherlands) and random hexamers (New England Biolabs, Inc, Ipswich, MA, USA), according to the manufacturer’s protocol.
The mRNA levels were quantified by 5× HOT FIREPol Probe qPCR Mix Plus (ROX) (Solis BioDyne, Tartu, Estonia) and by TaqMan Gene Expression Assays (Thermo Fisher Scientific, Waltham, MA, USA), labeled with FAM reporter dye specific to human β1-integrin ITGB1 (Hs00559595_m1) or to human collagen I COL1A1 (Hs00164004_m1) as a target gene, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Hs02758991_g1) as a reference gene, in a final reaction volume of 20 μL on a 96-well optical reaction plate using the iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories Inc., Hercules, CA, USA). The amplification conditions consisted of incubation with uracil-DNA glycosylase at 50°C for 2 minutes and initial DNA polymerase enzyme activation at 95°C for 10 minutes, followed by 50 cycles of denaturation at 95°C for 15 seconds and annealing/extension at 60°C for 1 minute.
The data are presented as the mean ± standard error of mean (SEM) from six to eight experimental measurements obtained from three to four independent experiments. Expression values were obtained from Ct numbers. The target gene levels are expressed as a relative value, the ratio of the target gene expression toward the reference GAPDH gene. The relative gene expression was calculated as 2−num, where ΔCt was determined in each sample by subtracting the Ct value of the target gene from the Ct value of the GAPDH.
Immunofluorescence staining
The morphology, stability, and degradation of fibrin nanocoating by the cells, distribution of integrin adhesion receptors with β1 chain, and the total collagen produced by fibroblasts on the PLA membranes were examined with immunofluorescence staining. The morphology of the fibrin nanocoating on the PLA membranes was examined on freshly prepared samples. The stability and degradation of fibrin nanocoating by the cells was evaluated in six time intervals (on days 1, 3, 5, 7, 10, and 14 after cell seeding). In each time interval, the fibrin nanocoating was stained on PLA membranes seeded with cells and also on membranes without cells, only immersed in the cell culture medium and incubated under the conditions that were used for cell cultivation. The nonmodified membranes incubated in the cell culture medium with and without cells were used as a control to show nonspecific binding of primary or secondary antibody and the cell morphology. The β1-integrins were visualized in the cells on the investigated substrates on days 3 and 7 after cell seeding. The visualization of total (intracellular and extracellular) collagen produced by fibroblasts on the PLA membranes was performed on days 7, 10, and 14. The cells were cultivated on PLA membranes trapped in CellCrowns and fitted into a 24-well plate. Microscopic glass coverslips were used as a control material. Two samples of each experimental group for each time interval were used.
The membranes cultivated with the cells were incubated with cold (−20°C) 70% ethanol for 10 minutes at room temperature to fix the cells. The membranes without cells or with fixed cells were treated with 1% bovine serum albumin in PBS (Sigma-Aldrich Co.; for the membranes without cells) or with 1% bovine serum albumin in 0.1% triton (Sigma-Aldrich Co.; for the membranes with the cells) for 20 minutes, and then with 1% Tween 20 (Sigma-Aldrich Co.) in PBS for 20 minutes at room temperature to block nonspecific binding sites. Subsequently, the samples were incubated overnight at 4°C with the following primary antibodies diluted in PBS in a ratio of 1:200: rabbit polyclonal antibody against human fibrinogen (Dako Denmark A/S, Glostrup, Denmark), a mouse monoclonal antibody against β1-integrin chain (Merck Millipore, Billerica, MA, USA), or mouse monoclonal antibody against collagen I (Sigma-Aldrich Co.). After rinsing with PBS, the secondary antibody goat anti-rabbit F(ab')2 fragments of IgG (H + L) or the secondary antibody goat anti-mouse F(ab')2 fragments of IgG (H + L), conjugated with Alexa Fluor® 488 (Molecular Probes; Thermo Fisher Scientific), were applied to the samples (diluted in PBS in a ratio of 1:400) for 1 hour at room temperature in the dark. The cells were rinsed with PBS and were scanned using a Leica TCS SPE DM2500 upright confocal microscope, obj 40×/1.15 NA oil or obj 63×/1.3 NA oil.
Immunofluorescence staining of extracellular collagen
The extracellular collagen, ie, collagen deposited by the cells on the membrane surface, was stained in live cells on days 7, 10, and 14. The cells were cultivated on PLA membranes trapped in CellCrowns and fitted into a 24-well plate. Microscopic glass coverslips were used as a control material.
To stain extracellular type I collagen, the cells were rinsed with a solution of 5% FBS in PBS. The samples were then incubated with primary mouse monoclonal antibody against collagen I (Sigma-Aldrich Co., diluted in PBS in a ratio of 1:200) for 30 minutes on ice. After rinsing the cells with 5% FBS in PBS, they were fixed with 2% paraformaldehyde dissolved in PBS for 20 minutes. The samples were rinsed with PBS, and the nonspecific binding sites were blocked with 1% FBS in PBS. The samples were rinsed with PBS and were incubated with secondary antibody anti-mouse F(ab')2 fragments of IgG (H + L), conjugated with Alexa Fluor® 488 (Molecular Probes, Thermo Fisher Scientific) for 2 hours in the dark. The secondary antibody was diluted in 1% FBS in PBS in a ratio of 1:1,000. The cells were rinsed in PBS, and the cell nucleus was stained with Hoechst #33342, which penetrates through the nonpermeabilized cell membrane.
The cells were scanned using a Leica TCS SPE DM2500 upright confocal microscope, obj 40×/1.15 NA oil.
Sircol™ soluble collagen assay
The total amount of collagen produced by the cells, ie, intracellular collagen and collagen deposited on the PLA membranes, was determined using a Sircol kit (Biocolor Ltd., Carrickfergus, UK) on day 14 after cell seeding. The Sircol assay was carried out only on samples incubated in the cell culture medium with AA. The cells cultivated in the cell standard cultivation medium (without AA) did not deposit collagen as ECM on surface samples in a sufficient amount to enable adequate measurements to be made (the amount of cell collagen was under the detection limit). The Sircol dye binds acid- and pepsin-soluble collagen synthesized by cells and deposited onto the surface as ECM. The Sircol reagent binds to the [Gly-X-Y] helical structure, as found in all types of collagen.
The PLA membranes with cells cultivated in CellCrowns fitted into a six-well plate were removed from the CellCrowns and were transferred into fresh cell culture plates. The polystyrene culture dishes were used as a control material. The membranes and the control polystyrene dishes without cells were used as a control to set a background. Four independent samples for each experimental group were used. The collagen was recovered by acid–pepsin digestion. The cells on the membranes and on the control polystyrene were rinsed with PBS, harvested with a cell scraper in 1 mL of pepsin solution (1 mg/mL dissolved in 0.5 M acetic acid), and lysed overnight at 4°C. The lysates were centrifuged and the supernatants were concentrated according to the Sircol kit manufacturer’s protocol. Finally, the Sircol dye was bound to the isolated collagen, was dissolved, and the absorbance of the colored solution was measured. The absorbance was measured using a VersaMax ELISA Microplate Reader (Molecular Devices LLC) in Nunc-Immuno MicroWell 96-well cell culture plates (Sigma-Aldrich Co.,) with wavelength at 555 nm. The amount of collagen on each type of sample was expressed in μg/cm2.
The amount of total collagen was adjusted to the cell number estimated by the amount of cellular DNA or to the cell proliferation estimated by the metabolic activity of the cells. The amount of cell DNA was determined using a Picogreen dsDNA assay kit (Thermo Fisher Scientific). The assay is based on measurements of the fluorescence of Picogreen, a nucleic acid stain for quantifying double-stranded DNA in solution. The assay was carried out according to the manufacturer’s protocol. Three independent samples of each experimental group were used for the measurements. Fluorescence was measured with the Synergy HT Multi-Mode Reader (BioTek, Winooski, VT, USA) in black 96-well cell culture plates (96 F Nunclon delta; Black Microwell SI) with excitation wavelength at 480 nm and emission wavelength at 520 nm. The amount of cell DNA on each sample was expressed in ng/cm2. The cell metabolic activity was measured by a CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS, Promega Corporation), as described in the “Cell mitochondrial activity” section.
Statistics
If not stated otherwise in the text earlier, the quantitative data are presented as mean ± standard deviation from four independent samples for each experimental group and time interval. Statistical significance was evaluated using the analysis of variance and Student–Newman–Keuls method. Values of P≤0.05 were considered significant.
Results
Morphology of PLA nanofibrous membranes and the fibrin nanocoating on a PLA membrane and its degradation during cell cultivation
The morphology of the nanofibrous membranes in the nonmodified state is shown in Figure 1. The nanofibrous membranes consisted of mostly straight and randomly oriented fibers (Figure 1A, B). The diameter of the fibers varied in a large range from tens of nanometer to >1 μm. The average fiber diameter was ~350 nm (Figure 1C). Thus, the fiber diameter was in the submicron scale rather than in the nanoscale, because the size of nanostructures is defined as ≤100 nm. However, in the scientific literature, submicron-scale fibers are commonly referred to as nanofibers.40
 | Figure 1 SEM images of nonmodified PLA membranes. |
The fibrin regularly coated each fiber in the membrane. Moreover, the fibrin randomly formed a thin fibrous mesh on the membrane surface (Figure 2). Tests on the durability of the fibrin nanocoating on the nanofibrous membrane without cells, immersed in the cell culture medium at 37°C for 14 days, showed that the fibrin nanocoating was stable and its morphology was almost unchanged (Figure 3A). The rate of degradation of the fibrin nanocoating on the membrane with cells correlated with the degree of cell proliferation (Figure 3B and C). After 10 days of cell cultivation, the fibrin was degraded on the membrane surface. In the deeper layers of the membrane, where the cells did not penetrate, the fibrin remained in unchanged form. The same results were obtained on the samples cultivated in the cell culture medium supplemented with AA (data not shown).
 | Figure 2 Morphology of the fibrin nanocoating (F) on PLA membranes. |
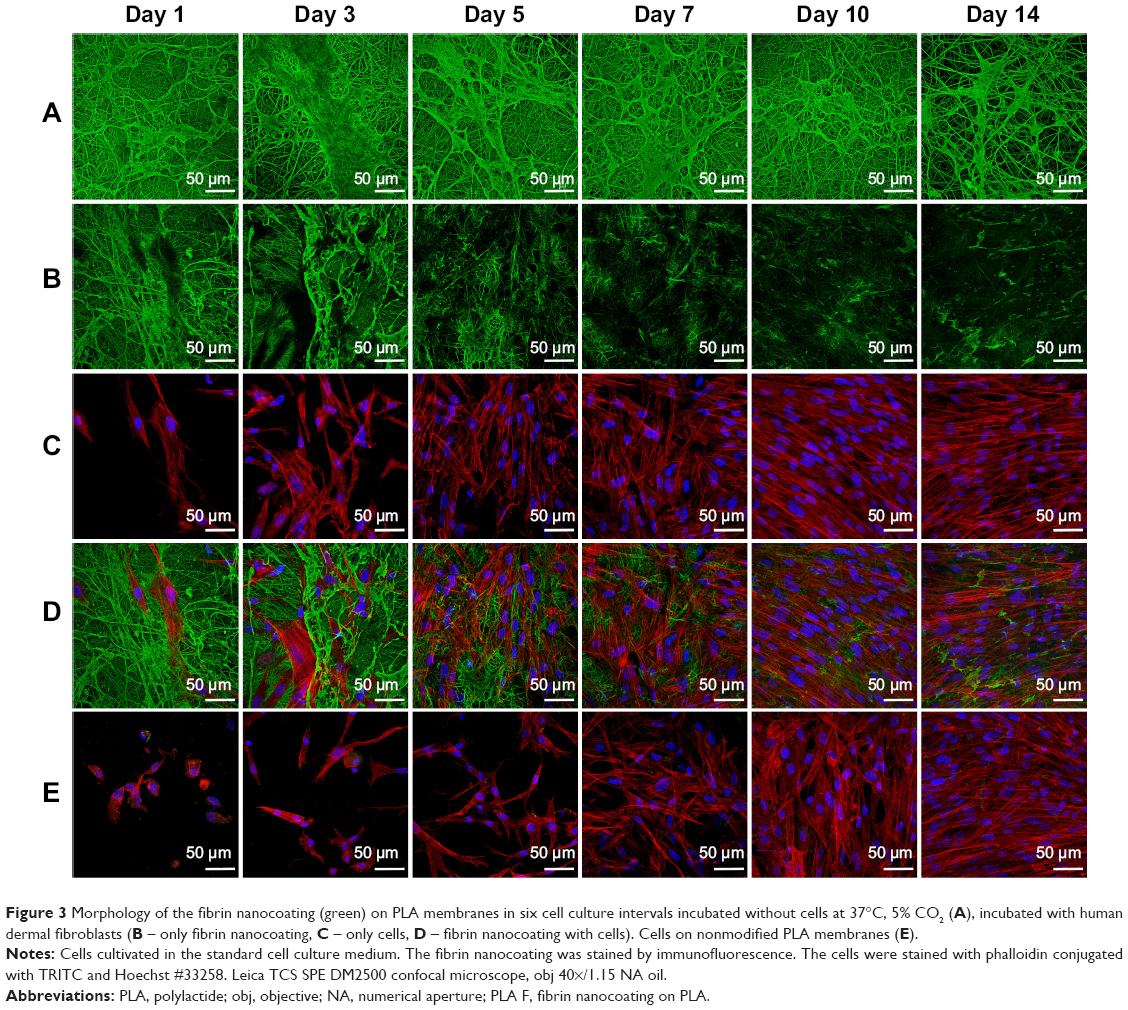 | Figure 3 Morphology of the fibrin nanocoating (green) on PLA membranes in six cell culture intervals incubated without cells at 37°C, 5% CO2 (A), incubated with human dermal fibroblasts (B – only fibrin nanocoating, C – only cells, D – fibrin nanocoating with cells). Cells on nonmodified PLA membranes (E). |
The morphology of the cells was different on nonmodified membranes and on membranes coated with fibrin. On the fibrin-modified membranes, the cells were well-spread already on day 1 after cell seeding, whereas on the nonmodified membrane, some cells were still round in shape even after 3 days of cell cultivation. The cell density was higher on the membranes coated with fibrin than on the nonmodified membranes (Figure 3C and D).
Cell metabolic activity, proliferation, adhesion, and morphology on PLA membranes
The cell mitochondrial metabolic activity, which correlates with cell proliferation, was significantly higher on the membranes coated with fibrin than on the nonmodified membranes from days 1 to 10 after cell seeding. These differences did not become nonsignificant until day 14. Differences in cell proliferation were found on the samples cultivated in the standard cell culture medium and also on the samples cultivated in the medium with AA. On the control polystyrene culture dishes, the cell metabolic activity decreased significantly after 1 week of cell cultivation and was lower than on the membranes coated with fibrin. Moreover, on day 14, the cell proliferation on the control polystyrene was significantly lower than on all types of nanofibrous membranes. AA added into the cell culture medium after 3 days of cell cultivation significantly increased the cell metabolic activity on all types of samples, mainly on days 5, 7, and 10 after cell seeding (Figure 4).
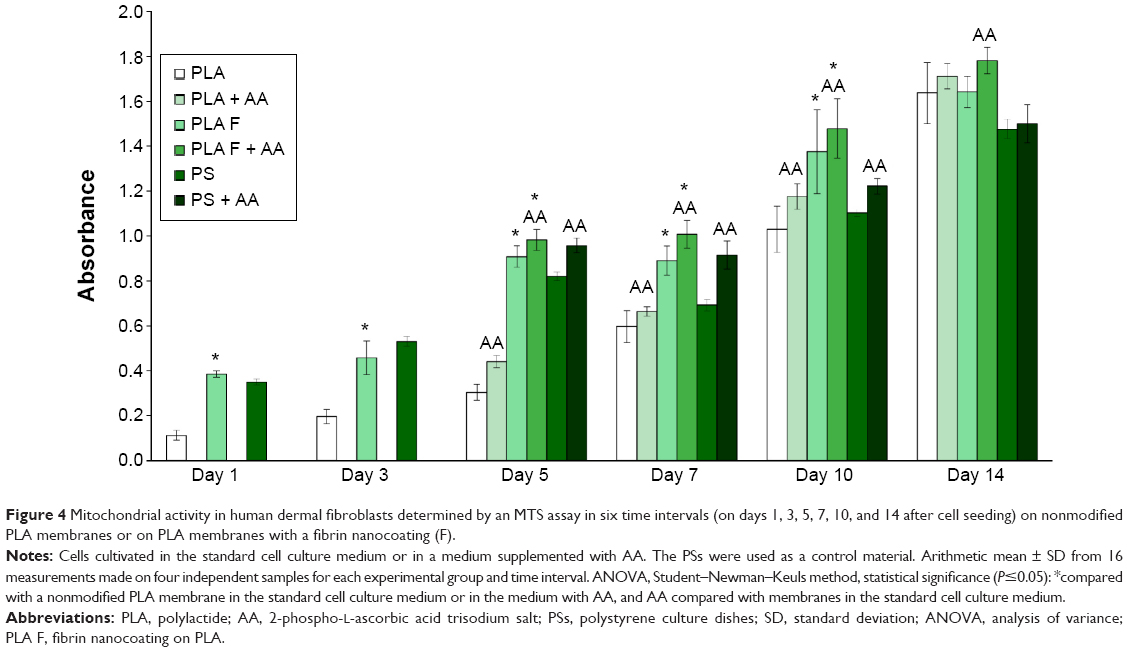 | Figure 4 Mitochondrial activity in human dermal fibroblasts determined by an MTS assay in six time intervals (on days 1, 3, 5, 7, 10, and 14 after cell seeding) on nonmodified PLA membranes or on PLA membranes with a fibrin nanocoating (F). |
The cell metabolic activity on nonmodified and modified membranes and on the control polystyrene increased with the time of cell cultivation (Figure 4), which indicates increasing density of the cells (Figures 5 and 6). The cells adhered in greater densities on membranes coated with fibrin than on nonmodified membranes (Figures 5 and 6, days 1–7). On day 7, the cells on fibrin were confluent, whereas there were free spaces among the cells on the nonmodified membranes. On day 14, the cells formed confluent monolayers on all types of samples. In addition, the cells on fibrin-coated membranes were better spread than the cells on nonmodified membranes (Figure 5). The addition of AA further stimulated the cell proliferation, which was manifested by their higher population density than that in the medium without AA. This was particularly apparent on the pure PLA (Figure 6).
 | Figure 5 Morphology of human dermal fibroblasts on days 1 and 3 after seeding on nonmodified PLA membranes, or on PLA membranes with a fibrin nanocoating (F). |
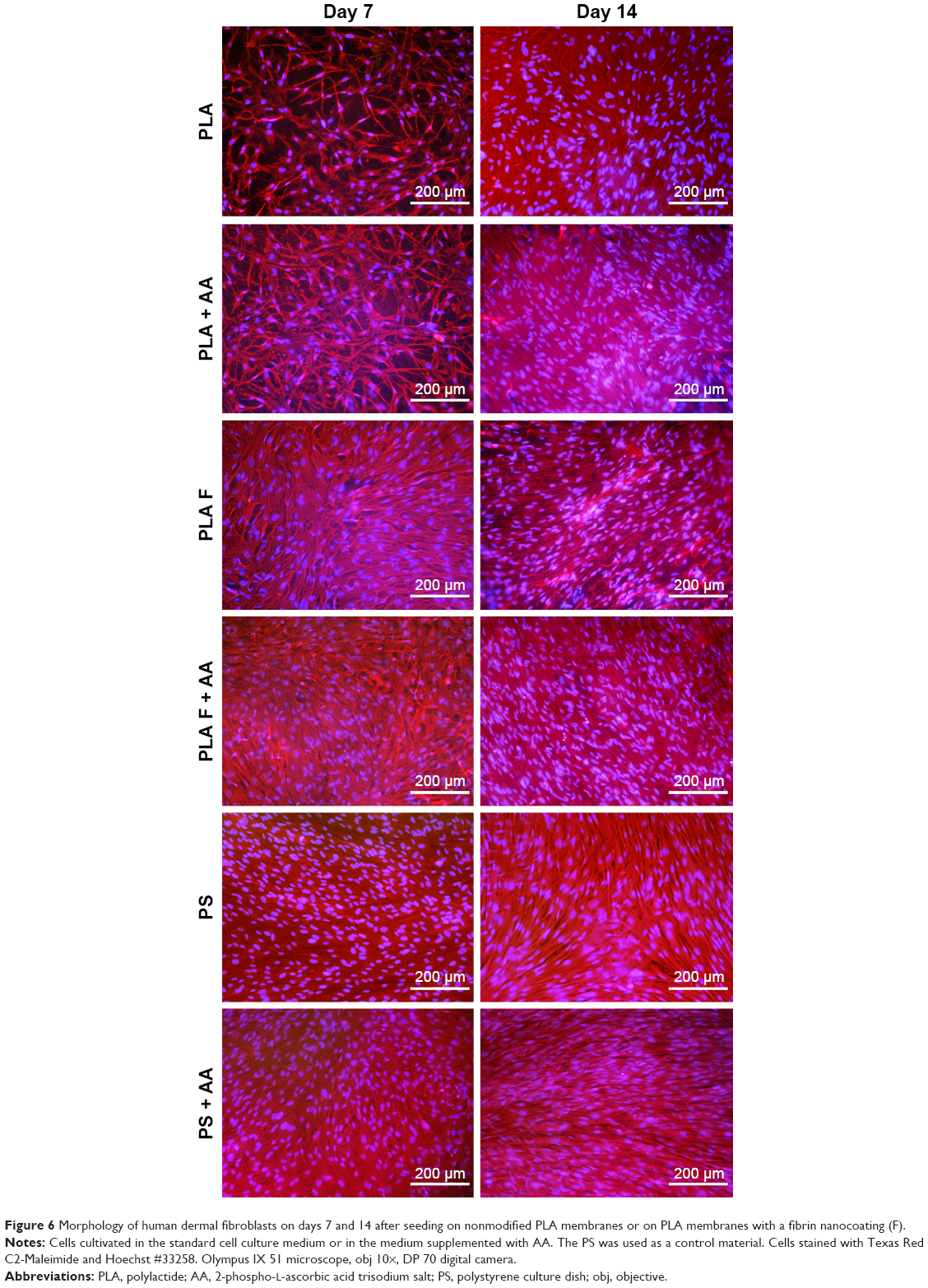 | Figure 6 Morphology of human dermal fibroblasts on days 7 and 14 after seeding on nonmodified PLA membranes or on PLA membranes with a fibrin nanocoating (F). |
β1-Integrins were evaluated as a molecular marker of cell adhesion. Relative mRNA expression of these integrins changed during cell cultivation. On day 7 after seeding, the integrin expression was lower on nonmodified membranes compared to membranes coated with fibrin or to control polystyrene. However, on day 10 after seeding, the expression of β1-integrins was significantly higher on nonmodified membranes than on fibrin-modified membranes. Moreover, on nonmodified membranes, the integrin expression was two times higher on day 10 than on day 7. On day 14 after seeding, the expression on nonmodified membranes, fibrin-modified membranes, and control polystyrene dishes was comparable. AA added into culture medium did not significantly influence β1-integrin expression. The most apparent (but not significant) positive effect of AA on the increase of β1-integrin expression was observed in cells on nonmodified membranes on day 10 (Figure 7).
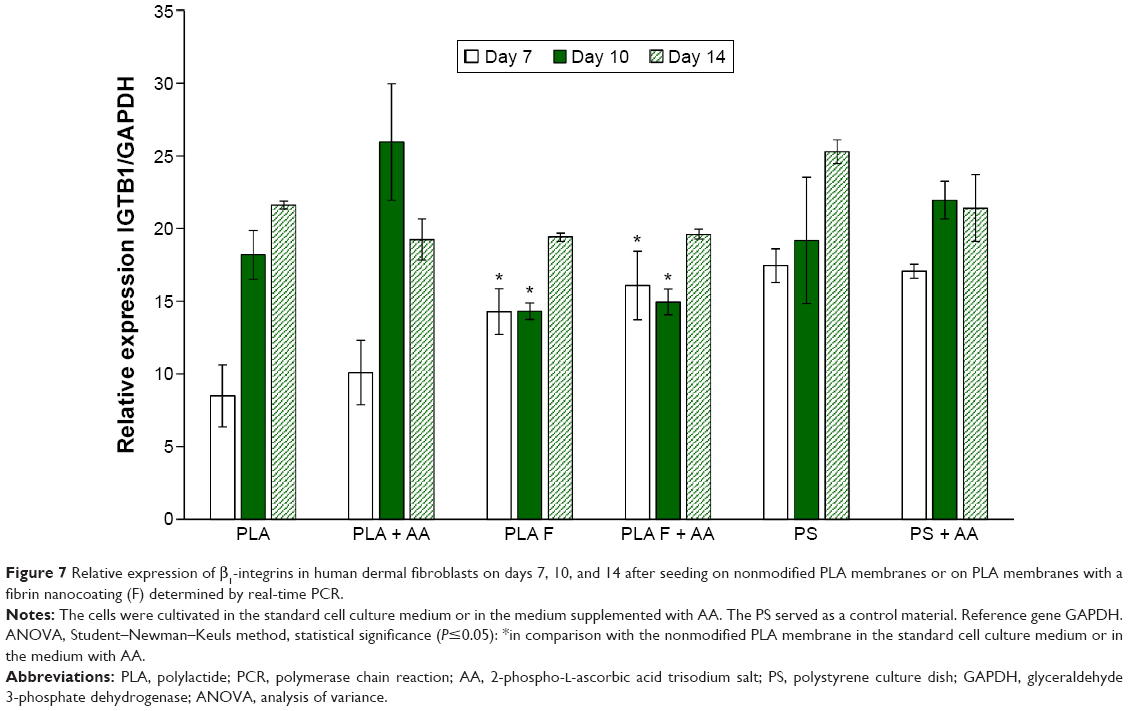 | Figure 7 Relative expression of β1-integrins in human dermal fibroblasts on days 7, 10, and 14 after seeding on nonmodified PLA membranes or on PLA membranes with a fibrin nanocoating (F) determined by real-time PCR. |
Focal adhesions (FA) containing β1-integrins were localized preferentially on the cell edges, and thus they were better observable in cells at earlier culture intervals (ie, on day 3, Figure 8) than at later intervals when the cells formed a confluent layer (ie, on day 7, data not shown). On day 3 after seeding (ie, 2 days after adding of AA), FA were better developed in cells on fibrin-modified membranes than on nonmodified membranes. Positive influence of AA added into the culture medium was slightly apparent on nonmodified membranes, where the FA were better visible in the cells cultured in medium supplemented with AA than in standard medium. On fibrin-modified membranes, the influence of AA was not observed (Figure 8).
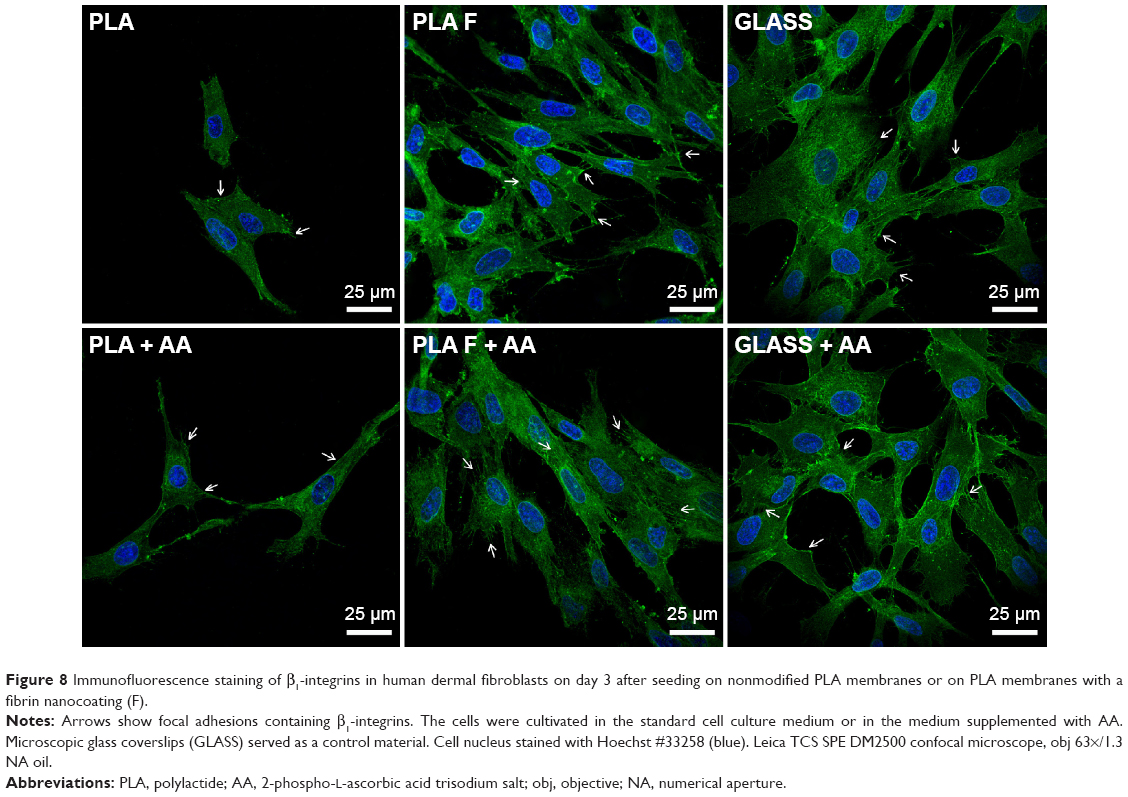 | Figure 8 Immunofluorescence staining of β1-integrins in human dermal fibroblasts on day 3 after seeding on nonmodified PLA membranes or on PLA membranes with a fibrin nanocoating (F). |
Collagen production by human dermal fibroblasts
The relative mRNA expression of collagen I, determined by real-time PCR, was estimated in three time cell culture intervals on days 7, 10, and 14. Collagen I expression was significantly higher on membranes coated with fibrin than on nonmodified membranes at all three time intervals of cell cultivation. After 10 days of cell cultivation, the positive effect of AA on collagen I expression began to be manifested. On day 10 after cell seeding, the expression of collagen I was significantly increased by adding AA into the cell culture medium only on the nonmodified membranes. On day 14, adding AA into the cell culture medium also increased collagen I expression on membranes coated with fibrin. The highest collagen I expression values of all the tested samples were reached on these membranes (Figure 9).
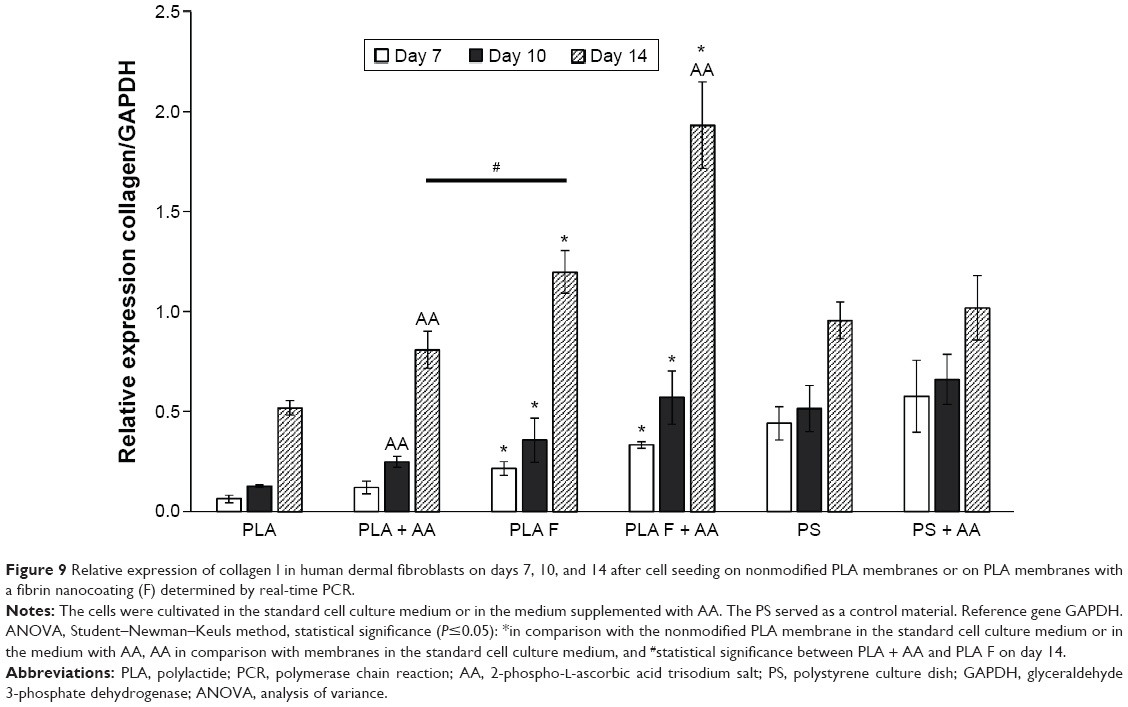 | Figure 9 Relative expression of collagen I in human dermal fibroblasts on days 7, 10, and 14 after cell seeding on nonmodified PLA membranes or on PLA membranes with a fibrin nanocoating (F) determined by real-time PCR. |
Collagen I expression was significantly lower on the nonmodified membranes than on the control polystyrene dishes. Collagen I expression on the membranes coated with fibrin was comparable to the expression on the control polystyrene dishes, or was slightly lower. However, the difference in expression was not statistically significant. On membranes coated with fibrin and incubated in the cell culture medium with AA for 14 days, the cells expressed the highest quantity of mRNA for collagen I, and this expression was significantly higher than that on the control polystyrene dishes (Figure 9). Collagen I expression increased significantly with the cell cultivation time, except in the case of the cells cultivated on the control polystyrene dishes (Figure 9).
Immunofluorescence staining of collagen I revealed that AA stimulated the cells to form extracellular collagen fibers deposited on the substrate surface after 1 week of cell cultivation (Figure 10). The cells cultivated in the standard cell culture medium without AA synthetized collagen but did not form a fibrous ECM (Figure 11). The collagen remained inside the cells or was probably released into the cell culture medium. After 2 weeks of cultivation, the cells started to deposit small amounts of collagen in ECM on the material surface (Figure 10). On day 14, the expression of collagen I in the cells cultivated in the standard cell culture medium on the membranes coated with fibrin was significantly higher than that in the cells cultivated in the medium with AA on the nonmodified membranes (Figure 9). However, the cells did not form well-developed fibrous collagen ECM on the membrane surface (Figure 10).
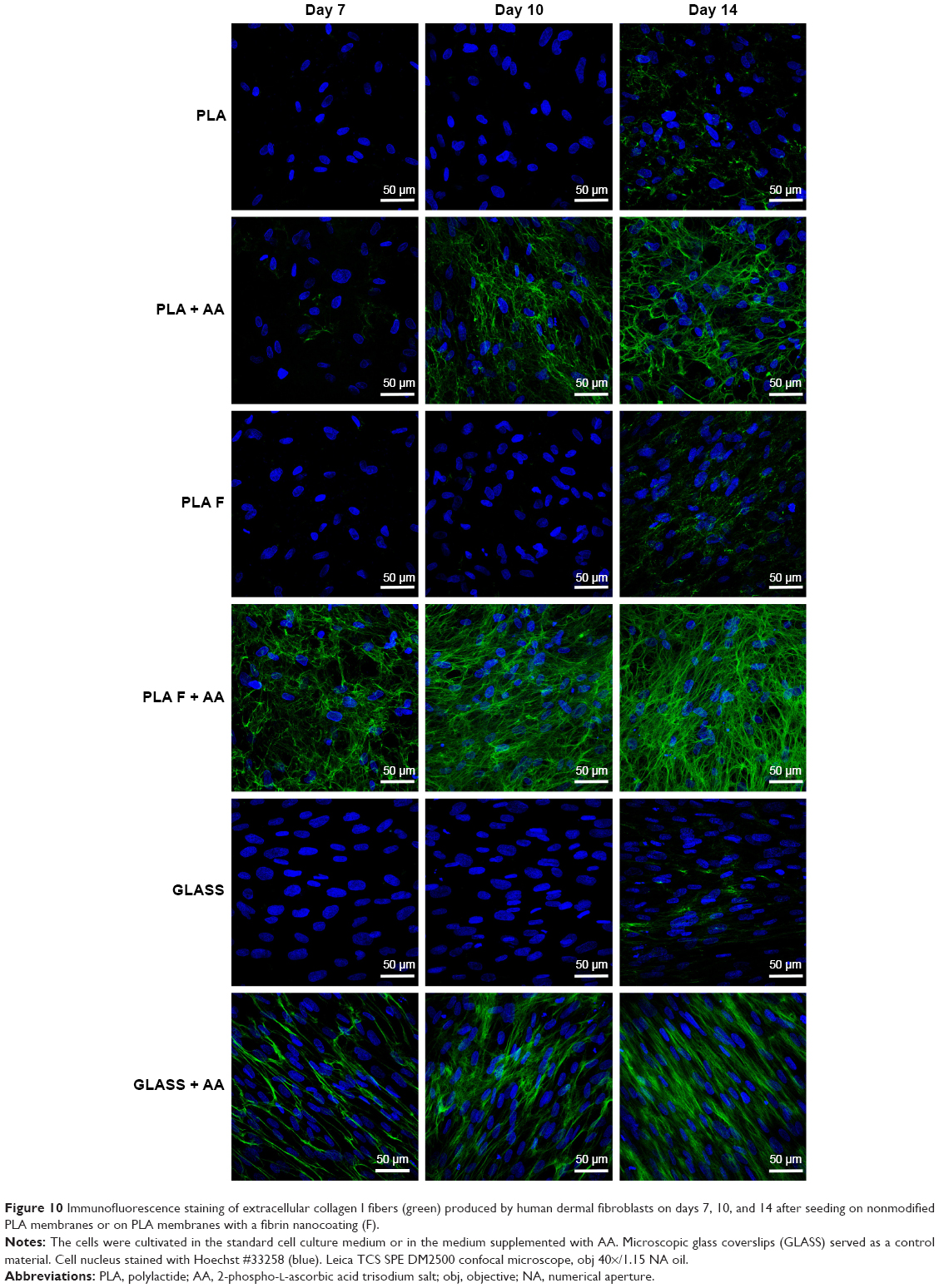 | Figure 10 Immunofluorescence staining of extracellular collagen I fibers (green) produced by human dermal fibroblasts on days 7, 10, and 14 after seeding on nonmodified PLA membranes or on PLA membranes with a fibrin nanocoating (F). |
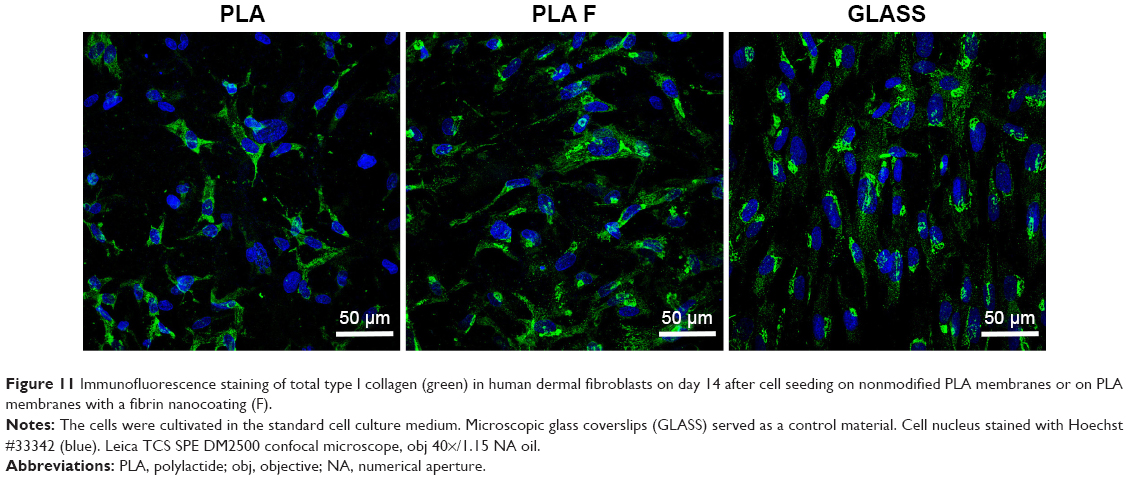 | Figure 11 Immunofluorescence staining of total type I collagen (green) in human dermal fibroblasts on day 14 after cell seeding on nonmodified PLA membranes or on PLA membranes with a fibrin nanocoating (F). |
On day 7 after cell seeding, the cells cultivated with AA on membranes coated with fibrin formed well-developed collagen fibers on the membrane surface. However, the cells cultivated with AA on nonmodified membranes formed only relatively small amounts of collagen ECM. On days 10 and 14 after cell seeding, the fibrin with AA continued to stimulate the cells to form collagen ECM.
The overall (total) collagen production by the cells cultivated in the medium with AA was measured after 14 days of cell cultivation, using a Sircol kit. The amount of collagen was adjusted to the amount of cell DNA (Figure 12A) or to the cell metabolic activity (Figure 12B). The amount of cell DNA was comparable on nonmodified membranes and on membranes coated with fibrin (Figure 12C), whereas the cell metabolic activity on the membranes coated with fibrin was slightly higher than that on the nonmodified membranes (Figure 12D). The amount of cell DNA and cell metabolic activity on the control polystyrene dishes was significantly lower than that on the PLA membranes (Figure 12C and D).
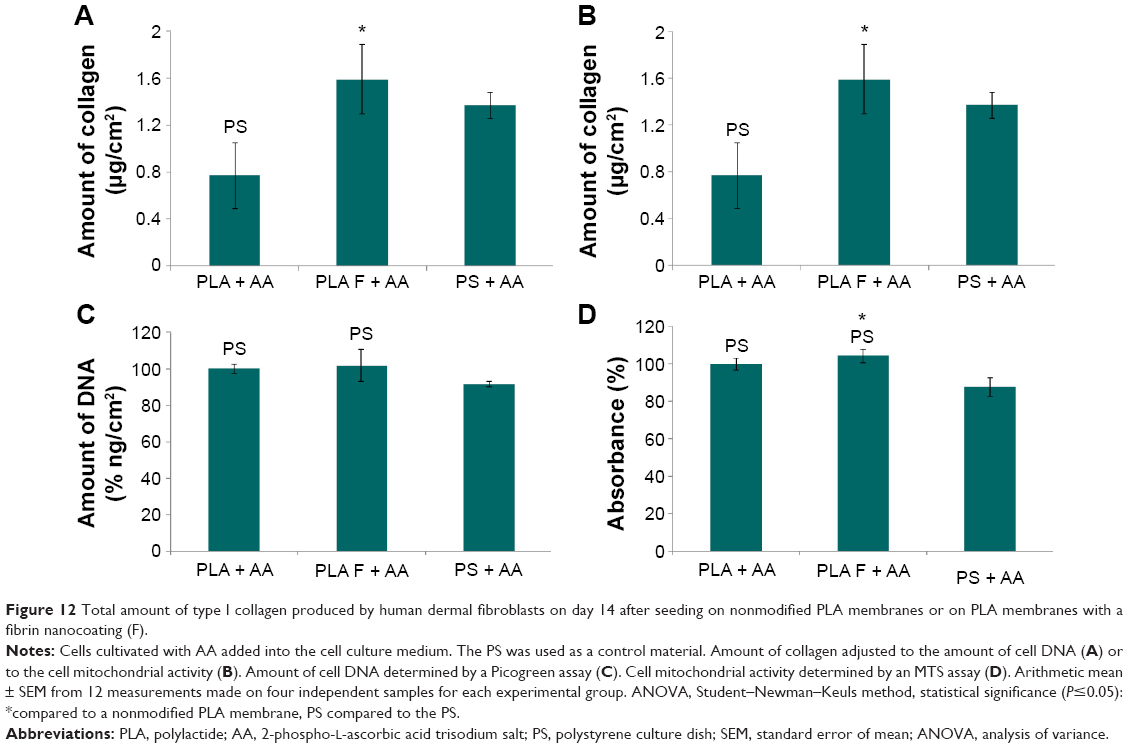 | Figure 12 Total amount of type I collagen produced by human dermal fibroblasts on day 14 after seeding on nonmodified PLA membranes or on PLA membranes with a fibrin nanocoating (F). |
Collagen production was significantly higher on membranes coated with fibrin than on nonmodified membranes. The amount of collagen on the control polystyrene dishes was significantly higher than that on the nonmodified membranes and was comparable to the amount on the membranes coated with fibrin (Figure 12A and B).
Discussion
The ideal skin substitute mimics the physiology and the mechanical properties of normal skin, enables nutrition supply, accelerates skin tissue regeneration, and is not subject to immune activation. Unfortunately, most of the clinically used skin substitutes have not yet achieved all of these properties. These substitutes mostly serve as a temporary coverage, and they are eventually replaced by scar tissue.42 A promising approach is to develop inexpensive biodegradable carriers of skin cells with desirable properties available in any quantity needed. This study, including our earlier study,7 proved that these carriers can be based on modified PLA nanofibrous scaffolds. These scaffolds can be expected to promote the formation of both epidermis and dermis layers. Modification of nanofibrous PLA membranes by plasma in our earlier study enhanced the adhesion and growth of human keratinocytes, while modification with fibrin in this study enhanced the adhesion and growth of human dermal fibroblasts and the synthesis of collagen I by these cells, which is necessary for the additional cell ingrowth.24 Our further plan is to develop keratinocyte and fibroblast layers on the opposite sides of the same nanofibrous membrane. This membrane will enable physical and biochemical communication between fibroblasts and keratinocytes together with the diffusion of nutrients, growth factors, and other biologically active molecules from the culture medium or from the surrounding tissue.15 It has been reported that bilayered tissue-engineered skin constructs were more efficient in healing full-thickness skin defects in sheep than single-layer constructs composed of only fibroblasts or keratinocytes.43
In the first part of this study, we investigated the influence of fibrin deposited on a nanofibrous membrane, and AA added into the cell culture medium, on the adhesion and proliferation of human dermal fibroblasts. Our results show that fibrin deposition led to markedly improved cell adhesion and spreading compared to the membrane without fibrin. On the membranes with fibrin, the cells were well-spread with well-developed typical FA containing β1-integrins and polygonal in shape, whereas on the nonmodified membranes, the cells were round until the third day of cell cultivation and with less developed β1-integrin-containing FA. β1-Integrins, namely α5β1 integrins (together with αvβ3 and αvβ3 integrins), mediate the adhesion of fibroblasts to fibrin molecules, in which these adhesion receptors recognize the RGD motifs.24,44 The group of β1-integrins also include receptors for collagen, namely α1β1, α2β1, and α3β1 integrins. The expression and production of collagen was increased on fibrin-coated membranes, particularly in the medium with AA, which could contribute to the improved cell adhesion on the modified PLA membranes. Moreover, through the αC domain, the fibrin attracts molecules of ECM from the cell culture medium, such as fibronectin and vitronectin that further support cell adhesion.24 On nonmodified membranes, cell adhesion is mediated practically only through ECM molecules spontaneously adsorbed from the cell culture medium or deposited on the membrane surface by the cells. In addition, these molecules could be adsorbed in an insufficient quantity and with an inappropriate spatial conformation for recognition by cell adhesion receptors.
The better developed β1-integrin-containing FA in cells on fibrin-coated membranes correlated with relative mRNA expression of this adhesion receptor. At earlier cell culture interval (on day 7 after seeding), the fibrin nanocoating increased relative β1-integrin expression. This could be explained by a more intense proliferation of cells on these coatings, which resulted in a higher formation of new cells and their need to express and synthesize integrin receptors in order to adhere to their growth support. In addition, as mentioned earlier, the fibrin coatings (together with increased amount of collagen deposited on these coatings) provided a higher number of adhesion ligands (eg, RGD motifs), which could also increase the β1-integrin expression. Nevertheless, on day 10, the β1-integrin expression became higher on nonmodified membranes than that on fibrin-coated membranes. An explanation could be that the cell proliferation and new cell formation, which were delayed on pure PLA membranes, reached their maximum on day 10, while the cells on fibrin-coated PLA membranes reached confluence and slowed down their proliferation. On day 14, the MTS assay indicated that the cell proliferation on fibrin-modified and nonmodified membranes was similar. The β1-integrin expression was also comparable in cells on both types of membranes. In addition, the β1-integrin expression in cells on pure PLA membranes was stimulated, although slightly, by addition of AA into the cell culture medium, which suggests that these cells started focusing on ECM synthesis rather than on cell proliferation.
Fibroblast proliferation was also markedly increased on the fibrin-coated membranes, which is in accordance with many studies published earlier.19,33 Fibrin can stimulate fibroblast proliferation via the alpha and beta chains present in fibrinogen and fibrin molecules,45 by the presence of thrombin, a potent mitogen for fibroblasts, in fibrin matrices,46 and also by stimulating the autocrine production of growth factors in cells by fibrin. In keratinocytes, fibrin stimulated the autocrine production of transforming growth factor alpha through the epidermal growth factor receptor.47 Fibrin also partially restored the ability of senescent and growth-arrested human dermal fibroblasts to continue replicating.48
However, after 2 weeks of cell cultivation, the cell proliferation (measured by cell metabolic activity) on membranes with fibrin became similar to the cell proliferation on the nonmodified membranes, which probably correlated with the degree of fibrin degradation. During cell cultivation, fibrin is physiologically reorganized and is degraded by the cells and replaced by newly synthesized ECM.28,49 On the membrane surface, the fibrin was degraded by the cells and remained only in the deeper layers of the membrane, where the cells did not penetrate. The degraded fibrin probably suppressed the cell proliferation and stimulated the cells to synthesize ECM molecules and to deposit them on the membrane surface. Similar results were obtained in a fibrin gel after it was contracted and partially degraded by human skin fibroblasts. These cells proliferated more slowly but deposited a greater amount of collagen than the cells in monolayers on culture dishes.28
After 1 week of cell cultivation, the cell proliferation on the control polystyrene dishes decreased significantly. On day 14, the cell proliferation on the control polystyrene dishes was even significantly lower than that on all types of samples of nanofibrous membrane. The decline in cell proliferation could be caused by cell contact inhibition on two-dimensional polystyrene dishes,28 whereas the three-dimensional structure of the nanofibrous membrane enabled cell penetration inside the membrane, ie, it provided more space for cell growth, and thus delayed contact inhibition of the cell growth.
We also found that AA added into the cell culture medium increased cell proliferation but did not significantly influence cell adhesion, as manifested by the expression of β1-integrins. Increased cell proliferation was apparent already 2 days after AA treatment and continued to day 10 after cell seeding. Previously published results showed that AA increased the proliferation of dermal fibroblasts,50,51 and this positive effect was more apparent in fibroblasts from elderly donors than in fibroblasts from newborn donors.52 On day 14, AA had no significant effect on increasing the cell proliferation. This was probably due to the fact that the cells concentrated on ECM synthesis and deposition and stopped proliferating.
In the second part of the study, we focused on collagen production by human dermal fibroblasts stimulated by AA added into the cell culture medium and cultivated on membranes coated with fibrin. We found that fibrin deposited on nanofibrous membranes increased the relative expression and synthesis of type I collagen and the formation of collagen ECM on the material surface. After 1 week of cell cultivation, the cells cultivated on nonmodified membranes deposited collagen ECM only slightly, while the cells cultivated on membranes with fibrin formed a well-developed network of collagen fibers. Sclafani and McCormick found that platelet-rich fibrin matrix injected into the dermis activated dermal fibroblasts to form new collagen.53 Mazlyzam et al constructed a fibrin scaffold seeded with skin cells. They found newly formed collagen fibers produced by dermal fibroblasts in the dermal layer of the scaffold. The dermal fibroblasts also showed expression of type I collagen at the mRNA level.19 As mentioned earlier, fibrin-based matrices deposited on poly(lactide-glycolide-caprolactone) nanofibrous scaffolds or on polystyrene dishes also stimulated the production of collagen in dermal fibroblasts.28,33
It has been reported repeatedly that ascorbic acid is important for collagen formation by cells. Ascorbic acid helps to catalyze the hydroxylation of lysine and proline amino acids, important components that form stable mature collagen with a triple-helical structure.25,54 Hydroxyproline stabilizes the collagen triple helix. Its absence results in unstable collagen, which cannot be secreted from the cells at a normal rate. Hydroxylysine is necessary for the crosslinking process of collagen.55
Our experiments proved that AA increased the expression of collagen I and stimulated the cells to deposit collagen fibers on the material surface. The cells cultivated in a standard cell culture medium (without AA) did not deposit a significant amount of collagen ECM on the material surface. The cells cultivated in the standard cultivation medium on the membranes coated with fibrin showed a higher expression of collagen I than the cells cultivated in the medium with AA on noncoated membranes. However, these cells did not deposit a considerable amount of fibrous collagen matrix on the material surface. This is in accordance with the study by Murad et al, mentioned earlier, which revealed that cells cultivated without ascorbic acid are incapable of forming stable collagen fibers deposited on the material surface, but form only unstable collagen that remains inside the cells.55 However, cells coated with fibrin secreted a greater amount of collagen fibers in the medium with AA than on uncoated membranes in the same medium (Figure 8).
The insoluble extracellular collagen fibers on the material surface did not form directly after the addition of AA into the cell cultivation medium, but formed after 1 week of cell cultivation (ie, after adding AA into the cell cultivation medium for 4 days). The explanation could be that this longer period is needed for the transition of newly synthesized procollagen into insoluble collagen fibrils.55
Similarly, the positive effect of AA on collagen I gene expression was not manifested until 10 days of cell cultivation. On day 10, this effect was significant only on nonmodified membranes. On day 14, increased collagen I expression with the addition of AA into the medium was also detected on membranes coated with fibrin. A possible explanation could be that on day 10, collagen I expression was predominantly stimulated by fibrin. However, on day 14, the fibrin nanocoating was almost degraded and AA played a major role in stimulating the cells to collagen expression.
The mRNA collagen I expression and the amount of collagen ECM that was formed increased significantly with the time of cell cultivation, except in the case of the control polystyrene dishes. de Clerck and Jones cultivated rat smooth muscle cells in a cultivation medium with AA and showed that the level of collagen expression increased with time of cultivation.54 Moreover, it is known that ascorbic acid increased the steady-state level of procollagen mRNA.25
Conclusion
The fibrin nanocoating on PLA nanofibrous membranes strongly influenced the behavior of human dermal fibroblasts. Fibrin promoted adhesion and spreading of cells and increased the expression of β1-integrins, cell proliferation, and collagen synthesis. The beneficial effect of fibrin on the adhesion and proliferation of cells was probably due to its ability to easily bind to cells through the integrin adhesion receptors, to attract the adhesive molecules from the cell culture medium. It was probably also due to its mitogenic effects on the cells. Fibrin increased the mRNA expression of collagen I, the total amount of synthesized collagen, and its deposition as ECM on the membrane surface. AA added into the cell culture medium also promoted cell proliferation, increased collagen I expression, and also slightly improved the expression of β1-integrins on the uncoated PLA membranes. In addition, ascorbic acid was important for collagen ECM deposition on the membrane surface due to its catalyzing effect for stable collagen fiber formation. Taken together, the combination of fibrin and AA was the most efficient way to stimulate the growth of dermal fibroblasts and the expression, synthesis, and deposition of collagen by these cells.
Acknowledgments
This study was supported by the Grant Agency of Charles University in Prague (Grant No 38214), by the Grant Agency of the Czech Republic (Grant No P108/12/G108) and by the project ‘‘BIOCEV – Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University’’ (CZ.1.05/1.1.00/02.0109). Mr Robin Healey (Czech Technical University in Prague) is gratefully acknowledged for his language revision of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Bacakova L, Filova E, Parizek M, Ruml T, Svorcik V. Modulation of cell adhesion, proliferation and differentiation on materials designed for body implants. Biotechnol Adv. 2011;29(6):739–767. | ||
Sun L, Stout DA, Webster TJ. The nano-effect: improving the long-term prognosis for musculoskeletal implants. J Long Term Eff Med Implants. 2012;22(3):195–209. | ||
Sridhar S, Venugopal JR, Ramakrishna S. Improved regeneration potential of fibroblasts using ascorbic acid-blended nanofibrous scaffolds. J Biomed Mater Res A. 2015;103(11):3431–3440. | ||
Seet WT, Manira M, Khairul Anuar K, et al. Shelf-life evaluation of bilayered human skin equivalent, MyDerm. PLoS One. 2012;7(8):e40978. | ||
Niu HT, Lin T. Fiber generators in needleless electrospinning. J Nanomater. 2012;2012:13. | ||
Yu DG, Zhu LM, Branford-White CJ, et al. Solid dispersions in the form of electrospun core-sheath nanofibers. Int J Nanomedicine. 2011;6:3271–3280. | ||
Yu DG, Zhou J, Chatterton NP, Li Y, Huang J, Wang X. Polyacrylonitrile nanofibers coated with silver nanoparticles using a modified coaxial electrospinning process. Int J Nanomedicine. 2012;7:5725–5732. | ||
Yu DG, Li XY, Wang X, Yang JH, Bligh SW, Williams GR. Nanofibers fabricated using triaxial electrospinning as zero order drug delivery systems. ACS Appl Mater Interfaces. 2015;7(33):18891–18897. | ||
Chen G, Xu Y, Yu DG, Zhang DF, Chatterton NP, White KN. Structure-tunable Janus fibers fabricated using spinnerets with varying port angles. Chem Commun (Camb). 2015;51(22):4623–4626. | ||
Garric X, Moles JP, Garreau H, Guilhou JJ, Vert M. Human skin cell cultures onto PLA50 (PDLLA) bioresorbable polymers: influence of chemical and morphological surface modifications. J Biomed Mater Res A. 2005;72(2):180–189. | ||
Hoveizi E, Nabiuni M, Parivar K, Rajabi-Zeleti S, Tavakol S. Functionalisation and surface modification of electrospun polylactic acid scaffold for tissue engineering. Cell Biol Int. 2014;38(1):41–49. | ||
Bacakova M, Lopot F, Hadraba D, et al. Effects of fiber density and plasma modification of nanofibrous membranes on the adhesion and growth of HaCaT keratinocytes. J Biomater Appl. 2015;29(6):837–853. | ||
Mo Y, Guo R, Liu J, et al. Preparation and properties of PLGA nanofiber membranes reinforced with cellulose nanocrystals. Colloids Surf B Biointerfaces. 2015;132:177–184. | ||
Garric X, Vert M, Moles JP. Development of new skin substitutes based on bioresorbable polymer for treatment of severe skin defects. Ann Pharm Fr. 2008;66(5–6):313–318. | ||
McMillan JR, Akiyama M, Tanaka M, et al. Small-diameter porous poly (epsilon-caprolactone) films enhance adhesion and growth of human cultured epidermal keratinocyte and dermal fibroblast cells. Tissue Eng. 2007;13(4):789–798. | ||
El-Ghalbzouri A, Lamme EN, van Blitterswijk C, Koopman J, Ponec M. The use of PEGT/PBT as a dermal scaffold for skin tissue engineering. Biomaterials. 2004;25(15):2987–2996. | ||
Altankov G, Albrecht W, Richau K, Groth T, Lendlein A. On the tissue compatibility of poly(ether imide) membranes: an in vitro study on their interaction with human dermal fibroblasts and keratinocytes. J Biomater Sci Polym Ed. 2005;16(1):23–42. | ||
Monteiro IP, Gabriel D, Timko BP, et al. A two-component pre-seeded dermal-epidermal scaffold. Acta Biomater. 2014;10(12):4928–4938. | ||
Mazlyzam AL, Aminuddin BS, Fuzina NH, et al. Reconstruction of living bilayer human skin equivalent utilizing human fibrin as a scaffold. Burns. 2007;33(3):355–363. | ||
Helmedag MJ, Weinandy S, Marquardt Y, et al. The effects of constant flow bioreactor cultivation and keratinocyte seeding densities on prevascularized organotypic skin grafts based on a fibrin scaffold. Tissue Eng Part A. 2015;21(1–2):343–352. | ||
Meana A, Iglesias J, Del Rio M, et al. Large surface of cultured human epithelium obtained on a dermal matrix based on live fibroblast-containing fibrin gels. Burns. 1998;24(7):621–630. | ||
Rajangam T, An SS. Fibrinogen and fibrin based micro and nano scaffolds incorporated with drugs, proteins, cells and genes for therapeutic biomedical applications. Int J Nanomedicine. 2013;8:3641–3662. | ||
Mutsaers SE, Bishop JE, McGrouther G, Laurent GJ. Mechanisms of tissue repair: from wound healing to fibrosis. Int J Biochem Cell Biol. 1997;29(1):5–17. | ||
Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost. 2006;4(5):932–939. | ||
Tajima S, Pinnell SR. Ascorbic acid preferentially enhances type I and III collagen gene transcription in human skin fibroblasts. J Dermatol Sci. 1996;11(3):250–253. | ||
Park HJ, Ock SM, Kim HJ, et al. Vitamin C attenuates ERK signalling to inhibit the regulation of collagen production by LL-37 in human dermal fibroblasts. Exp Dermatol. 2010;19(8):E258–E264. | ||
Geesin JC, Gordon JS, Berg RA. Regulation of collagen synthesis in human dermal fibroblasts by the sodium and magnesium salts of ascorbyl-2-phosphate. Skin Pharmacol. 1993;6(1):65–71. | ||
Tuan TL, Song A, Chang S, Younai S, Nimni ME. In vitro fibroplasia: matrix contraction, cell growth, and collagen production of fibroblasts cultured in fibrin gels. Exp Cell Res. 1996;223(1):127–134. | ||
Mazzone L, Pontiggia L, Reichmann E, Ochsenbein-Kolble N, Moehrlen U, Meuli M. Experimental tissue engineering of fetal skin. Pediatr Surg Int. 2014;30(12):1241–1247. | ||
Ahmed TA, Dare EV, Hincke M. Fibrin: a versatile scaffold for tissue engineering applications. Tissue Eng Part B Rev. 2008;14(2):199–215. | ||
Han CM, Zhang LP, Sun JZ, Shi HF, Zhou J, Gao CY. Application of collagen-chitosan/fibrin glue asymmetric scaffolds in skin tissue engineering. J Zhejiang Univ Sci B. 2010;11(7):524–530. | ||
Gorodetsky R, Clark RA, An J, et al. Fibrin microbeads (FMB) as biodegradable carriers for culturing cells and for accelerating wound healing. J Invest Dermatol. 1999;112(6):866–872. | ||
Nair RP, Joseph J, Harikrishnan VS, Krishnan VK, Krishnan L. Contribution of fibroblasts to the mechanical stability of in vitro engineered dermal-like tissue through extracellular matrix deposition. Biores Open Access. 2014;3(5):217–225. | ||
Paxton JZ, Wudebwe UN, Wang A, Woods D, Grover LM. Monitoring sinew contraction during formation of tissue-engineered fibrin-based ligament constructs. Tissue Eng Part A. 2012;18(15–16):1596–1607. | ||
Rampichova M, Buzgo M, Krizkova B, Prosecka E, Pouzar M, Strajtova L. Injectable hydrogel functionalised with thrombocyte-rich solution and microparticles for accelerated cartilage regeneration. Acta Chir Orthop Traumatol Cech. 2013;80(1):82–88. | ||
Gurevich O, Vexler A, Marx G, et al. Fibrin microbeads for isolating and growing bone marrow-derived progenitor cells capable of forming bone tissue. Tissue Eng. 2002;8(4):661–672. | ||
Adebayo O, Hookway TA, Hu JZ, Billiar KL, Rolle MW. Self-assembled smooth muscle cell tissue rings exhibit greater tensile strength than cell-seeded fibrin or collagen gel rings. J Biomed Mater Res A. 2013;101(2):428–437. | ||
Williams C, Johnson SL, Robinson PS, Tranquillo RT. Cell sourcing and culture conditions for fibrin-based valve constructs. Tissue Eng. 2006;12(6):1489–1502. | ||
Kubies D, Rypacek F, Kovarova J, Lednicky F. Microdomain structure in polylactide-block-poly(ethylene oxide) copolymer films. Biomaterials. 2000;21(5):529–536. | ||
Novotna K, Zajdlova M, Suchy T, et al. Polylactide nanofibers with hydroxyapatite as growth substrates for osteoblast-like cells. J Biomed Mater Res A. 2014;102(11):3918–3930. | ||
Riedel T, Brynda E, Dyr JE, Houska M. Controlled preparation of thin fibrin films immobilized at solid surfaces. J Biomed Mater Res A. 2009;88(2):437–447. | ||
Eisenbud D, Huang NF, Luke S, Silberklang M. Skin substitutes and wound healing: current status and challenges. Wounds. 2004;16(1):2–17. | ||
Idrus RB, Rameli MA, Low KC, et al. Full-thickness skin wound healing using autologous keratinocytes and dermal fibroblasts with fibrin: bilayered versus single-layered substitute. Adv Skin Wound Care. 2014;27(4):171–180. | ||
Gailit J, Clarke C, Newman D, Tonnesen MG, Mosesson MW, Clark RA. Human fibroblasts bind directly to fibrinogen at RGD sites through integrin alpha(v)beta3. Exp Cell Res. 1997;232(1):118–126. | ||
Gray AJ, Bishop JE, Reeves JT, Laurent GJ. A alpha and B beta chains of fibrinogen stimulate proliferation of human fibroblasts. J Cell Sci. 1993;104(pt 2):409–413. | ||
Dawes KE, Gray AJ, Laurent GJ. Thrombin stimulates fibroblast chemotaxis and replication. Eur J Cell Biol. 1993;61(1):126–130. | ||
Yamamoto M, Yanaga H, Nishina H, Watabe S, Mamba K. Fibrin stimulates the proliferation of human keratinocytes through the autocrine mechanism of transforming growth factor-alpha and epidermal growth factor receptor. Tohoku J Exp Med. 2005;207(1):33–40. | ||
Acevedo CA, Brown DI, Young ME, Reyes JG. Senescent cultures of human dermal fibroblasts modified phenotype when immobilized in fibrin polymer. J Biomater Sci Polym Ed. 2009;20(13):1929–1942. | ||
Nguyen DT, Orgill DP, Murphy GF. The pathofysiologic basis for wound healing regeneration. In: Orgill D, Bianco C, editors. Biomaterials for Treating Skin Loss. Cambridge: Woodhead Publishing; 2009:25–58. | ||
Lima CC, Pereira AP, Silva JR, et al. Ascorbic acid for the healing of skin wounds in rats. Braz J Biol. 2009;69(4):1195–1201. | ||
Taniguchi M, Arai N, Kohno K, Ushio S, Fukuda S. Anti-oxidative and anti-aging activities of 2-O-alpha-glucopyranosyl-L-ascorbic acid on human dermal fibroblasts. Eur J Pharmacol. 2012;674(2–3):126–131. | ||
Phillips CL, Combs SB, Pinnell SR. Effects of ascorbic acid on proliferation and collagen synthesis in relation to the donor age of human dermal fibroblasts. J Invest Dermatol. 1994;103(2):228–232. | ||
Sclafani AP, McCormick SA. Induction of dermal collagenesis, angiogenesis, and adipogenesis in human skin by injection of platelet-rich fibrin matrix. Arch Facial Plast Surg. 2012;14(2):132–136. | ||
de Clerck YA, Jones PA. The effect of ascorbic acid on the nature and production of collagen and elastin by rat smooth-muscle cells. Biochem J. 1980;186(1):217–225. | ||
Murad S, Grove D, Lindberg KA, Reynolds G, Sivarajah A, Pinnell SR. Regulation of collagen synthesis by ascorbic acid. Proc Natl Acad Sci U S A. 1981;78(5):2879–2882. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.