Back to Journals » Clinical Interventions in Aging » Volume 17
The Mechanism of Bone Remodeling After Bone Aging
Authors Fang H, Deng Z ![]() , Liu J, Chen S, Deng Z, Li W
, Liu J, Chen S, Deng Z, Li W
Received 16 November 2021
Accepted for publication 29 March 2022
Published 5 April 2022 Volume 2022:17 Pages 405—415
DOI https://doi.org/10.2147/CIA.S349604
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Zhi-Ying Wu
Huankun Fang,1,2,* Zhiqin Deng,1,* Jianquan Liu,1,* Siyu Chen,3 Zhenhan Deng,3 Wencui Li1
1Hand and Foot Surgery Department, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital, Shenzhen, Guangdong, 518035, People’s Republic of China; 2Medical College, Shantou University, Shantou, Guangdong, 515041, People’s Republic of China; 3Department of Sports Medicine, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital, Shenzhen, Guangdong, 518035, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhenhan Deng, Department of Sports Medicine, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital, 3002 Sungang West Road, Shenzhen City, 518025, People’s Republic of China, Tel +86 13928440786, Fax +86 755-83366388, Email [email protected] Wencui Li, Department of Hand and Foot Surgery, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital, 3002 Sungang West Road, Shenzhen City, 518025, People’s Republic of China, Tel +86 13923750767, Email [email protected]
Abstract: Senescence mainly manifests as a series of degenerative changes in the morphological structure and function of the body. Osteoporosis is a systemic bone metabolic disease characterized by destruction of bone microstructure, low bone mineral content, decreased bone strength, and increased brittleness and fracture susceptibility. Osteoblasts, osteoclasts and osteocytes are the main cellular components of bones. However, in the process of aging, due to various self or environmental factors, the body’s function and metabolism are disordered, and osteoporosis will appear in the bones. Here, we summarize the mechanism of aging, and focus on the impact of aging on bone remodeling homeostasis, including the mechanism of ion channels on bone remodeling. Finally, we summarized the current clinical medications, targets and defects for the treatment of osteoporosis.
Keywords: aging, osteoporosis, bone formation, epigenetics, autophagy, chlorine channel
Introduction
At present, human life expectancy is significantly longer than ever before, the process of aging is still not considered to be healthy; instead, the proportion of an individual’s healthy life expectancy within their total life expectancy remains low. The World Health Organization (WHO) defines healthy ageing as the process of maintaining the functional ability of the elderly to enable them to achieve wellbeing. The incidence of many age-related diseases, including cardiovascular, cerebrovascular, and neurodegenerative conditions, is increasing year by year, bringing about suffering and a great economic burden to many elderly people.1 Moreover, aging has become a primary risk factor of certain chronic and devastating diseases. Therefore, in the pursuit of longevity, attention to aging has become an important part of people’s pursuit of healthy aging process.2 In this review, we will first explain the mechanism of aging, and then focus on the effect of aging on the homeostasis of bone remodeling, and summarize the pathological mechanism of bone remodeling after aging including some recent evidence of ion channels in bone remodeling.
Mechanism of Aging
Aging is considered in the literature to be the main cause of many chronic diseases in the body, yet its mechanism is still unclear. The mechanism of cell senescence can inhibit the proliferation of damaged cells but, with the advancement of age, senescent cells accumulate in various tissues and organs and the substances they secrete may destroy the structure and function of tissues.3 As Berdasco reported, aging is a complex process that leads to complex biological changes in the body and increases an organism’s susceptibility to disease and death.4 Several hypotheses about the mechanism of aging exist at this time. Unraveling the mechanism of aging can explain many age-related diseases and provide theoretical basis for the research and development of anti-aging drugs. Cellular senescence is a basic activity of cells, along with proliferation, differentiation, and apoptosis. Cellular senescence is a sign of aging and the accumulation of senescence cells in senescence tissues is considered to be the driving factor of the senescence process.5 Cell senescence has been shown to consist of three stages—namely, early, complete, and late.6 Here, in the early stage, the response to stress is manifested by morphological and functional changes, while the second stage is the main senescence-associated secretory phenotype (SASP) response. The late stage involves interferon type I reactions and the upregulation of L1 (line-1) retrotransposable elements.7 In general, the aging process encompasses genomic damage and telomere reduction epigenetic changes that alter the protein balance and trigger nutrient perception disorders, mitochondrial dysfunction, stem-cell pool collapse, intercellular communications disorders, and cellular senescence.8
Free Radical Theory and Senescence
Harman, a British molecular biologist, first proposed the free radical theory of aging: he believed that “oxidative stress determines the life expectancy.” Aging is the result of the destruction of cells and tissues by oxygen free radicals in a process closely related to the aging process controlled by the environment, disease, and heredity factors. With aging, the metabolic mechanism maintaining the balance in vivo weakens and the concentration of free radicals increases, facilitating irreversible changes to the biofilm, amino acid chain, and DNA molecular structure and accelerating the aging process. Free radical inhibitors and antioxidants extend cell life. Some people believe that free radicals and their associated oxidants are only part of the stressor. The body can adjust the homeostasis range of stress defense and repair system through adaptive homeostasis and produce the most suitable complex structure for induction and specific requirements of enzyme protection.9
Telomere and Telomerase Theory in Aging
Telomere is a complex of telomere DNA and binding proteins located at the end of chromosomes in eukaryotic cells. Telomerase can regulate the replication of telomeres under specific conditions and keep them short in chromosome replication. Maintaining the stability of the effect of telomerase on the regulation of telomeres is an important consideration in the process of aging. It has been shown that overexpression of telomerase can rejuvenate tissues and prolong life span.10 Telomerase may be a target to treat some senile diseases and even delay senescence. However, DNA damage spreads the consequences of aging throughout the body, and it is the cause of age-related diseases. Some genetic syndromes that encompass defective DNA repair are associated with premature aging.11
Autophagy and Senescence
Autophagy is one of the important regulatory mechanisms in the aging process that can regulate cell growth and realize the homeostasis of substances and energy in cells. Three types of autophagy exist: macrophage autophagy (MA), chaperon-mediated autophagy (CMA), and microautophagy, with MA being the main form among the three. It has been reported that aging leads to decreased autophagy levels. Autophagy can be either nonselective or cargo-specific, with mitochondria-specific autophagy known as mitochondrial autophagy. Increasing evidence supports the critical role of autophagy and mitochondrial autophagy in regulating baseline homeostasis and stress. The decrease in autophagy level leads to the aging of many organs such as bone, muscle and kidney.12 Autophagy theory explains the phenomenon of slow metabolism and homeostasis destruction in the elderly.
Cell Senescence and Epigenetic Mechanisms of Senescence
Epigenetic factors, including histone modification, DNA methylation and microRNA (miRNA) expression, may play a key role in regulating gene expression changes and genomic instability during aging. Epigenetic modifications can regulate gene expression and participate in the occurrence and development of many diseases.8 Moreover, particularly in the mammalian system, global and local changes in DNA methylation, heterochromatin site-specificity of loss and gain, and significant restructuring occur.13 Prior research suggests that cytosine-methylation patterns of cytosine–guanine dinucleotide (located at the CpG site) in the mouse and human genomes change with age.14 On this basis, an epigenetic clock reflecting the CpGs methylation level was established15 and used to measure the age of cells in different tissue types; notably, research has suggested its accuracy was significantly higher than that of classical telomere length. This could become a new standard for judging aging.16 DNA methylation plays an important role in maintaining DNA stability, cell proliferation, and senescence. Evidences show that most methylation changes are programmed and occur in subsets of tissue cells during normal aging.17 It has been reported that age-related diseases are often accompanied by a drop in DNA methylation levels. Although DNA methylation clocks were initially considered molecular biomarkers of real age, DNA methylation is now being used as a marker of disease risk.18
Aging Caused by Inflammation
The immune system can recognize and remove senescent cells, while the progenitor cells in the tissue can differentiate to form young cells to replace senescent cells, preventing the spread of harmful cells such as a decaying or potential tumor cell. With the aging of the body, however, cell senescence is accelerated and the immune system slows down the elimination of senescence cells, resulting in more senescence cells remaining in the body. Senescence secretion of proinflammatory cytokines and matrix metal albumin by senescent cells on peripherals is referred to as the SASP. SASP and nuclear factor kappa B overactivate immune system dysfunction and reduce autophagy function and, together with other factors, act on the body to cause inflammatory aging. This provides new ideas for the development of anti-aging drugs. Aging releases many inflammatory factors, such as interleukin-1 and tumor necrosis factors (Figure 1). Reducing the inflammatory factors that the body releases as it ages may delay aging. There have been reported that the inhibition of nuclear factor kappa B by a drug or substrate led to a trend toward youth on the aging spectrum in aging mice.19
 |
Figure 1 Mechanism of aging. Notes: Superoxide dismutase, glutathione peroxidase, and catalase in the body eliminate oxygen free radicals. These enzyme’s activity is inhibited while aging and too many oxygen free radicals appear, leading to cell senescence. The autophagic lysosome formed by autophagic vesicles and lysosomes can encapsulate the molecules that need to be degraded in cells for degradation. The level of autophagy decreases with age. Telomerase can regulate the replication of telomeres and prevent them from shortening in chromosome replication, thus affecting aging. DNA methylation can reflect aging. The activation of the DNA damage response (DDR) in senescent cells promotes the attainment of proinflammatory secretory phenotype (SASP), which, in turn, triggers the activation of DDR and SASP in adjacent cells, thus creating a proinflammatory environment and accelerating senescence both locally and at the systemic level. |
The Epidemiology of Bone Aging and Senile Osteoporosis
Human beings are gradually moving towards an aging society, which has become a consensus. For example, China is facing significant medical challenges brought on by the aging problem; by 2050, the country will have 400 million people older than 65 years, including 150 million over the age of 80 years.20 The 2015 United Nations (UN) report on the ageing of the world’s population claimed that in the next 35 years the number of people aged 60 years or over will more than double to 2.1 billion.21
Osteoporosis is a systemic metabolic bone disease defined by the World Health Organization (WHO). When bone mass is reduced and the microstructure of bone tissue is impaired, it is characterized by fragility and increased fracture susceptibility. With more and more of the population aging, osteoporosis has gradually become a global health problem for people older than 50 years of age. As highlighted earlier, the proportion of the elderly in China is increasing and osteoporosis has become a serious challenge faced by Chinese society.22 The annual number and cost of osteoporosis-related fractures are expected to double by 2035 and increase to 5.99 million cases by 2050 at a cost of $25.43 billion per year.23 At the level of clinical research and medical policy-making, assessing the vulnerability of older individuals contributes to the assessment, management, and decision-making of osteoporosis and osteoporotic fractures.24 Osteoporosis caused by aging has become a social problem that cannot be ignored.
Marrow Stem Cell Depletion in Bone Aging Breaks the Homeostasis of Bone Remodeling
Aging leads to the decline of the function of every tissue in the body and lead to age-related bone dysfunction.8 Bone aging is characterized by massive loss of bone mass and accumulation of bone marrow adipose tissue (BMAT), which is related to the exhaustion and abnormal differentiation of stem cells.25 Mesenchymal stem cells (MSCs) are found widely in the bone marrow and spongy bone and play an important role in maintaining the dynamic balance of bone resorption and formation.26 In older people, the ability of osteoblasts to differentiate and proliferate is reduced, while that of fat cells is increased. This results in accumulation of fat in the bone marrow cavity, which threatens the survival of osteoblasts.27 Abnormal pedigree specification of bone stem cells contributes to decreased bone mass and increased bone marrow adipose tissue in the context of osteoporosis and bone aging. Some studies have suggested that peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) may act as a key switch that determines the fate of cells. PGC-1α expression levels decrease with age in bone stem cells in both humans and mice; thus, PGC-1α can be a potentially important therapeutic target in the treatment of osteoporosis and bone aging.28 In animal experiments, it was found that miR-188 expression in bone marrow–derived MSCs (BMSCs) significantly increased during the aging process, which resulted in increased conversion of BMSCs to adipocytes and reduced differentiation to osteoblasts, culminating in reduced fat accumulation; bone formation in bone marrow; and, eventually, age-related bone loss and osteoporosis. However, the injection of a miR-188–specific antagonist into the bone marrow cavity reduced the degree of fat accumulation and increased bone formation in the bone marrow of aged mice.29 A peroxisome proliferation-activated receptor is the main regulatory factor of adipogenic differentiation and can inhibit the activity of osteoblasts by blocking the expression of core-binding factor 1, thus playing an important role in the adipogenic differentiation of BMSCs.30 Aging increases the adipogenic differentiation of BMSCs, which may be related to the decrease of PGC-1α and the increase of miR-188. Inhibiting the adipogenic differentiation of BMSCs may be a new way to treat senile osteoporosis.
Telomere dysfunction caused by aging of mesenchymal stem cells may also be another important reason for the imbalance of bone remodeling. Through in vitro culturing of MSCs in a mouse model, Wang et al found that telomere dysfunction could induce senescence and apoptosis in MSCs by stimulating the P53/P21 pathway, downregulating the expression of osteoblastic transcription factor Runx2 in MSCs, and inhibiting the transformation and differentiation of MSCs into osteoblasts, resulting in a reduced bone mass.31 Telomere dysfunction may be one of the causes of senile osteoporosis.
The Accumulation of Oxidative Stressors in Aging Accelerates the Imbalance of Bone Remodeling
A lot of evidence suggests that aging and the associated increase in reactive oxygen species (ROS) are the main culprits of osteoporosis.32 The decrease in estrogen or androgen levels reduces the bone’s defenses against oxidative stress. The accumulation of ROS simultaneously produces and survives osteoclasts, osteoblasts and osteocytes.33–35 Aging can lead to the accumulation of oxidative stress products and the inhibition of bone formation, which may be related to the blocking of the Wnt signal and the transfer of β-catenin from T-cell–specific transcription factors to the transcription-factor binding site mediated by Forkhead box O.32 The important role of advanced protein-oxidation products in many age-related diseases has been widely noted.36 As mentioned above, aging is closely related to free radicals, which can also damage bone cells and reduce bone mass, leading to osteoporosis. Zeng et al found that advanced protein-oxidation products accelerated bone deterioration in elderly rats.37
Decreased Estrogen Levels Caused by Aging Lead to Bone Loss
The ovarian function declines in postmenopausal women, and the decline in estrogen levels is an irreversible fact. Estrogen protects the remodeling homeostasis of adult bones by slowing down the speed of bone remodeling while maintaining the local balance between bone formation and resorption.38,39 Kyunghee’s study found that RANKL caused a decline in ovarian function. Decreased estrogen levels in vivo promote osteoclast differentiation by inducing the phosphorylation of ERK1 and ERK2.40 Experiments have shown that estrogen decline is associated with an increased OPG/RANKL ratio. Estrogen inhibits osteoclasts through OPG/RANKL pathway in early menopause, thus reducing bone loss. Moreover, with greater estrogen doses, local vascular bone formation increases, suggesting that estrogen is related to angiogenic osteoporosis.41 miRNA is a class of uncoded single-stranded RNA commonly found in eukaryotic organisms, with a length of about 21 to 25 nucleotides, which is used for regulatory purposes after radical transcription. The abnormal expression of miRNA is closely related to the occurrence of osteopenia. Zhang found that miR-221 can directly target and inhibit Runx-2–mediated osteoblast differentiation,42 while Chen found that miR-125b inhibited the differentiation and proliferation of bone marrow MSCs by downregulating the osterix expression.43 In addition, miR-145 can inhibit the expression of OPG, while estrogen can increase the expression of OPG by reducing the expression of miR-145.44 Estrogen levels fall as women age, a risk factor for postmenopausal osteoporosis.
Immune System and Its Imbalance with Aging
The clearance of senescent cells is obviously a programmed process in which cellular immunity and humoral immunity participate together.45 Immune senescence refers to a decline in the vitality and precision of the adaptive immune system in old age, mainly due to thymus degeneration and changes in T lymphocyte subsets.46 Furthermore, senescent cells appear to escape immune suppression, such as paracrine suppression of CD8+ cells by natural killer factors and proinflammatory cytokines. RANKL is a member of the tumor necrosis factor superfamily that is secreted by T-cells and B-cells and which binds to the biological receptor RANK on the surface of osteoclasts, thus stimulating the differentiation of osteoclasts into mature multinucleated osteoclasts. B-cells are the main source of OPG in the bone microenvironment. Activated B-cells can also overexpress RANKL. Studies have found that the number of B-cells expressing RANKL in the bone marrow of ovoidless mice is significantly increased, while the loss of RANKL in B-cells can protect from bone loss after ovoidectomy.47 With age, the immune system’s ability to recognize senescent cells decreases, leading to the differentiation and maturation of osteoclasts, leading to osteoporosis.
The decrease in CD4+ T and CD8+ T lymphocytes leads to a decrease in OPG production.48–50 Surface T cells also have anti-osteoclast properties. It shows that T cells play an important role in bone remodeling. In the aging immune system, the decreased diversity of T cells may lead to aging osteoporosis.51
Autophagy and Senile Osteoporosis
Autophagy is a process in which the substrates in the cytoplasm are phagocytic by assembling the membrane structure derived from the cell’s own organelles, such as misfolded or senescent proteins, while damaged organelles are combined with lysosomes to degrade the ingestion phagocytes and recycle them.52 Studies have shown that, as the human body ages, the autophagy level of bone cells gradually decreases, which increases the secretion of proinflammatory factors such as interleukin-1β (IL-1β), accelerates bone loss, and affects bone metabolism, leading to osteoporosis.53 Almeida demonstrated that autophagy can prevent bone loss in bone cells and reduce the apoptosis of bone cells with aging.54 A loss of autophagy may lead to increased dysfunction of bone cells.55 In addition, autophagy induction can lead to the survival response of BMSCs against oxidative stress.56 Studies have shown that BMSCs in osteoporosis patients present a senescent phenotype with significantly lower autophagy levels and significantly reduced osteogenic potential as compared with those in healthy subjects.57 Autophagy plays a role in downregulating oxidative stress in osteoblasts and osteocytes. In a mouse model of osteoblast-specific autophagy, bone loss because of aging and estrogen deficiency worsened significantly.58 Autophagy also regulates the homeostasis of the miRNA network, which can regulate bone formation through the Wnt signaling pathway and bone resorption through the RANKL/RANK pathway.59 In the aging process, osteocyte autophagy is not correlated with osteocyte apoptosis, suggesting that osteocyte autophagy may be related to bone loss. Senescence can cause a decrease of autophagy in cells and the decrease of autophagy in osteoblasts and osteocytes related to senescence is considered to be another hotspot in the theory of osteoporosis. The decreased autophagy activity of bone cells may be one of the causes of bone loss in senile osteoporosis.52
Vitamin D and Senile Osteoporosis
Studies have shown that 1,25-hydroxyvitamin D promotes osteogenic differentiation of human BMSCs. Senescence can downregulate the expression and activity of 1α-hydroxylase in human BMSCs. Parathyroid hormone can promote the activity of 1α-hydroxylase to repair bone defects, and can rejuvenate the osteogenesis of BMSCs in the elderly. Moreover, aging affects the skin’s ability to make vitamin D. Senescence is associated with decreased kidney function, reduced vitamin D receptors, a lack of intestinal responsiveness to active vitamin D, and limitations on calcium absorption, which affects the production of active vitamin D.60
The Role of Ion Channels in Bone Remodeling
The transport of ions across cell membranes can participate in the basic life processes of cells through ion channels.61–65 The process of bone remodeling is finely regulated by various ion channels. For example, the mechanically controlled ion channel Piezo1, Piezo1 is expressed in osteoblasts and osteocytes and regulates mechanical load-dependent bone formation.66–68 Conditional knockout of Piezo1 in osteoblasts and osteocytes will result in a significant reduction in bone mass and damage to bone structure and bone strength.69 In addition, abnormal subchondral osteoblastic metabolism changes the NaV1.8 for osteoarthritis.70 Purinergic P2X receptors and their family proteins play a role in osteoclast bone resorption, bone pain and inflammation, and may be promising potential therapeutic targets for osteoporosis.71
Five families of chloride ion channels exist: ligand-gated anion channels, CFTR, ClCs, bestrophins, and anoctamins.72 When the CFTR chloride channel is missing, it can cause OPG to decrease, RANK to increase, and RANKL/OPG ratio to decrease, thereby enhancing bone resorption.73 Chlorine channels may affect bone remodeling by controlling acid secretion. When the proton pump and chlorine channel are activated, the pH in the local absorption cavity of osteoclasts drops to about 4.5.74 Other research also shows osteoclasts secrete hydrogen ions through proton pumps, while chlorine ions dissolve minerals in bone through chloride ion channels on the cell membrane near the bottom of calcified bone.75 It is speculated that the opening of the chloride channel may be a key step of osteoclast acid secretion and bone resorption. In addition, the chlorine channel also has a regulatory effect on bone formation. On the one hand, the expression of the ClC-3 chloride channel in osteoblasts is conducive to the stimulation of PTH by osteoblasts, thus mediating the differentiation of osteoblasts.76 The ClC-3 chloride channel regulates the bone formation function of osteoblasts by regulating the estrogen a receptor.77,78 The increased expression levels of ClC-3 chlorine channel proteins can upregulate the expression of the downstream Runx2 gene, thus promoting osteogenic differentiation.79 The lack of ClC-7 function can lead to bone sclerosis (Figure 2).80,81
 |
Figure 2 Mechanism of SOP. Notes: With aging, the ability of BMSCs to differentiate into osteoblasts decreases and that of adipocytes increases. A decrease in peroxisome proliferator-activated receptor-γ coactivator 1 increases bone loss and fat accumulation during bone aging. Aging leads to the increase in reactive oxygen species (ROS), the accumulation of oxygen free radicals, the destruction of bone cells, and the inhibition of bone formation. With the growth of age, estrogen synthesis in vivo is reduced, CFTR chloride channel is lost, OPG is decreased, and overexpression of RANKL leads to osteoporosis. ClC-3 chloride channel in osteoblasts regulates the bone formation function of osteoblasts by regulating estrogen A receptor. In addition, the decrease in immune cells during aging leads to the decrease of OPG production and the increase in secretion of pro-inflammatory factors, which accelerates bone loss, affects bone metabolism and leads to osteoporosis. MiRNA networks can regulate bone formation through Wnt signaling pathway and bone resorption through RANKL/RANK pathway. In aging, autophagy level decreases and homeostasis of miRNA networks is broken. Finally, aging can down-regulate the expression and activity of 1α-hydroxylase in human bone marrow mesenchymal stem cells and inhibit the osteogenic differentiation of human bone marrow mesenchymal stem cells. |
Treatment Strategies for Senile Osteoporosis
The drugs currently used to treat osteoporosis are mainly based on the principle of inhibiting bone resorption or promoting bone formation. Commonly used anti-osteoporotic drugs include bisphosphonates, parathyroid hormone, vitamin D, calcium and estrogen. Among them, calcium, vitamin D, simvastatin and parathyroid hormone mainly promote bone formation, and estrogen, bisphosphonates and calcitonin mainly function by inhibiting bone resorption. In Table 1,82–89 we have summarized the current methods, shortcomings and mechanisms of clinical medications for the treatment of osteoporosis. Traditional drugs such as raloxifene and tamoxifen bind to estrogen α or β receptors in osteoblasts and osteoclasts, promoting osteoclast apoptosis and inhibiting bone resorption. Teriparatide as well as calcium tablet and vitamin D promote calcium and phosphorus metabolism and increase bone mineralization. Bisphosphonates and salmon calcificin inhibit osteoclast activity and formation to reduce bone resorption. In addition, some new drugs like simvastatin can improve the high metabolic state of bone, while monoclonal antibody drugs can affect osteoclast activity and have a positive effect on bone reconstruction. A good diet that includes calcium-rich foods can help to prevent osteoporosis. Aging and SASP play a mechanistic role in age-related and obesity-related fat deterioration, including inflammation and metabolic defects, which can be prevented or attenuated by exercise.90 Mechanical signaling promotes bone and muscle anabolism through exercise, while limiting the formation and expansion of fat. By regulating the function of cells in bone tissue, machinery adjustment of bone and bone marrow fat can be achieved while preserving bias in selection of bone marrow mesenchymal stem cells lineages.91 Calcium and vitamin D are critical in the treatment of osteoporosis in older adults. Increased antiresorption or anabolic therapy or combination therapy, at least in older adults with osteoporosis, may be effective in reducing the risk of vertebral fractures.92 Aging is closely related to osteoporosis. The study of the pathogenesis of aging can provide theoretical basis for the treatment of senile osteoporosis.
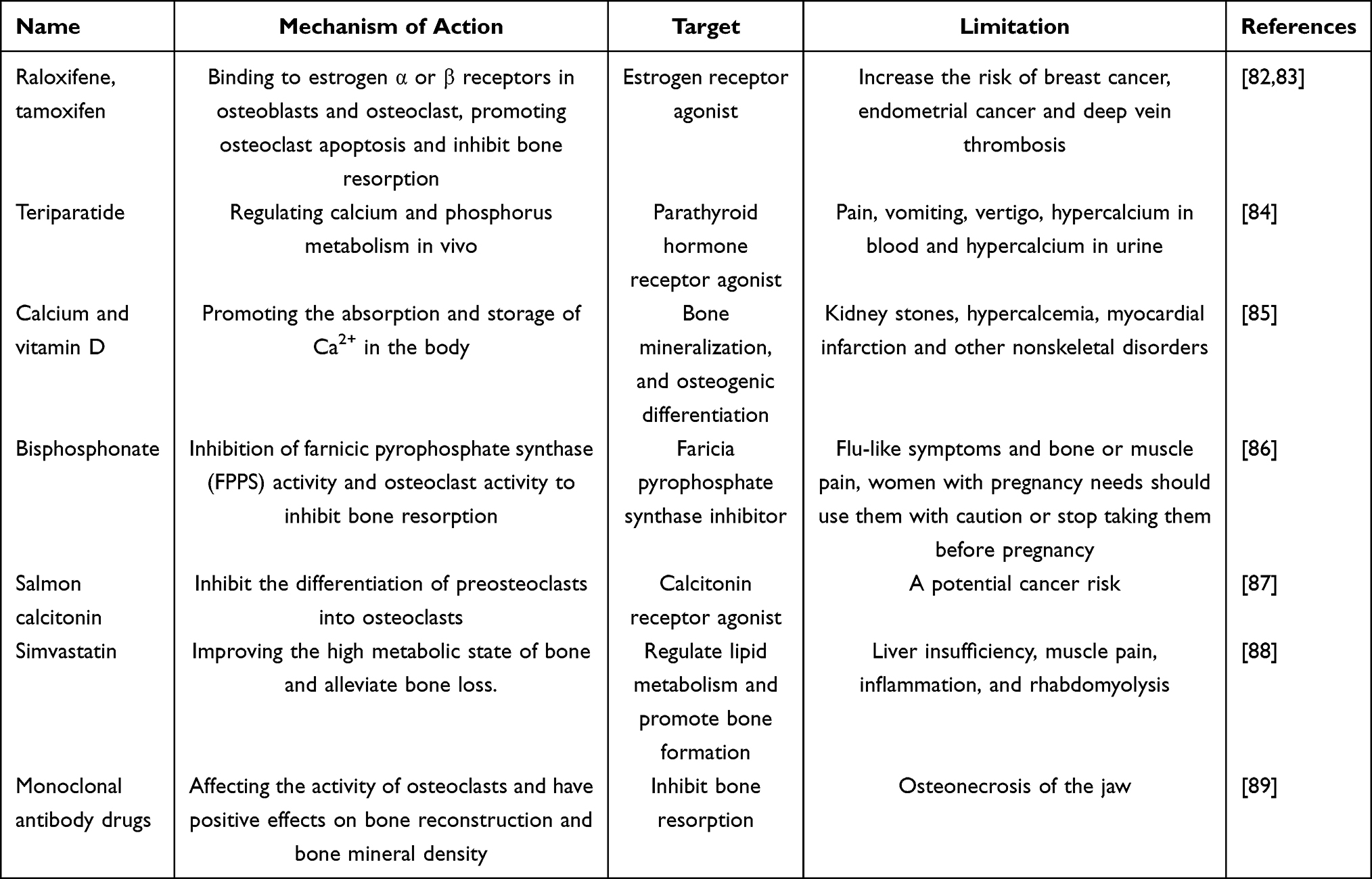 |
Table 1 Common Clinical Medications and Limitations in the Treatment of Osteoporosis |
Conclusion
To sum up, aging is a complex biological problem: the pathogenesis of aging is multicausative and comprehensive and complex aging is also a process in which various factors participate in and act together and various mechanisms interact with each other to jointly promote aging of the body. To explore the various mechanisms of aging in many aspects is helpful for us to fully understand the essence of aging and even delay the progress of aging. In the aging process, autophagy levels decrease and proinflammatory factors are produced, thus increasing the incidence of osteoporosis. Osteoporosis is a common metabolic disease, the incidence of which is increasing year by year in the population, seriously affecting the global economy and health. However, few studies on the relevant mechanism that connects aging and osteoporosis exist. It is believed that improving the level of autophagy, enhancing immune defense, limiting adipogenic differentiation, antioxidant-free radicals, slowing down the decline of estrogen, can delay the occurrence of senile osteoporosis. Therefore, we can enhance the immunity through daily exercise and limit the intake of fatty foods to strengthen bone. In addition, in terms of drug research, we can design new antioxidation or ion channel-based estrogen drugs to reduce bone loss.
Acknowledgments
The authors thank Dr Murad Alahdal for discussion and critical reading of the article.
Funding
This study was supported by the National Natural Science Foundation of China (81800785, 81972085, 81902303), the Natural Science Foundation of Guangdong Province (2018A0303100027, 2020A151501048), Medical Science and Technology Research Foundation of Guangdong Province (A2018262), the Sanming Project of Shenzhen Health and Family Planning Commission (SZSM201612086), Shenzhen Science and Technology Planning (JCYJ20180228163401333, JCYJ20190806170612680, JCYJ20190806164216661, RCBS20200714114856299), and the Doctor Innovation Project of Shenzhen Health System (SZBC2018015), Shenzhen Peacock Project (KQTD20170331100838136), Shenzhen Key Medical Discipline Construction Fund (SZXK025), and discipline construction Capacity Improvement project of Shenzhen Municipal Health Commission (SZXJ2018065).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kim HJ, Min JY, Min KB. Successful aging and mortality risk: the Korean longitudinal study of aging (2006–2014). J Am Med Dir Assoc. 2019;20:1013–1020. doi:10.1016/j.jamda.2018.12.010
2. Aramillo Irizar P, Schäuble S, Esser D, et al. Transcriptomic alterations during ageing reflect the shift from cancer to degenerative diseases in the elderly. Nat Commun. 2018;9:327. doi:10.1038/s41467-017-02395-2
3. Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi:10.1038/nature10600
4. Berdasco M, Esteller M. Hot topics in epigenetic mechanisms of aging: 2011. Aging Cell. 2012;11:181–186. doi:10.1111/j.1474-9726.2012.00806.x
5. Schmeer C, Kretz A, Wengerodt D, Stojiljkovic M, Witte OW. Dissecting aging and senescence-current concepts and open lessons. Cells. 2019;8:1446. doi:10.3390/cells8111446
6. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128:1238–1246. doi:10.1172/JCI95148
7. De Cecco M, Ito T, Petrashen AP, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019;566:73–78. doi:10.1038/s41586-018-0784-9
8. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi:10.1016/j.cell.2013.05.039
9. Pomatto LCD, Davies KJA. Adaptive homeostasis and the free radical theory of ageing. Free Radic Biol Med. 2018;124:420–430. doi:10.1016/j.freeradbiomed.2018.06.016
10. Jaskelioff M, Muller FL, Paik JH, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469:102–106. doi:10.1038/nature09603
11. de Boer J, Andressoo JO, de Wit J, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–1279. doi:10.1126/science.1070174
12. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res. 2016;118:1563–1576. doi:10.1161/CIRCRESAHA.116.307474
13. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. 2016;166:822–839. doi:10.1016/j.cell.2016.07.050
14. Horvath S, Zhang Y, Langfelder P, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97. doi:10.1186/gb-2012-13-10-r97
15. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi:10.1186/gb-2013-14-10-r115
16. Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19:371–384. doi:10.1038/s41576-018-0004-3
17. Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016;76:3446–3450. doi:10.1158/0008-5472.CAN-15-3278
18. Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA methylation clocks in aging: categories, causes, and consequences. Mol Cell. 2018;71:882–895. doi:10.1016/j.molcel.2018.08.008
19. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol a Biol Sci Med Sci. 2014;69(Suppl 1):S4–S9. doi:10.1093/gerona/glu057
20. Fang EF, Scheibye-Knudsen M, Jahn HJ, et al. A research agenda for aging in China in the 21st century. Ageing Res Rev. 2015;24:197–205. doi:10.1016/j.arr.2015.08.003
21. Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539:180–186. doi:10.1038/nature20411
22. Lin X, Xiong D, Peng YQ, et al. Epidemiology and management of osteoporosis in the People’s Republic of China: current perspectives. Clin Interv Aging. 2015;10:1017–1033. doi:10.2147/CIA.S54613
23. Si L, Winzenberg TM, Jiang Q, Chen M, Palmer AJ. Projection of osteoporosis-related fractures and costs in China: 2010–2050. Osteoporos Int. 2015;26:1929–1937. doi:10.1007/s00198-015-3093-2
24. Li G, Thabane L, Papaioannou A, Ioannidis G, Levine MA, Adachi JD. An overview of osteoporosis and frailty in the elderly. BMC Musculoskelet Disord. 2017;18:46. doi:10.1186/s12891-017-1403-x
25. Farr JN, Khosla S. Cellular senescence in bone. Bone. 2019;121:121–133. doi:10.1016/j.bone.2019.01.015
26. Idris AI, Sophocleous A, Landao-Bassonga E, et al. Cannabinoid receptor type 1 protects against age-related osteoporosis by regulating osteoblast and adipocyte differentiation in marrow stromal cells. Cell Metab. 2009;10:139–147. doi:10.1016/j.cmet.2009.07.006
27. Mirsaidi A, Genelin K, Vetsch JR, et al. Therapeutic potential of adipose-derived stromal cells in age-related osteoporosis. Biomaterials. 2014;35:7326–7335. doi:10.1016/j.biomaterials.2014.05.016
28. Yu B, Huo L, Liu Y, et al. PGC-1α controls skeletal stem cell fate and bone-fat balance in osteoporosis and skeletal aging by inducing TAZ. Cell Stem Cell. 2018;23:615–623. doi:10.1016/j.stem.2018.09.001
29. Li CJ, Cheng P, Liang MK, et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J Clin Invest. 2015;125:1509–1522. doi:10.1172/JCI77716
30. Xu C, Wang J, Zhu T, et al. Cross-talking between PPAR and WNT signaling and its regulation in mesenchymal stem cell differentiation. Curr Stem Cell Res Ther. 2016;11:247–254. doi:10.2174/1574888X10666150723145707
31. Wang H, Chen Q, Lee SH, Choi Y, Johnson FB, Pignolo RJ. Impairment of osteoblast differentiation due to proliferation-independent telomere dysfunction in mouse models of accelerated aging. Aging Cell. 2012;11:704–713. doi:10.1111/j.1474-9726.2012.00838.x
32. Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266–300. doi:10.1210/er.2009-0024
33. Liu Y, Wang C, Wang G, et al. Loureirin B suppresses RANKL-induced osteoclastogenesis and ovariectomized osteoporosis via attenuating NFATc1 and ROS activities. Theranostics. 2019;9:4648–4662. doi:10.7150/thno.35414
34. Callaway DA, Jiang JX. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J Bone Miner Metab. 2015;33:359–370. doi:10.1007/s00774-015-0656-4
35. Rharass T, Lucas S. High glucose level impairs human mature bone marrow adipocyte function through increased ROS production. Front Endocrinol. 2019;10:607. doi:10.3389/fendo.2019.00607
36. Zhang YB, Zhong ZM, Hou G, Jiang H, Chen JT. Involvement of oxidative stress in age-related bone loss. J Surg Res. 2011;169:e37–e42. doi:10.1016/j.jss.2011.02.033
37. Zeng JH, Zhong ZM, Li XD, et al. Advanced oxidation protein products accelerate bone deterioration in aged rats. Exp Gerontol. 2014;50:64–71. doi:10.1016/j.exger.2013.11.014
38. Khosla S, Monroe DG. Regulation of bone metabolism by sex steroids. Cold Spring Harb Perspect Med. 2018;8:a031211. doi:10.1101/cshperspect.a031211
39. Compston JE. Sex steroids and bone. Physiol Rev. 2001;81:419–447. doi:10.1152/physrev.2001.81.1.419
40. Lee K, Seo I, Choi MH, Jeong D. Roles of mitogen-activated protein kinases in osteoclast biology. Int J Mol Sci. 2018;19:3004. doi:10.3390/ijms19103004
41. Zhang Y, Hua F, Ding K, Chen H, Xu C, Ding W. Angiogenesis changes in ovariectomized rats with osteoporosis treated with estrogen replacement therapy. Biomed Res Int. 2019;2019:1283717. doi:10.1155/2019/1283717
42. Zhang Y, Gao Y, Cai L, et al. MicroRNA-221 is involved in the regulation of osteoporosis through regulates RUNX2 protein expression and osteoblast differentiation. Am J Transl Res. 2017;9:126–135.
43. Chen S, Yang L, Jie Q, et al. MicroRNA-125b suppresses the proliferation and osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. Mol Med Rep. 2014;9:1820–1826. doi:10.3892/mmr.2014.2024
44. Jia J, Zhou H, Zeng X, Feng S. Estrogen stimulates osteoprotegerin expression via the suppression of miR-145 expression in MG-63 cells. Mol Med Rep. 2017;15:1539–1546. doi:10.3892/mmr.2017.6168
45. Prata LG, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin Immunol. 2018;40:101275. doi:10.1016/j.smim.2019.04.003
46. Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci. 2019;76:1901–1918. doi:10.1007/s00018-019-03048-x
47. Onal M, Xiong J, Chen X, et al. Receptor activator of nuclear factor κB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem. 2012;287:29851–29860. doi:10.1074/jbc.M112.377945
48. Choi Y, Woo KM, Ko SH, et al. Osteoclastogenesis is enhanced by activated B cells but suppressed by activated CD8(+) T cells. Eur J Immunol. 2001;31:2179–2188. doi:10.1002/1521-4141(200107)31:7<2179:AID-IMMU2179>3.0.CO;2-X
49. Toraldo G, Roggia C, Qian WP, Pacifici R, Weitzmann MN. IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor kappa B ligand and tumor necrosis factor alpha from T cells. Proc Natl Acad Sci U S A. 2003;100:125–130. doi:10.1073/pnas.0136772100
50. Srivastava RK, Dar HY, Mishra PK. Immunoporosis: immunology of osteoporosis-role of T cells. Front Immunol. 2018;9:657. doi:10.3389/fimmu.2018.00657
51. Müller L, Di Benedetto S, Pawelec G. The immune system and its dysregulation with aging. Subcell Biochem. 2019;91:21–43.
52. Chen K, Yang YH, Jiang SD, Jiang LS. Decreased activity of osteocyte autophagy with aging may contribute to the bone loss in senile population. Histochem Cell Biol. 2014;142:285–295. doi:10.1007/s00418-014-1194-1
53. Pierrefite-Carle V, Santucci-Darmanin S, Breuil V, Camuzard O, Carle GF. Autophagy in bone: self-eating to stay in balance. Ageing Res Rev. 2015;24:206–217. doi:10.1016/j.arr.2015.08.004
54. Almeida M, Han L, Martin-Millan M, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282:27285–27297. doi:10.1074/jbc.M702810200
55. Abrigo J, Rivera JC, Aravena J, et al. High fat diet-induced skeletal muscle wasting is decreased by mesenchymal stem cells administration: implications on oxidative stress, ubiquitin proteasome pathway activation, and myonuclear apoptosis. Oxid Med Cell Longev. 2016;2016:9047821. doi:10.1155/2016/9047821
56. Song C, Song C, Tong F. Autophagy induction is a survival response against oxidative stress in bone marrow-derived mesenchymal stromal cells. Cytotherapy. 2014;16:1361–1370. doi:10.1016/j.jcyt.2014.04.006
57. Wan Y, Zhuo N, Li Y, Zhao W, Jiang D. Autophagy promotes osteogenic differentiation of human bone marrow mesenchymal stem cell derived from osteoporotic vertebrae. Biochem Biophys Res Commun. 2017;488:46–52. doi:10.1016/j.bbrc.2017.05.004
58. Camuzard O, Santucci-Darmanin S, Breuil V, et al. Sex-specific autophagy modulation in osteoblastic lineage: a critical function to counteract bone loss in female. Oncotarget. 2016;7:66416–66428. doi:10.18632/oncotarget.12013
59. Shen G, Ren H, Qiu T, et al. Implications of the interaction between miRNAs and autophagy in osteoporosis. Calcif Tissue Int. 2016;99:1–12. doi:10.1007/s00223-016-0122-x
60. Gallagher JC. Vitamin D and aging. Endocrinol Metab Clin North Am. 2013;42:319–332. doi:10.1016/j.ecl.2013.02.004
61. Sims NA, Ng KW. Implications of osteoblast-osteoclast interactions in the management of osteoporosis by antiresorptive agents denosumab and odanacatib. Curr Osteoporos Rep. 2014;12:98–106. doi:10.1007/s11914-014-0196-1
62. Deng Z, Lin Z, Zhong Q, et al. Interleukin 1 beta-induced chloride currents are important in osteoarthritis onset: an in vitro study. Acta Biochim Biophys Sin. 2021;53:400–409. doi:10.1093/abbs/gmab010
63. Liang Y, Duan L, Xiong J, et al. E2 regulates MMP-13 via targeting miR-140 in IL-1β-induced extracellular matrix degradation in human chondrocytes. Arthritis Res Ther. 2016;18:105. doi:10.1186/s13075-016-0997-y
64. Zhang H, Deng Z, Yang L, et al. The AQP-3 water channel is a pivotal modulator of glycerol-induced chloride channel activation in nasopharyngeal carcinoma cells. Int J Biochem Cell Biol. 2016;72:89–99. doi:10.1016/j.biocel.2016.01.009
65. Zhang N, Deng Z, Li W, et al. Expression of LRRC8A is elevated in the cytoplasm of osteosarcoma tissues: an immunohistochemical study with tissue microarrays. Exp Ther Med. 2021;21:71. doi:10.3892/etm.2020.9503
66. Sun W, Chi S, Li Y, et al. The mechanosensitive Piezo1 channel is required for bone formation. Elife. 2019;8:e47454. doi:10.7554/eLife.47454
67. Li X, Han L, Nookaew I, et al. Stimulation of Piezo1 by mechanical signals promotes bone anabolism. Elife. 2019;8:e49631. doi:10.7554/eLife.49631
68. Hendrickx G, Fischer V, Liedert A, et al. Piezo1 inactivation in chondrocytes impairs trabecular bone formation. J Bone Miner Res. 2021;36:369–384. doi:10.1002/jbmr.4198
69. Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat Commun. 2020;11:282. doi:10.1038/s41467-019-14146-6
70. Zhu J, Zhen G, An S, et al. Aberrant subchondral osteoblastic metabolism modifies NaV1.8 for osteoarthritis. Elife. 2020;9:e57656. doi:10.7554/eLife.57656
71. Jørgensen NR. Role of the purinergic P2X receptors in osteoclast pathophysiology. Curr Opin Pharmacol. 2019;47:97–101. doi:10.1016/j.coph.2019.02.013
72. Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol. 2010;72(1):95–121. doi:10.1146/annurev-physiol-021909-135811
73. Stalvey MS, Clines KL, Havasi V, et al. Osteoblast CFTR inactivation reduces differentiation and osteoprotegerin expression in a mouse model of cystic fibrosis-related bone disease. PLoS One. 2013;8:e80098. doi:10.1371/journal.pone.0080098
74. Du J, Wang Q, Hu F, et al. Effects of estradiol on voltage-gated potassium channels in mouse dorsal root ganglion neurons. J Membr Biol. 2014;247:541–548. doi:10.1007/s00232-014-9670-z
75. Boyce BF, Li J, Xing L, Yao Z. Bone remodeling and the role of TRAF3 in osteoclastic bone resorption. Front Immunol. 2018;9:2263. doi:10.3389/fimmu.2018.02263
76. Lu X, Ding Y, Niu Q, et al. ClC-3 chloride channel mediates the role of parathyroid hormone [1–34] on osteogenic differentiation of osteoblasts. PLoS One. 2017;12:e0176196. doi:10.1371/journal.pone.0176196
77. Deng Z, Li W, Xu J, et al. ClC-3 chloride channels are involved in estradiol regulation of bone formation by MC3T3-E1 osteoblasts. J Cell Biochem. 2018;10:8366–8375.
78. Deng Z, Peng S, Zheng Y, et al. Estradiol activates chloride channels via estrogen receptor-α in the cell membranes of osteoblasts. Am J Physiol Cell Physiol. 2017;313:C162–C172. doi:10.1152/ajpcell.00014.2017
79. Wang H, Mao Y, Zhang B, et al. Chloride channel ClC-3 promotion of osteogenic differentiation through Runx2. J Cell Biochem. 2010;111:49–58. doi:10.1002/jcb.22658
80. Tolar J, Teitelbaum SL, Orchard PJ. Osteopetrosis. N Engl J Med. 2004;351:2839–2849. doi:10.1056/NEJMra040952
81. Jentsch TJ, Pusch M. CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev. 2018;98:1493–1590. doi:10.1152/physrev.00047.2017
82. Lee J, Alqudaihi HM, Kang MS, et al. Effect of tamoxifen on the risk of osteoporosis and osteoporotic fracture in younger breast cancer survivors: a nationwide study. Front Oncol. 2020;10:366. doi:10.3389/fonc.2020.00366
83. Olevsky OM, Martino S. Randomized clinical trials of raloxifene: reducing the risk of osteoporosis and breast cancer in postmenopausal women. Menopause. 2008;15(4 Suppl):790–796. doi:10.1097/gme.0b013e31817e6683
84. Anagnostis P, Gkekas NK, Potoupnis M, Kenanidis E, Tsiridis E, Goulis DG. New therapeutic targets for osteoporosis. Maturitas. 2019;120:1–6. doi:10.1016/j.maturitas.2018.11.010
85. Capozzi A, Scambia G, Lello S. Calcium, vitamin D, vitamin K2, and magnesium supplementation and skeletal health. Maturitas. 2020;140:55–63. doi:10.1016/j.maturitas.2020.05.020
86. Zhou W, Liu Y, Guo X, Yang H, Xu Y, Geng D. Effects of zoledronic acid on bone mineral density around prostheses and bone metabolism markers after primary total hip arthroplasty in females with postmenopausal osteoporosis. Osteoporos Int. 2019;30:1581–1589. doi:10.1007/s00198-019-05005-7
87. Chen C, Alqwbani M, Zhao J, Yang R, Wang S, Pan X. Effects of teriparatide versus salmon calcitonin therapy for the treatment of osteoporosis in Asia: a meta-analysis of randomized controlled trials. Endocr Metab Immune Disord Drug Targets. 2021;21:932–942. doi:10.2174/1871530320999200817114817
88. Wu T, Sun J, Tan L, et al. Enhanced osteogenesis and therapy of osteoporosis using simvastatin loaded hybrid system. Bioact Mater. 2020;5:348–357. doi:10.1016/j.bioactmat.2020.03.004
89. Deeks ED. Denosumab: a review in postmenopausal osteoporosis. Drugs Aging. 2018;35:163–173. doi:10.1007/s40266-018-0525-7
90. Schafer MJ, Miller JD, LeBrasseur NK. Cellular senescence: implications for metabolic disease. Mol Cell Endocrinol. 2017;455:93–102. doi:10.1016/j.mce.2016.08.047
91. Pagnotti GM, Styner M, Uzer G, et al. Combating osteoporosis and obesity with exercise: leveraging cell mechanosensitivity. Nat Rev Endocrinol. 2019;15:339–355. doi:10.1038/s41574-019-0170-1
92. Vandenbroucke A, Luyten FP, Flamaing J, Gielen E. Pharmacological treatment of osteoporosis in the oldest old. Clin Interv Aging. 2017;12:1065–1077. doi:10.2147/CIA.S131023
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.