Back to Journals » International Journal of Nanomedicine » Volume 11
The influence of different long-circulating materials on the pharmacokinetics of liposomal vincristine sulfate
Authors Zhang J ![]() , Chen Y, Li X, Liang X
, Chen Y, Li X, Liang X ![]() , Luo X
, Luo X
Received 30 March 2016
Accepted for publication 18 June 2016
Published 26 August 2016 Volume 2016:11 Pages 4187—4197
DOI https://doi.org/10.2147/IJN.S109547
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Jing Zhang,1,* Yingchong Chen,1,* Xiang Li,1,2 Xinli Liang,1 Xiaojian Luo2
1Key Laboratory of Modern Preparation of TCM, Ministry of Education, 2State Key Laboratory of Innovative Drug and Efficient Energy-Saving Pharmaceutical Equipment, Jiangxi University of Traditional Chinese Medicine, Nanchang, People’s Republic of China
*These authors contributed equally to this work
Purpose: This study was designed to improve the in vivo pharmacokinetics of long-circulating vincristine sulfate (VS)-loaded liposomes; three different long-circulating materials, chitosan, poly(ethylene glycol)-1,2-distearoyl sn-glycero-3-phosphatidylethanolamine (PEG-DSPE), and poly(ethylene glycol)-poly-lactide-co-glycolide (PEG-PLGA), were evaluated at the same coating molar ratio with the commercial product Marqibo® (vincristine sulfate liposome injection [VSLI]).
Materials and methods: VS-loaded liposomes were prepared by a pH gradient method and were then coated with chitosan, PEG-DSPE, or PEG-PLGA. Physicochemical properties, including the morphology, particle size, zeta potential, encapsulation efficiency (EE%), pH, drug loading, and in vitro release, were determined. Preservation stability and pharmacokinetic studies were performed to compare the membrane-coated liposomes with either commercially available liposomes or the VS solution.
Results: The sphere-like morphology of the vesicles was confirmed by transmission electron microscope. Increased particle size, especially for the chitosan formulation, was observed after the coating process. However, the EE% was ~99.0% with drug loading at 2.0 mg/mL, which did not change after the coating process. The coating of long-circulation materials, except for chitosan, resulted in negatively charged and stable vesicles at physiological pH. The near-zero zeta potential exhibited by the PEG-DSPE formulation leads to a longer circulation lifetime and improved absorption for VS, when compared with the PEG-PLGA formulation. Compared with the commercial product, PEG was responsible for a higher plasma VS concentration and a longer half-life.
Conclusion: PEG-DSPE coating may be related to better absorption, based on the stability and a pharmacokinetic improvement in the blood circulation time.
Keywords: vincristine sulfate, long-circulating materials, zeta potential, stability, pharmacokinetics
Introduction
Cancer has become a leading cause of death, and it is reported that by 2020, 15 million new cases will be diagnosed every year.1 A primary issue for cancer treatment is achieving efficient targeting for therapeutic agents to reach their intended site in vivo. Nanotechnology is an advanced technology allowing for design of a safe and efficient drug delivery system that could pave the way to overcome the physical barriers to drug targeting. The enhanced permeation and retention effect, which makes use of the difference in capillary structure between normal and cancerous tissues, is responsible for the achievement of targeting delivery.2 Neoplastic tissues usually have “leaky” vasculature and limited lymphatic drainage, which will lead to enhanced permeation for nanoparticles to across the vascular endothelium and achieve improved accumulation in the tumor. It was reported that the concentration of drug in the vessel should be high enough for at least 6 hours in order for the vehicle to be transported to the targeted tissue over time.3,4 However, the major obstacle for prolonging the retention time of the drug to achieve effective targeting is the rapid opsonization by the reticuloendothelial system (RES).5 An “invisible” surface of long-circulating nanoparticles (NPs) that can avoid opsonic adsorption and recognition by phagocytic cells is related to the physicochemical properties of NPs, including size distribution, surface charge, and chemistry.6–9
For many therapeutic drugs, liposomes are reported to effectively prolong the circulation time and reduce toxicity, and they can also improve drug accumulation in the targeted tissue.10 However, their application is strongly limited by their rapid uptake by the RES.11 Various methods have been used to avoid RES uptake, such as the modification of the liposomal membrane with incorporation of poly(ethylene glycol) (PEG), which conjugates to various hydrophobic moieties in the liposomal membrane.12 For a series of PEG derivatives (MW 120–5,000 Da) used in hydrogenated soybean phosphatidylcholine (HSPC):cholesterol:poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) liposomes with a size of 100 nm, it was reported that PEG with a molecular weight of <750 Da promoted blood clearance and PEG with a molecular weight of >1,000 Da did not further increase the residence time of the vehicles in the blood.12–15 Maruyama et al reported that liposomes (200 nm diameter) with PEG1000 or PEG2000 led to higher blood levels compared to liposomes with PEG5000.16 The liposome composition, including the lipids, PEG, and PEG–lipids, is likely to influence the performance of colloidal particles in the blood and the tissue distribution.4,16–18
In addition to PEG, long-circulating liposomes could also be obtained by incorporating ganglioside GM1, phosphatidylinositol, polysaccharides, or polyvinyl alcohol into the surface.19–21 Currently, polysaccharides are assumed to have potential for developing a long-circulating system.22 Increased attention has been paid to chitosan because of its attractive biodegradability, biocompatibility, nontoxicity, and low cost.23 The water-soluble chitosan has a low molecular weight and exhibits excellent hydrophilicity without impairing the native biological properties. It was reported that a combination of PEG and water-soluble chitosan could strongly avoid macrophage uptake and prolong the in vivo half-life of liposomes.7 It was also reported that prolonged half-life of liposomes could also be achieved by the incorporation of cholesterol and neutral long-chain saturated phospholipids. Liposomes composed of sphingomyelin (SM) and cholesterol (55:45, molar ratio) showed improved longevity in blood when vincristine, vinorelbine, and ciprofloxacin were encapsulated, owing to a strong resistance to plasma lipoproteins.24–27 In fact, the commercially available vincristine sulfate liposome injection ([VSLI], Marqibo®) contains this SM/cholesterol formulation-encapsulated vincristine sulfate (VS) to overcome the pharmacokinetic and dosing limitations of nonliposomal VS.28,29 VSLI exhibited greater anticancer activity and a larger maximum tolerated dose than free VS. However, the VSLI Kit was designed to contain three vials, which include sodium phosphate injection, sphingomyelin/cholesterol liposome injection, and VinCRIStine sulfate injection. The complicated preparation steps of VSLI involve mixing the three vials.
To facilitate the protocol for using VS injection and at least maintain its clinical efficacy, liposomal vesicles encapsulated with VS and modified with long-circulation materials were designed and studied in this article.
Although there is a large amount of literature concerning long-circulation modified liposomes for cancer treatment, few studies have investigated the relationship between coating composition and performance. Gaining a better understanding about this relationship will facilitate improved design and formulation for targeted drug delivery. The purpose of this study is to develop VS liposomes using different pharmaceutically acceptable excipients and adjust the properties of the liposomes with blood residence time. We investigated the influence of PEG-DSPE and poly(ethylene glycol)-poly-lactide-co-glycolide (PEG-PLGA), or chitosan with low molecular weight for prolonged residence time in blood. The impact of different modifying materials on the liposomal physical stability and circulation longevity was explored for the obtained formulations.
Materials and methods
Materials
VS (purity >99.1%) was obtained from Guangzhou Hanfang Pharmaceutical Co., Ltd (Guangzhou, People’s Republic of China). The internal standard (IS) epirubicin (purity >99.0%) was obtained from Shanghai Tauto Biotechnology Co., Ltd (Shanghai, People’s Republic of China). HSPC (>99% phosphatidylcholine), cholesterol, and PEG2000-DSPE (>99%) were purchased from Shanghai Advanced Vehicle Technology Pharmaceutical, Ltd (Shanghai, People’s Republic of China). PEG-PLGA (MW 20,000) with a 50:50 molar ratio of lactic to glycolic acid moieties and 2 kDa molecular weight PEG was supplied by Jinan Daigang Biomaterial Co., Ltd (Jinan, People’s Republic of China). Chitosan (MW 100 kDa) was purchased from Zhejiang Golden Shell Pharmaceutical Co., Ltd (Zhejiang, People’s Republic of China). High-pressure liquid chromatography (HPLC)-purity grade methanol was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Distilled water was produced by a Milli-Q purification system (EMD Millipore, Billerica, MA, USA).
Animals
Male Sprague Dawley rats (200±20 g) were obtained from Hunan SJA Laboratory Animal Co., Ltd (Hunan, People’s Republic of China; SYXK 2011-0003). All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the National Research Council. The protocol was approved by the Institutional Animal Care and Use Committee of Jiangxi University of Traditional Chinese Medicine. Rats were given a commercial diet and water ad libitum. The animal room was well ventilated and had a regular 12:12-hour light–dark cycle throughout the experimental period.
Preparation of VS-loaded liposomal vesicles
The preparation of VS-loaded liposomes was described previously.10,30 Briefly, lipid films (PEG-DSPE:HSPC:cholesterol =1:22:10, molar ratio) were prepared by drying the lipid solution (3%, w/v) in chloroform under a vacuum. These lipid films were hydrated with 0.3 mM citrate (pH 4.0) to obtain multilamellar vesicles that were sonicated at 300 W for 8 minutes. The obtained suspension was then extruded through two stacked 0.45 μm and 0.22 μm filters. The loading of VS into these vesicles was conducted using a pH gradient.31 A solution of VS (4 g/L) and sodium carbonate (500 mM, pH 11.0) was added into the liposomes. VS-loaded liposomes modified with PEG-DSPE (PEG-DSPE formulation) were obtained after 30 minutes at 60°C. To achieve a uniform size distribution of vesicles, the suspensions were extruded by a Nano DeBEE High Pressure Homogenizer (BEE International Inc., South Easton, MA, USA). The PEG-PLGA formulation was prepared by a similar process, in which PEG-PLGA was added to the lipids with the same molar ratio as PEG-DSPE.
As for the chitosan formulation, chitosan was dissolved in pH 6.7 phosphate buffer (0.1%, w/v) and was added dropwise to the HSPC formulation (1:1, v/v) without the addition of PEG-PLGA or PEG-DSPE under magnetic stirring. The mixture was then incubated at 10°C for 60 minutes.32
Morphology analysis
The morphological examination of liposomes was performed by a JEM-1200 EX electron microscope (JEOL, Tokyo, Japan) by conventional negative staining methods using 0.3% phosphotungstic acid buffer (pH =6.0) as a staining agent.33
Particle size and zeta potential analysis
The size and zeta potentials of the liposomes were measured by dynamic light scattering and electrophoretic light scattering, respectively, both using a PSA NANO2590 apparatus (Malvern Instruments, Malvern, UK) at 25°C.23
Determination of encapsulation efficiency
Encapsulation efficiency (EE%) was determined using the ultrafiltration technique for separating the nonentrapped drug from liposomes.33 For this, a 500 μL drug-loaded liposomal dispersion was placed in an ultrafiltration tube (Nanosep MF; Pall Corporation, Port Washington, NY, USA), which was fitted with a filter membrane (MW cutoff: 10 kDa). The free drug in the underlying solution was collected by centrifugation at 8,000 rpm for 15 minutes (3–18 K high-speed refrigerated centrifuge; Sigma-Aldrich Co.), and the drug content in the ultrafiltrate (cfree) was determined by HPLC on a C18 Agilent column (Agilent Technologies, Santa Clara, CA, USA; 250×4.6 mm2, 5 μm) at 297 nm. The mobile phase was composed of a 4.5% ethanediamine solution (pH 7.0)–methanol (1:7, v:v), and the flow rate was 1.0 mL/min. The column temperature was maintained at 25°C, and the injection volume was 20 μL. Then, 0.5 mL of liposomal suspension was diluted with 2.0 mL of a mixture (acetone:chloroform =2:1, v:v) to determine the total drug concentration (ctotal) by HPLC. The EE% was calculated using the following equation:
|
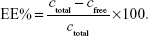
Stability
The physical stability of the products at 4°C was assessed by evaluation of the suspensions at predetermined time points.
In vitro release
In vitro release analysis was performed by a modified US Pharmacopoeia apparatus 4 with flow-through cells having a 12 mm diameter (Sotax CE7 smart; Sotax, Horsham, PA, USA) packed with glass beads (1 mm) in a closed system mode at 37°C.34 The release of VS from different liposomal preparations was assessed in both saline and 50% human plasma in saline. The HSPC formulation, PEG-DSPE formulation, PEG-PLGA formulation, and chitosan formulation were all evaluated. The preparations were dispersed in the cells using 100 mL of dissolution medium at a flow rate of 4 mL/min. One milliliter samples were withdrawn and replenished with an equal volume of blank fresh media. For the release evaluation in saline, the samples were then directly analyzed by HPLC. For the release evaluation in 50% plasma, due to the high plasma protein-binding rate of VS, the samples were treated as follows. Two-hundred microliters of the samples were added with 10 μL 0.1 M NaOH solution to alkalize the sample, then 2 mL of chloroform was added, and the sample was vortexed for 5 minutes. Afterward, the mixture was centrifuged for 10 minutes at 5,000 rpm, and the chloroform phase was separated before being dried under N2. The residue was resolved in the mobile phase. After vortexing for 2 minutes, the sample solution was ready for HPLC analysis. The media replenishment was taken into account in the calculation of the cumulative percent release. All measurements were conducted in triplicate, and the mean values and standard deviations are reported.
The values of f1 and f2 were obtained by a mathematical comparison and calculated according to the method previously reported.35 Values for f1 (0–15) and f2 (50–100) verify the equivalence between the two tested dissolution curves.36 VS release kinetics within 24 hours were then analyzed by four mathematical models.37–39
Pharmacokinetics studies
Thirty-six healthy Sprague Dawley rats (200±20 g) were randomly divided into six groups and were fasted overnight prior to the experiment. The rats in the control groups were injected with native VS solution and VSLI (1.2 mg of VS/kg, 0.6 mL/kg) intravenously into the tail vein.40 The other three groups (the HSPC formulation, PEG-DSPE formulation, and PEG-PLGA formulation) were injected into the tail vein with a dose of 0.6 mL/kg and a concentration of 2.0 mg/mL. For the 1.0 mg/mL chitosan formulation, the treated rats were injected at 1.2 mL/kg. After intravenous injection at designated time points (0.0167 hour, 0.05 hour, 0.083 hour, 0.25 hour, 0.5 hour, 1 hour, 2 hours, 4 hours, 8 hours, 12 hours, and 24 hours), 200 μL blood samples were withdrawn from the retro-orbital plexus. The blood samples were centrifuged at 2,300× g for 5 minutes, and 100 μL of the separated plasma was kept at −80°C for analysis. Twenty microliters of the IS working solution was added to the sample before the addition of methanol (300 μL) to precipitate the protein. The mixture was centrifuged at 16,000 rpm (HC-3018R High Speed Refrigerated Centrifuge; Anhui USTC Zonkia Scientific Instruments Co., Ltd, People’s Republic of China) for 10 minutes to dissolve the drugs in the organic solvent. The obtained supernatant was subjected to HPLC/mass spectrometry analysis for the detection of VS by an LCMS-8040 Liquid Chromatograph triple quadrupole mass spectrometer equipped with an electronic spray ion source in the positive mode (Shimadzu Corporation, Kyoto, Japan).
Chromatographic conditions
The chromatographic separation was determined on a C18 Venusil MP column (150×4.6 mm2, 3 μm) at 297 nm. Gradient elution was used using solvent A (1% formic acid in water) and solvent B (acetonitrile) at 40°C. The gradient program used was as follows: initial phase from 0 minute to 0.2 minute, a linear change from A-B (90:10, v:v) to A-B (75:25, v:v); from 0.2 minute to 1 minute, a linear change to A-B (60:40, v:v); from 1 minute to 2.5 minutes, a linear change to A-B (10:90, v:v); from 2.5 minutes to 2.51 minutes, linear change to A-B (90:10, v:v). The flow rate was maintained at 0.6 mL/min.
Mass spectrometry conditions
The mass spectrometer was operated in multiple reaction monitoring mode. The compounds were detected as doubly charged ions, and the MRM transitions of VS and IS were m/z 825.2 → 765.4 and m/z 544.1 → 397.1, respectively. The ionization source conditions included a capillary voltage of 4 kV, a cone voltage of 50 V, and a desolvation temperature of 350°C. The optimized collision energy was 25 V for both VS and IS. The cone and desolvation gas flow rates were 50 L/h and 500 L/h, respectively. Argon was used as collision gas at a pressure of ~35 psi.
The area under the concentration-time curve (AUC0–t) from zero to the last time point, the distribution phase half-life (T1/2α), and the elimination phase half-life (T1/2β) of the drug were all obtained.
Statistical analysis
All data were expressed as the mean ± standard deviation. Statistical analysis was conducted using a one-way analysis of variance with SPSS software (SPSS Inc., Chicago, IL, USA). Probability values 0.05 were considered significant.
Results and discussion
Characterization of liposomes
The features of VS-loaded liposomes with different modifying materials are summarized in Figure 1 and Table 1. In the morphological characterization, spherical-shaped particles were found. The inner particles were observed in a lighter region surrounded by a dark coating, such as chitosan and PEG. Similar observations were reported by Prego et al41 and Zahr et al.42
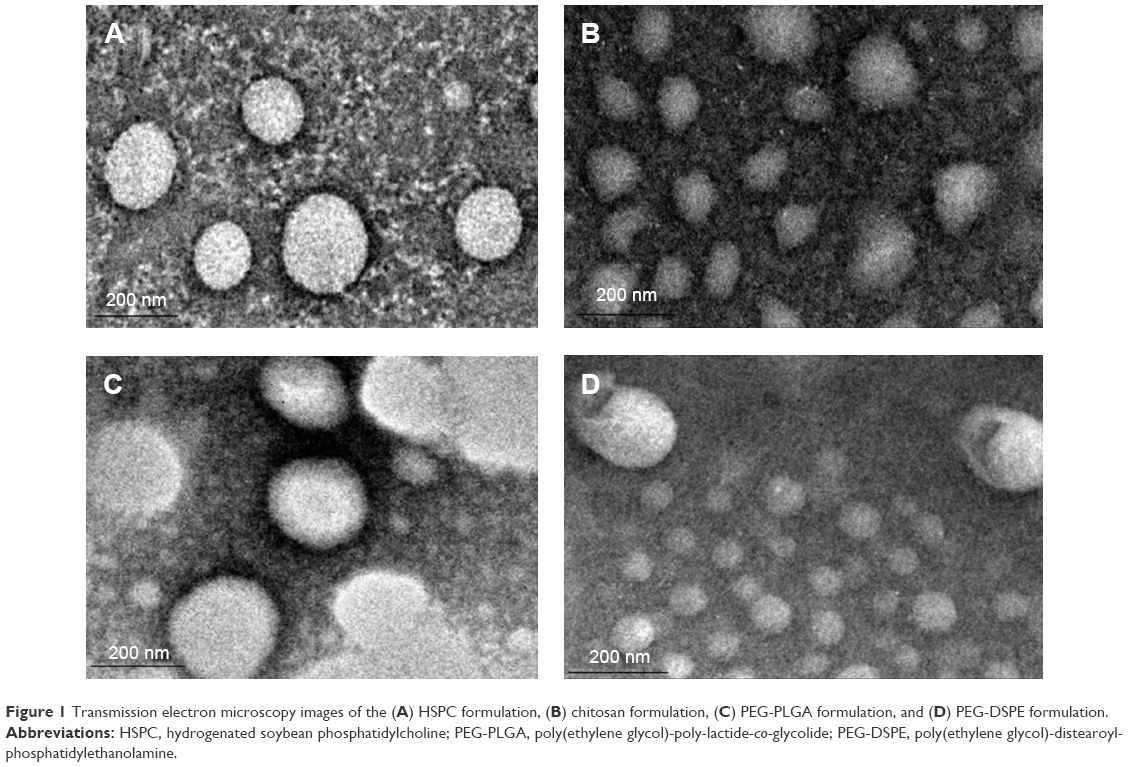 | Figure 1 Transmission electron microscopy images of the (A) HSPC formulation, (B) chitosan formulation, (C) PEG-PLGA formulation, and (D) PEG-DSPE formulation. |
 | Table 1 Characterization of VS-loaded liposomes with different modifying materials |
For all liposomes, the size was uniformly distributed so that the polydispersity index was <0.2. The increase in particle size could be explained by the presence of long-circulation materials surrounding the vesicles, which led to the appearance of larger vesicles. Higher surface negativity of the VS-loaded liposomes is attributed to the carboxyl acid groups of the PEG-capped PLGA chains and the use of HSPC in the lipid composition. The zeta-potential decreased to ~−4 mV when the liposomes were modified by PEG-DSPE. This negative charge rises from the incorporation of PEG-DSPE, which is considered to also be influenced by the shielding effect of the hydrophilic PEG moiety.43
The size and zeta potential increased from 110.5 nm to 140.9 nm and from −1.4 mV to 18.4 mV, respectively, for the uncoated HSPC liposomes (the HSPC formulation) compared to chitosan-coated liposomes (the chitosan formulation). The EE% was determined by HPLC. As shown in Table 1, the addition of chitosan and PEG-conjugated polymers did not influence the entrapment efficiency and drug loading of the liposomes.
Stability
For the conventional liposomal suspension of the HSPC formulation, in the stability study, the EE% of VS was constant in the first 5 months, but decreased by 11.2% in the sixth month, which also resulted in an increased particle size and decreased drug loading. The EE% of the PEG-DSPE formulation and PEG-PLGA formulation decreased <1% by the sixth month. Moreover, the EE% for the chitosan formulation dropped within 60 days with significantly increased particle size. No significant changes in the vesicle mean size or drug concentration were found for the PEG-PLGA and PEG-DSPE formulations during the long-term stability study (Table 2), and the vesicles had a semitransparent, homogeneous, and blue-opalescent appearance. Hence, the chitosan formulation should be considered unstable. The decreased stability of the chitosan formulation compared to the other three preparations is assumed to be related to the zeta potential difference between them. Compared to the other preparations, the chitosan formulation is positively charged, while the others are negatively charged. The negatively charged liposomes exhibited a slower leakage behavior. This may be attributed to the positive charge of vincristine, which might bind strongly with the negative charges of the liposomal membrane, thereby improving the stability of the liposomes.44 Thus, the formulation charge appeared to affect the leakage behavior and stability of vincristine in liposomes.
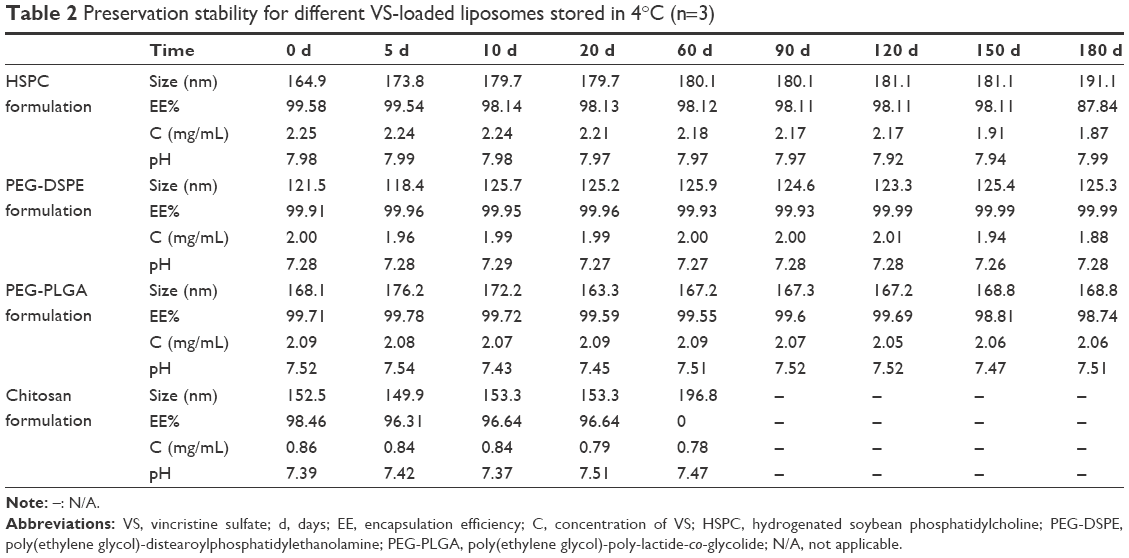 | Table 2 Preservation stability for different VS-loaded liposomes stored in 4°C (n=3) |
In vitro release
The cumulative release of VS (%) in saline versus the time plots of different preparations is shown in Figure 2.
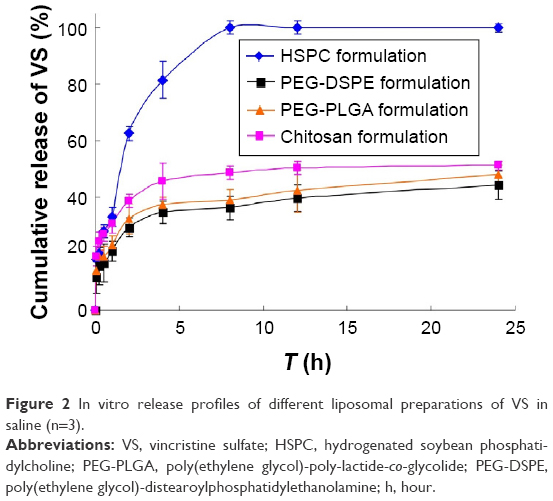 | Figure 2 In vitro release profiles of different liposomal preparations of VS in saline (n=3). |
The values of f1 and f2 for formulations were calculated by the mean values of the cumulative release of VS (%) at every time point. It showed that all preparations for liposomes showed a faster release at the beginning and a slower release thereafter, especially for the coated liposomes when compared to the HSPC formulation. We observed that the dissolution profile of conventional liposomes differed from all of the other preparations (Table 3). PEG-DSPE and chitosan formulations can be considered non-equivalent to each other. The formulation of PEG-DSPE is similar to that of PEG-PLGA. Furthermore, we also performed the linearization of VS dissolution profiles by using four mathematical models, which will help us to better understand the differences between all of the tested preparations. Table 4 shows the related parameters and coefficients of these models. Considering the R2 values, the calculated first-order model failed to fit the PEG-DSPE, PEG-PLGA, and chitosan formulations; however, it could fit the HSPC formulation (R=0.996), which indicates that the release profiles of VS from these liposomal preparations are different from that observed for the HSPC formulation. The Higuchi model was found to fit the PEG-DSPE, PEG-PLGA, and chitosan formulations, but not the HSPC formulation. The release behavior of VS in the Higuchi model may be square root time dependent, which is often exhibited for some types of formulations, such as plastic, wax matrices, and matrix tablets.35,45,46 The zero-order model did not seem to adequately fit each preparation. The Weibull model was able to fit the dissolution data of the PEG-DSPE and Chitosan formulation.45 The cumulative release of VS (%) in plasma versus the time plots of different preparations is shown in Figure 3. The release behavior of all preparations in plasma proved to be faster to different degrees compared with those in saline, especially for the HSPC formulation and chitosan formulation. This observation was likely be related to the interaction between high-density lipoprotein and the lipids, which would sabotage the stability of the vesicles and accelerate the leakage of the drug. However, the release behaviors of the PEG-DSPE formulation are similar to that of the PEG-PLGA formulation according to Tables 5 and 6.
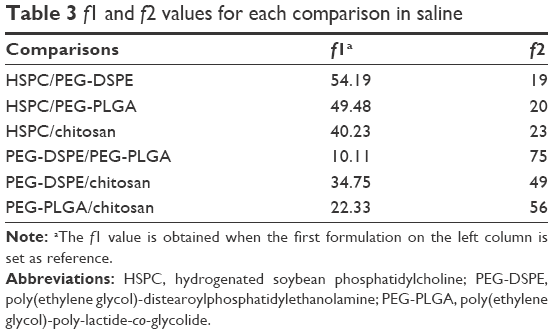 | Table 3 f1 and f2 values for each comparison in saline |
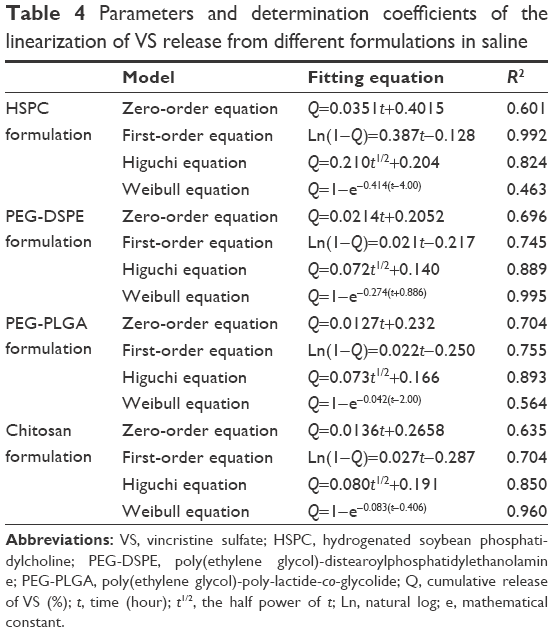 | Table 4 Parameters and determination coefficients of the linearization of VS release from different formulations in saline |
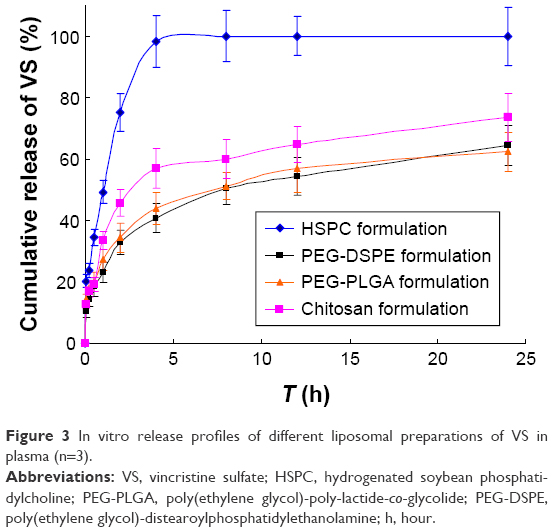 | Figure 3 In vitro release profiles of different liposomal preparations of VS in plasma (n=3). |
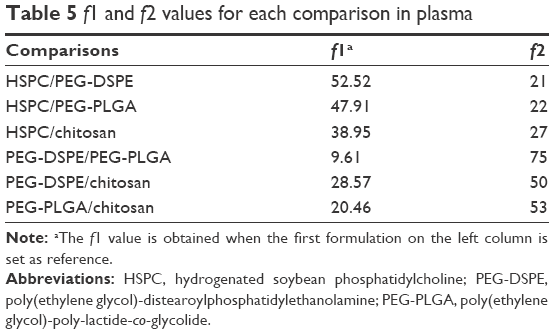 | Table 5 f1 and f2 values for each comparison in plasma |
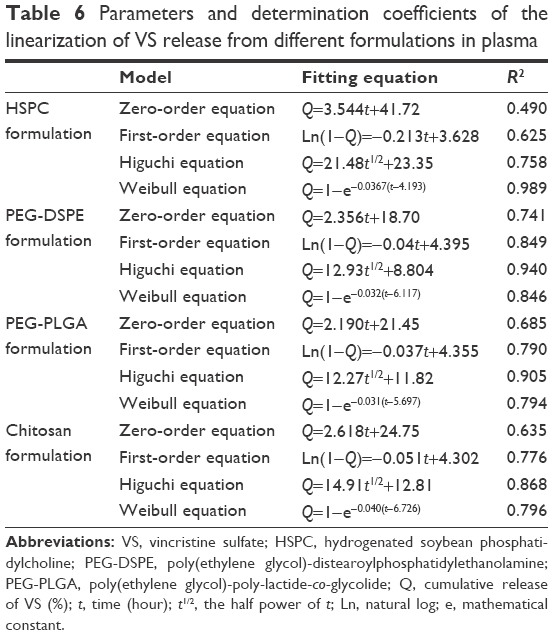 | Table 6 Parameters and determination coefficients of the linearization of VS release from different formulations in plasma |
Pharmacokinetics of VS preparations
To fully describe how the body handles the foreign liposomes, the in vivo blood clearance of the formulated liposomes were monitored and compared. VS was reported to be membrane permeable and the lipid composition would alter its leakage profiles.10,47 Therefore, the effect of lipid composition on the pharmacokinetics of VS was evaluated. The blood concentration-time profiles after the iv administration of different formulations in rats are shown in Figure 4. PKslover software was used for various compartmental analyses of all curves, and the results are listed in Table 7. The profile for VS solution is a three-compartmental model. The HSPC, PEG-DSPE, PEG-PLGA formulations, and the commercially available liposomes are two-compartmental models, and the chitosan formulation is a one-compartmental model.
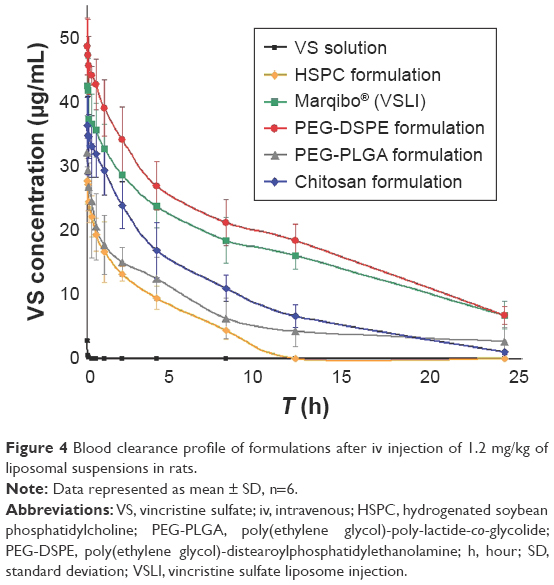 | Figure 4 Blood clearance profile of formulations after iv injection of 1.2 mg/kg of liposomal suspensions in rats. |
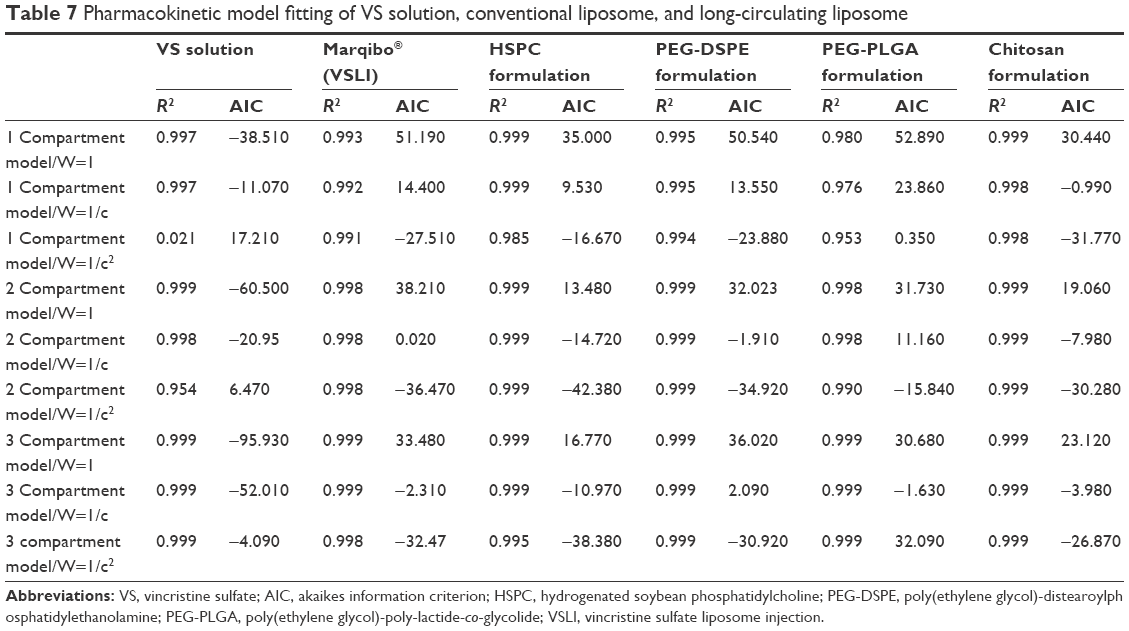 | Table 7 Pharmacokinetic model fitting of VS solution, conventional liposome, and long-circulating liposome |
The relevant pharmacokinetic parameters, including the AUC, T1/2α, T1/2β, and T1/2γ were determined using the corresponding compartmental analysis and are listed in Table 8. It could be observed that the VS solution, HSPC, and commercial formulations exhibited a rapid elimination from the bloodstream and the remaining doses at 4 hours post-injection were 0.65% and 34.2% and 55.9%, respectively. After the liposomes were coated with PEG-DSPE or PEG-PLGA, a significant prolongation of liposomes in the blood occurred. The AUC0→∞ calculated for the PEG-DSPE formulation was 2.3-fold as high as that of the PEG-PLGA and chitosan formulations. Every half-life (T1/2) for the prepared VS preparations was at least 1.4-fold as long as that of the marketed VS liposomes. In contrast, the marketed preparation did not show the same long-circulating property as that of the PEG-DSPE and PEG-PLGA formulation, disappearing from the blood with a T1/2 of 14.9 hours and 18.2 hours, respectively. Furthermore, most of the preparations showed a biphasic clearance profile. Especially for PEG-DSPE, an obvious retardation in the clearance from the blood happened within the initial 12 hours after iv administration.
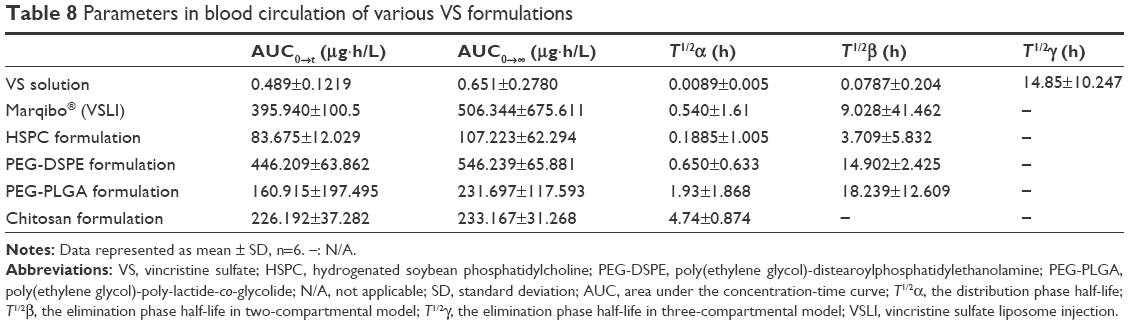 | Table 8 Parameters in blood circulation of various VS formulations |
The physical properties of these liposomes may be contributing to the changes in circulation lifetimes of VS or its liposomes. There were no clear differences in drug loading or pH for all liposomes used in this study. However, the aforementioned zeta potential analysis of conventional liposomes or liposomes containing PEG-DSPE was slightly, but significantly, different. Although negatively charged liposomes tend to be easily removed after administration,48 the addition of 3 mol% of PEG-DSPE to conventional liposomes increased both the AUC and the circulation lifetimes of VS. This proved that besides a near-zero zeta potential, the composition of lipids can also induce slow clearance from the circulation for liposomal preparation. That result was assumed to be caused by the long-circulation effect of 2,000 Da PEG. The same trend happened for the PEG-PLGA formulations. This result can be explained by the fact that the existence of PEG-DSPE could increase the hydrodynamic radius because of PEG involvement. This change can shield and effectively reduce the negative charge to a near-zero zeta potential.49 PEG-coated nanoparticles were found to be useful for shielding the nanoparticles from the RES because of steric repulsion.50,51 A hydrated outer shell could be constructed by PEG, which could also shield the nanoparticles from being recognized and taken by the RES. All of the effects of PEG result in an extended drug half-life and improved tissue distribution.52,53
It was reported that the pharmacokinetic profiles of liposomal vesicles with modified PEG-hydrophobic lipid are influenced mostly by the PEG moiety. It is usually unaffected by the conjugated lipids or the surface charge induced by the involvement of PEG-hydrophobic lipid.10 However, there were clear differences in the zeta potential between the PEG-DSPE and PEG-PLGA formulations. Under such circumstances, the in vivo drug concentration–time curves for these two preparations were significantly different, with 2.3-fold different AUC values. These results were unusual, and we assumed that there was a complicated reason for this observation. There are two possible reasons for this. First, the difference in the inner membrane permeability reported by the incorporation of the membrane modifying material PEG. Second, the uniformity of PEG-hydrophobic lipids could also play a role. Further studies are necessary to understand this phenomenon more completely.
Chitosan has been used as a pharmaceutical excipient for drug delivery because of its favorable biological properties.54 Similar to PEG, chitosan can also attenuate the phagocytic effects and enhance the longevity of NPs. When compared with the PEG-DSPE formulation, the relatively larger particle size and extremely positive charge of the chitosan formulation was supposed to lead to a reduced long-term circulation ability for the in vivo pharmacokinetics profiles, which may relate to RES recognition.55,56
Although the obtained results might result from various other more complicated mechanisms, the pharmacokinetic work showed that the PEG-DSPE formulation had a longer term blood circulation when it was compared to the PEG-PLGA, chitosan, and commercial formulations. Further studies are being performed to investigate these factors.
Conclusion
Through a series of comparative studies with the chitosan, PEG-PLGA formulation, and VSLI, it was demonstrated that 2 kDa PEG conjugated to DSPE very effectively increases the circulation and absorption of VS. Especially, when compared with VSLI, the PEG-DSPE formulation exhibited better clearance and higher AUC values, which demonstrates its potential to serve as a promising drug delivery system for VS with efficient targeted performance.
Acknowledgments
Thanks to American Journal Experts for editing (Certificate Verification Key: 9883-3148-AE38-E7E5-4C98). The study was supported by the Natural Science Fund of Jiangxi Province (20123BBG70181 and 20151BAB215040), the Scientific Research Foundation of Traditional Chinese Medicine of Jiangxi Provincial Health Department (2014A018), National Natural Science Fund of China (81202927 and 81560575), and the Open fund of collaborative innovation center of Jiangxi University of Traditional Chinese Medicine (JXXT201403013).
Disclosure
The authors report no conflicts of interest in this work.
References
Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancertherapy. Adv Drug Deliv Rev. 2004;56(11):1649–1659. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Greish K, Fang J, Inutsuka T, Nagamitsu A, Maeda H. Macromolecular therapeutics: advantages and prospects with special emphasis on solid tumour targeting. Clin Pharmacokinet. 2003;42(13):1089–1105. | ||
Rossi J, Giasson S, Khalid MN, Delmas P, Allen C, Leroux JC. Long-circulating poly(ethylene glycol)-coated emulsions to target solid tumors. Eur J Pharm Biopharm. 2007;67(2):329–338. | ||
Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263(5153):1600–1603. | ||
Owens DE 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. | ||
Sheng Y, Liu CS, Yuan Y, et al. Long-circulating polymeric nanoparticles bearing a combinatorial coating of PEG and water-soluble chitosan. Biomaterials. 2009;30(12):2340–2348. | ||
Stolnik S, Illum L, Davis SS. Long circulating microparticulate drug carriers. Adv Drug Deliv Rev. 1995;16(2–3):195–214. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Webb MS, Saxon D, Wong FMP, et al. Comparison of different hydrophobic anchors conjugated to poly(ethylene glycol): effects on the pharmacokinetics of liposomal vincristine. Biochim Biophys Acta. 1998;1372(2):272–282. | ||
Senior JH. Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst. 1987;3(2):123–193. | ||
Allen TM, Hansen C, Martin F, Redemann C, Yau-Young A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim Biophys Acta. 1991;1066(1):29–36. | ||
Charrois GJ, Allen TM. Rate of biodistribution of STEALTH liposomes to tumor and skin: influence of liposome diameter and implications for toxicity and therapeutic activity. Biochim Biophys Acta. 2003;1609(1):102–108. | ||
Maruyama K, Yuda T, Okamoto A, Ishikura C, Kojima S, Iwatsuru M. Effect of molecular weight in amphipathic polyethyleneglycol on prolonging the circulation time of large unilamellar liposomes. Chem Pharm Bull (Tokoyo). 1991;39(6):1620–1622. | ||
Woodle MC, Matthay KK, Newman MS, et al. Versatility in lipid composition showing prolonged circulation with sterically stabilized liposomes. Biochim Biophys Acta. 1992;1105(2):193–200. | ||
Maruyama K, Yuda AO, Kojima S, Suginaka A, Iwatsuru M. Prolonged circulation time in vivo of large unilamellar liposomes composed of distearoylphosphatidylcholine and cholesterol containing amphipathic poly(ethylene glycol). Biochim Biophys Acta. 1992;1128(1):44–49. | ||
Allen TM, Hansen CB, Lopes de Menezes DE. Pharmacokinetics of long-circulating liposomes. Adv Drug Deliv Rev. 1995;16(1–2):267–284. | ||
Allen C, Dos Santos N, Gallagher R, et al. Controlling the physical behavior and biological performance of liposome formulations through use of surface grafted poly(ethylene glycol). Biosci Rep. 2002;22(2):225–250. | ||
Wang XY, Ishida T, Ichihara M, Kiwada H. Influence of the physicochemical properties of liposomes on the accelerated blood clearance phenomenon in rats. J Control Release. 2005;104(1):91–102. | ||
Shehata T, Ogawara K, Higaki K, Kimura T. Prolongation of residence time of liposome by surface-modification with mixture of hydrophilic polymers. Int J Pharm. 2008;359(1–2):272–279. | ||
Zant-Przeworska E, Stasiuk M, Gubernator J, Kozubek A. Resorcinolic lipids improve the properties of sphingomyelin-cholesterol liposomes. Chem Phys Lipids. 2010;163(7):648–654. | ||
Sihorkar V, Vyas SP. Potential of polysaccharide anchored liposomes in drug delivery, targeting and immunization. J Pharm Pharm Sci. 2001;4(2):138–158. | ||
Zhang J, Wang SL. Topical use of coenzyme Q10-loaded liposomes coated with trimethyl chitosan: tolerance, precorneal retention and anti-cataract effect. Int J Pharm. 2009;372(1–2):66–75. | ||
Ellens E, Morselt HWM, Scherphof GL. In vivo fate of large unilamellar sphingomyelin-cholesterol liposomes after intraperitoneal and intravenous injection into rats. Biochim Biophys Acta. 1981;674(1):10–18. | ||
Webb MS, Harasym TO, Masin D, Bally MB, Mayer LD. Sphingomyelin-cholesterol liposomes significantly enhance the pharmacokinetic and therapeutic properties of vincristine in murine and human tumour models. Br J Cancer. 1995;72(4):896–904. | ||
Semple SC, Leone R, Wang J, et al. Optimization and characterization of a sphingomyelin/cholesterol liposome formulation of vinorelbine with promising antitumor activity. J Pharm Sci. 2005;94(5):1024–1038. | ||
Fenske DB, Wong KF, Maurer E, et al. Ionophore-mediated uptake of ciprofloxacin and vincristine into large unilamellar vesicles exhibiting transmembrane ion gradients. Biochim Biophys Acta. 1998;1414(1–2):188–204. | ||
Silverman JA, Deitcher SR. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71(3):555–564. | ||
Deitcher OR, Glaspy J, Gonzalez R, et al. High-dose vincristine sulfate liposome injection (Marqibo) is not associated with clinically meaningful hematologic toxicity. Clin Lymphoma Myeloma Leuk. 2014;14(3):197–202. | ||
Zhigaltsev IV, Maurer N, Akhong QF, et al. Liposome-encapsulated vincristine, vinblastine and vinorelbine: a comparative study of drug loading and retention. J Control Release. 2005;104(1):103–111. | ||
Mayer LD, Nayar R, Thies RL, Boman NL, Cullis PR, Bally MB. Identification of vesicle properties that enhance the antitumour activity of liposomal vincristine against murine L1210 leukemia. Cancer Chemother Pharmacol. 1993;33(1):17–24. | ||
Zhang J, Liang XL, Li X, et al. Ocular delivery of cyanidin-3-glycoside in liposomes and its prevention of selenite-induced oxidative stress. Drug Dev Ind Pharm. 2016;42(4):546–553. | ||
Zhang J, Han XZ, Li X, et al. Core-shell hybrid liposomal vesicles loaded with panax notoginsenoside: preparation, characterization and protective effects on global cerebral ischemia/reperfusion injury and acute myocardial ischemia in rats. Int J Nanomedicine. 2012;7:4299–4310. | ||
Zolnik BS, Burgess DJ. Evaluation of in vivo-in vitro release of dexamethasone from PLGA microspheres. J Control Release. 2008;127(2):137–145. | ||
Koester LS, Ortega GG, Mayorga P, Bassani VL. Mathematical evaluation of in vitro release profiles of hydroxypropylmethylcellulose matrix tablets containing carbamazepine associated to beta-cyclodextrin. Eur J Pharm Biopharm. 2004;58(1):177–179. | ||
Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms. Rockville, MD: FDA; 1997. | ||
Langenbucher F. Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol. 1972;24(12):979–981. | ||
Gibaldi M, Feldman S. Establishment of sink conditions of dissolution rate determinations. J Pharm Sci. 1967;56(10):1238–1242. | ||
Wagner JG. Interpretation of percent dissolved-time plots derived from in vitro testing of conventional tablets and capsules. J Pharm Sci. 1969;58(10):1253–1257. | ||
Chen JN, He HJ, Li SS, Shen Q. An HPLC method for the pharmacokinetic study of vincristine sulfate-loaded PLGA–PEG nanoparticle formulations after injection to rats. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(21):1967–1972. | ||
Prego C, Torres D, Fernandez-Megia E, Novoa-Carballal R, Quinoa E, Alonso MJ. Chitosan–PEG nanocapsules as new carriers for oral peptide delivery. Effect of chitosan pegylation degree. J Control Release. 2006;111(3):299–308. | ||
Zahr AS, Davis CA, Pishko MV. Macrophage uptake of core-shell nanoparticles surface modified with poly(ethylene glycol). Langmuir. 2006;22(19):8178–8185. | ||
Nakamura K, Kamashita K, Itoh Y, Yoshino K, Nozawa S, Kasukawa H. Comparative studies of polyethylene glycol-modified liposomes prepared using different PEG-modification methods. Biochim Biophys Acta. 2012;1818(11):2801–2807. | ||
Manosroi A, Podjanasoonthon K, Manosroi J. Stability and release of topical tranexamic acid liposome formulations. J Cosmet Sci. 2002;53(6):375–386. | ||
Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. | ||
Desai SJ, Singh P, Simonelli AP, Higuchi WI. Investigation of factors influencing release of solid drug dispersed in inert matrices. II. Quantitation of procedures. J Pharm Sci. 1996;55(11):1224–1229. | ||
Boman NL, Cullis PR, Mayer LD, Bally MB, Webb MS. Liposomal vincristine: the central role of drug retention in defining therapeutically optimized anticancer formulations. In: Woodle MC, Storm G, editors. Long Circulating Liposomes: Old Drugs, New Therapeutics. Georgetown, TX: Landes Bioscience; 1997:29–49. | ||
Chonn A, Semple SC, Cullis PR. Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J Biol Chem. 1992;267(26):18759–18765. | ||
Woodle MC, Collins LR, Sponsler E, Kossovsky N, Papahadjopoulos D, Martin FJ. Sterically stabilized liposomes. Reduction in electrophoretic mobility but not electrostatic surface potential. Biophys J. 1992;61(4):902–910. | ||
Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Deliv Rev. 2003;55(3):403–419. | ||
Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials. 2006;27(24):4356–4373. | ||
Hu Y, Jiang X, Ding Y, et al. Preparation and drug release behaviors of nimodipine-loaded poly(caprolactone)-poly (ethylene oxide)-polylactide amphiphilic copolymer nanoparticles. Biomaterials. 2003;24(13):2395–2404. | ||
Parveen S, Sahoo SK. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur J Pharmacol. 2011;670(2–3):372–383. | ||
Parveen S, Mitra M, Krishnakumar S, Sahoo SK. Enhanced antiproliferative activity of carboplatin-loaded chitosan-alginate nanoparticles in a retinoblastoma cell line. Acta Biomater. 2010;6(8):3120–3131. | ||
Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42(6):463–478. | ||
Rigotti A, Acton SL, Krieger M. The class B scavenger receptors SR-BI and CD36 are receptors for anionic phospholipids. J Biol Chem. 1995;270(27):16221–16224. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.