Back to Journals » Cancer Management and Research » Volume 12
The Dynamic Process and Its Dual Effects on Tumors of Therapy-Induced Senescence
Authors Liao C, Xiao Y ![]() , Liu L
, Liu L ![]()
Received 2 October 2020
Accepted for publication 8 December 2020
Published 31 December 2020 Volume 2020:12 Pages 13553—13566
DOI https://doi.org/10.2147/CMAR.S285083
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Chenxi Liao,1,* Yin Xiao,2,* Lingbo Liu1
1Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, People’s Republic of China; 2Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lingbo Liu
Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
Tel +8613387536308
Fax +86278572257
Email [email protected]
Yin Xiao
Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
Tel +8613871482376
Fax +862785726006
Email [email protected]
Abstract: Cellular senescence is traditionally considered as stable cell cycle arrest state with other phenotypic alterations including the production of an array of cytokines and growth factors. Cancer cells undergo senescence in response to chemotherapeutic agents, radiotherapy and molecular targeted therapy. This form of senescence is termed therapy-induced senescence (TIS) and represents a desirable target in cancer therapy. Recent studies have shown that cellular senescence is a highly heterogeneous and dynamic process. Apart from being cleared by the immune system, the senescent cancer cells may survive for a long time and escape from senescence state. Notably, these cells even have the potential to regain stem-like state with high aggressiveness that eventually facilitates cancer recurrence. Furthermore, the senescence-associated secretory phenotype (SASP) of senescent cells is not always the same, and could establish immunosuppression and a protumor microenvironment. Given these detrimental effects, senescence-inducing chemotherapy followed by senotherapy (the “one–two punch” approach), has emerged. This combined therapy could mitigate unnecessary side effects of the persistent senescent cells, reduce the toxicity of pro-senescence therapy and prolong the survival of cancer patients, and it has a potential future in the precise treatment of cancer. Herein, we review the complex effects of therapy-induced senescence in cancer and highlight the great promise of two-step strategies in anticancer therapies.
Keywords: cellular senescence, SASP, cancer therapy, reversibility, senotherapy
Introduction
Cellular senescence is a stable state of growth arrest induced by different stresses and plays a key role in developmental processes.1,2 This phenomenon was first observed by Hayflick and Moorhead and dates back to the 1960s.3 Through a range of experiments, they observed that human diploid fibroblasts in culture would enter a senescent phase wherein the mitotic activity was degenerated and lessened.3 This phenomenon of “Hayflick limit” is currently characterized as replicative senescence (RS), which is largely a result of telomere shortening upon each cell division.3 This senescent physiological response contributes to preventing genomic instability and the accumulation of DNA damage, representing a fundamental tumor-suppressor mechanism.4 That is, senescence-associated growth arrest is a barrier against malignant transformation and the development of cancer. Apart from this form of senescence, later studies revealed that cellular senescence could be induced by diverse stimuli.2 The activation of oncogenes is found to result in senescence induction (oncogene-induced senescence (OIS)).1 In addition, exposure to DNA damage agents such as ionizing radiation and various anticancer chemotherapy agents are also known to mediate a senescent phenotype in both normal and cancer cells, which is often termed “therapy-induced senescence” (TIS).5,6 Inducing cancer cell senescence by chemotherapy may provide an effective means to limit cancer progression and to improve the prognosis of various solid and hematological malignancies.7–9
While the defining features of senescence include the establishment of a stable cell cycle arrest, many other cellular and molecular alterations are observed in senescent cells which contributes to a better understanding of the pathophysiological functions of senescent cells. For example, senescent cells undergo global epigenetic changes and metabolic reprogramming, and secrete senescence-associated secretory phenotype (SASP).1,2 SASPs, a complex mix of proinflammatory cytokines and chemokines, play a crucial role in crosstalk with neighboring cells, which could alter the tissue environment and affect tissue homeostasis.10,11 Of note, the qualitative and quantitative compositions of the SASPs are not always the same. They depend on senescence-inducing factors, cell type and the elapsed time since senescence initiation, which could be the reasons for the dual role of SASPs in tumor development.12–14
Cellular senescence is traditionally considered an “irreversible” mechanism by which tumor proliferation can be limited for a long time.4 Therefore, senescence may be a favorable goal for cancer treatment. However, in recent years, several studies have highlighted an unexpected dark side of TIS cells. This body of research showed that TIS cells could remain for a prolonged period of time and amplify many side effects of cancer therapy, such as bone marrow suppression, bone loss, and cancer metastasis and relapse.15–17 Long-term senescent cells can shape their surroundings toward a tumor-permissive and chronic inflammatory microenvironment by releasing SASP, which contributes to therapeutic resistance, malignant cell spreading and cancer relapse.10,12,18 Recent studies also showed that some of the senescence tumor cells induced by chemotherapy may escape cycle rest and recover proliferation in vivo and in vitro, which indicates that senescent cancer cells may be one form of tumor dormancy and contribute to tumor recurrence.19–22 Therefore, senescence induction can be considered as a “double-edged sword”. On the one hand, cellular senescence might limit the propagation of damaged cells and malignant transformation. On the other hand, senescent cells might induce a tumor-promoting microenvironment and maintain a tendency of cell proliferative reprogramming. To overcome the side effects of cellular senescence, the use of senotherapy to target senescent cells may be a potential anticancer adjuvant therapy. Indeed, more recent studies suggest that killing senescent cells in mice could extend their life span.23,24 When combined with currently used cancer therapies,25,26 this approach may achieve a better purpose of extending the short-term and long-term survival of cancer patients. In this review article, we describe the regulatory mechanism, the fate and biological function of therapy-induced senescent cells in cancer. Furthermore, we highlight the vast potential of the combination therapy of pro-senescence chemotherapy and senotherapy in cancer.
Cancer Cellular Senescence Following Therapy
Cellular senescence, is an important mechanism that maintains the normal physiological functions of the body. It affects many physiological and pathological processes, and promotes aging and aging-related diseases, even including cancer. A variety of stressors trigger senescence such as oncogene activation, telomere erosion and chemoradiotherapy-induced DNA damage. The stressors activate the p53/p21CIP1 and p16INK4a/Rb tumor suppressor networks, resulting in hypophosphorylation of Rb and blocking cycle arrest in G1/G2 phase.1 Cellular senescence could not be characterized by universal or specific biomarkers. In contrast, it has a multiplicity of biomarkers that are characterized by a number of nonexclusive characteristics: stable cell cycle and growth arrest; morphological changes such as increased cell size; chromatin remodeling; metabolic reprogramming; senescence-associated β-galactosidase (SA-β-gal) activity; resistance to apoptosis; and secretion of SASP.27
The aim of many chemotherapies is to kill cancer cells by damaging DNA; however, they also induce senescence. The list of drugs is long and still expanding, including: doxorubicin, cisplatin, taxol, etoposide (ETO), cyclophosphamide (CTX), and many others.6,28–31 Upon the treatment with doxorubicin, most of the tested cell lines derived from 10 different types of human solid tumors exhibited a senescent-like phenotype, which included enlarged and flattened morphology, increased granularity and positive SA-β-gal.6 When exposed to the DNA-damaging agents ETO or SN-38, various human cancer cell lines undergo terminal growth arrest and express of SA-β-gal.30 Apart from in vitro conditions, the phenomenon of TIS in vivo has been repeatedly identified in various animal models and human tumor tissues. For instance, in primary murine lymphomas treated with CTX, upregulation of SA-β-gal activity, p53, and p16INK4a could be found.31 Forty-one percent of breast tumors from patients who had received neoadjuvant chemotherapy had SA-β-gal positive staining.30 Of note, the majority of these agents can force tumor cells into senescence via the induction of DNA damage.5
Apart from senescence, cancer cells can also experience necrosis or apoptosis in response to chemotherapy. The outcome partly depends on the magnitude of stress that is applied to cancer cells: lower levels of stressors cause senescence, whereas stronger stimuli lead to apoptosis. Chang et al identified that senescence was more common than apoptosis among human solid tumor cell lines at moderate doses of doxorubicin.6 Such a dichotomy was also observed in prostate cancer cells, the cancer cells exhibited SA-β-gal activity and characteristic morphology at 25 nM doxorubicin, but underwent apoptotic cell death and cytotoxicity at 100–250 nM increased concentrations of doxorubicin.32,33 These observations suggest that lower drug doses are more likely to induce cancer cell senescence rather than cause cell death, which may minimalize treatment-related toxic side effects, when administered chronically.
The Intracellular Mechanism of Therapy-Induced Cell Senescence
There are numerous drugs and other therapies that can robustly induce cancer cell senescence, and the majority of these agents and therapies directly or indirectly act by inducing DNA damage, mainly by single- and double-strand breaks. The DNA damage response (DDR) activates p53/p21 and/or p16INK4a, and then results in a senescent phenotype by improving the relative abundance of hypophosphorylated Rb.30,31 These results imply that the mechanism of TIS in tumor cells may have the same core as the classic pattern of senescence (Figure 1A).
 |
Figure 1 The mechanism and function of therapy-induced senescence. (A) Therapy induce senescence-associated growth arrest by generating DNA damage or targeting reinitiation of senescent pathway signaling. (B) The senescent cells could reprogram with stemness by Suv39 h1 or p53 inactivation, Wnt stem signaling activation and/or derepression of the H3K9 me3 mark. (C) The SASP is an important mediator of cross-talk between senescent cells and nonsenescent cells. Scheme summarizing some of the functions associated with the SASP. |
The DDR pathway starts from the activation of the Ataxia Telangiectasia–Mutated (ATM), Ataxia Telangiectasia and Rad3-related (ATR) kinases. This reaction leads to the phosphorylation of numerous proteins, including γ-H2Ax (histone H2AX phosphorylated at Ser139), p53-binding protein 1(53BP1), and kinases Chk1 and Chk2. As a consequence, p53 and various biologic cyclin-dependent kinase inhibitors (CDKis) are triggered and eventually increase the expression of phase-specific CDKis, resulting in irreversible exit from the cell cycle at the G1 or G2/M stage.34 For example, the DNA-methylating drug temozolomide (TMZ) leads to ATM/ATR kinase mediated cellular senescence. Functional p53 and persistent p21 induction are required for TMZ-induced senescence.35 Our group has demonstrated that miR-34c-5p is a key regulator in senescence induction pathways. Increasing the expression of miR-34c-5p could enhance DA (daunorubicin and cytarabine)-induced leukemia stem cell (LSC) senescence by activating p53-p21CIP1-cyclin-dependent kinase (CDK)/cyclin or p53-independent CDK/cyclin pathways.36 Only miR-34c-5p overexpression can induce LSC senescence, which could also promote the senescence induction of chemotherapy.36 Importantly, it has been demonstrated that elevated p16INK4a expression leads to persistent cell cycle arrest.36 Induction of p16INK4a by TIS agents selectively inhibits CDK4 and CDK6, thus leading to hypophosphorylated Rb and cell cycle exit. Moreover, p16INK4a was observed that it markedly increased in various therapy-induced senescent cells such as doxorubicin-induced breast cancer cells and cisplatin-induced liver cancer cells.37–39 Furthermore, rather than acting separately, there are is complicated crosstalk between p53 and p16INK4a to achieve senescence activation in response to chemotherapy. A study in primary murine lymphomas showed that CTX-induced senescence was controlled by p53 and p16INK4a.29 These data suggest that the involvement of certain tumor suppressor genes, such as p21, p16INK4a, and p53, is highly cell-specific during the executionary phases of senescence. Of note, the observations indicate that p53, p21 and p16INK4a expression is necessary, but is now known to, the absence of these proteins does not mean that cells cannot achieve TIS.30,40
In addition, TIS could occur independently of the DNA damage response or classical senescence pathways. In a prostate mouse model, TIS was also induced in PTEN hemizygous cancer cells without a DNA damage response.41 One important p53 target gene is cyclin-dependent kinase inhibitor (CDKi) p21, the upregulation of which blocks CDK2 activity and the reinitiation of cell division. However, another report showed that 6-anilino-5,8-quinolinequinone treatment induced the transcription of p21 rather than p53 and subsequently resulted in growth arrest and senescence in colorectal cancer, which indicated that p21 may serve as a senescence promoter in a p53-independent manner.42 There is a report that in a mouse model of acute promyelocytic leukemia (APL), retinoic acid (RA) and/or arsenic trioxide could mediate PML-RARA degradation, activate p53 and induce leukemia cell senescence.7 Lapatinib-induced breast cancer cell senescence was correlated with increased levels of p15 and p27 rather than the expression of p16INK4a or p2143 (Figure 1A). Another study suggested that an S phase kinase-associated protein 2 (Skp2) inhibitor could drive growth arrest and cellular senescence, which was associated with PTEN or p19ARF mutations. This chemotherapy-induced tumor cell senescence was identified not to involve p53 and DNA damage but instead critically depends on p21, p27, and ATF4 induction.40 These reports suggest that some cancers may undergo TIS by particular stress-signaling responses.
The Fate of Chemotherapy-Induced Senescent Cells
Immune Cell-Mediated Clearance
Senescent cells play a key role in immune surveillance by stimulating both innate and adaptive immune responses. This immunosurveillance of senescent cells consequently leads to the clearance of senescent tumor cells, suggesting that the cellular senescence is a general mechanism that limits cancer development.18 Therapy induced senescent cells with upregulation and secretion of SASPs leads to the activation of an innate immune system by recruiting neutrophils, macrophages and natural killer (NK) cells. Immune cells, especially NK cells, play a crucial role in immune surveillance by recognizing senescent tumor cells and leading to specific cytotoxicity toward senescent tumor cells. Xue et al showed that p53 reactivation in tumor-bearing mice can trigger the senescence response in vivo, accompanied by tumor clearance mediated by activated NK cells.44 Lymphomas from lymphoma-bearing mice treated with CTX underwent stable cell cycle arrest and displayed key SASP components that triggered NK cell-mediated immune clearance of senescent cells.45 In another study, senescent multiple myeloma cells induced by low doses of chemotherapeutics were able to increase NK cell receptor ligand (NKG2D) and DNAM-1 ligand expression, which enhanced NK cell-mediated immune responses leading to increased tumor clearance.46 In a mouse model with orthotopic implantation of melanoma patient tumors, Liu et al observed that chemotherapy-induced senescent melanocyte cells released SASP factors including IL-6 and IL-8. These factors promote macrophage and neutrophil recruitment into the tumor, where they exhibit anti-tumor activity and consequently limit tumor growth.47
Moreover, senescent cells can activate the adaptive immune response to support an antitumor microenvironment. In a patient-derived xenograft mouse model of melanoma tumor, chemotherapy-induced senescent cells secreted the chemotactic protein CCL5, which promotes immune cell homing into melanoma tumors to support a tumor-suppressive immune response.48 A series of experiments in the mouse liver revealed that the clearance of premalignant senescent hepatocytes strictly depended on the adaptive immune response mediated by CD4+ T cells, and the immunosurveillance of senescent cells inhibited the development of hepatocellular carcinomas. In contrast, a lack of CD4+ T cells impairs immune clearance of senescent hepatocytes, which contributes to liver cancer growth.49 In addition, monocytes/macrophages are required for senescence surveillance mediated by CD4+ T cells.49
Long-Term Survival and Immune Escape
One of the characteristics of senescent cells is growth arrest at the G1 or G2/M phases of the cell cycle, but senescent cells were identified to be able to survive for a long time.1 Senescent melanocytes were observed to remain indolent for years in benign nevi in vivo.50 In another study, a subpopulation of senescent prostate cancer cells induced by doxorubicin treatment persisted for at least 5 weeks in mouse tumor xenograft models.51 The immunosuppressive microenvironment created by senescent cells could protect senescent cancer cells from being cleared and permit tumor cell residues,10,18 which may be one of the most important reasons for the long-term survival of senescent cells. For instance, fibroblasts treated with palbociclib promoted Gr-1+ myeloid cell-mediated immunosuppression in melanoma mouse models.21 Toso et al found that senescent prostate cells shaped an immunosuppressive tumor microenvironment by activating the Jak2/Stat3 pathway. In contrary, a JAK2 inhibitor could enhance the efficacy of docetaxel-induced senescence and promote the clearance of senescent cells by triggering the immune system.52 The senescent cancer cells induced by chemotherapy (mitoxantrone (MIT), ETO) shed NKG2D-Ls to promote their immunoevasion and persist.8
Senescence-Associated Reprogramming Inducing Cancer Relapse
In the past, cellular senescence was considered to be a static, irreversible state. However, recent studies have found that cellular senescence is not a one-way street, but a dynamic process. It was observed in vivo and in vitro that some of the senescent tumor cells induced by chemotherapy could restore proliferation and tumorigenesis. In this case, the tumor cells that escape senescence may contribute to tumor recurrence and have a higher potential for invasive growth.2
A study conducted by Was et al showed the reversibility of senescence in human colon cancer HCT116 cells. In their study, HCT116 cells were exposed to repeated cycles of doxorubicin, which mimics clinical chemotherapy regimens. On the 13th day, the characteristics of senescent cells, including a flat morphology, increased activity of SA-β-gal and polyploidy in these doxorubicin-induced HCT116 cells, were observed. However, two weeks after the removal of doxorubicin, the number of cells increase 6-fold. Time-lapse imaging confirmed the presence of highly proliferative progeny in senescent HCT116 cells.53 In another study, a subpopulation of colon cancer cells that treated with chemotherapy showed a combination of senescent cells and stem-like characteristics.54 Through analysis of the microarray signature, senescent cells were found to express excessive expression of many stem-related genes, including CD34, Lgr6, Prom1, CD44, Ngfr, and Nestin.11 Similarly, the correlative observations have also been reported in which stem cell markers were observed in OIS or TIS in recent years.53–55 Indeed, the senescence melanocyte induced by the oncogene N-RAS61K showed stem-like features, in which the expression of Pdpn and Nanog was markedly increased. Nanog is a master transcription factor of cellular self-renewal.55 Moreover, senescent human and mouse cancer cells could also exhibit proliferation potential and senescence characteristics at the same time.11,19 In epithelial ovarian cancer (EOC), Nacarelli et al found that cisplatin-induced senescence upregulated ALDH1 protein levels and the expression of ALDH1A1 along with CD133 mRNA, which is also the cancer stem-like cell (CSC) signature. When cisplatin-treated EOC cells rather than EOC cells were inhibited, the EOC CSC signature was suppressed and the survival of EOC-bearing mice was improved.56 By lineage tracing the p21+ senescent cells in the p21-CreER mouse model, some of the previously labeled senescent cells were found to re-entered the cell cycle and proliferate rather than being cleared at the late embryonic stage.57 In agreement with previous observations, the researchers from Charité University Medicine found that Eμ-Myc-Bcl2-overexpressing lymphoma cells induced senescence by chemotherapy exhibited both senescence and stem cell-like characteristics. Based on single-cell tracking experiments, SA-β-gal-positive cells were found to incorporate 5-ethynyl-2′-deoxyuridine (EdU), which suggested that senescent cells restarted DNA synthesis. More importantly, the senescent tumor cells of mouse and human origin were allowed to escape cellular senescence and resume sustainable proliferation when senescence-essential mediators such as Suv39 h1 or p53 were acutely inactivated. Strikingly, compared to cells that are never senescent, tumor cells that escape from the senescent state can re-enter the cell cycle with higher aggressive growth and tumor-initiation potential by activating the WNT pathway.19 In addition, Yu et al showed that loss of the H3K9 me3 mark, such as elevate activity of the H3K9 demethylase JMJD2C, could make OIS cell re-entry into cell cycle in melanocyte models.58 These findings indicate that senescent cells have the potential to repossess their self-renewal capability. TIS may result in senescence-associated reprogramming, which promotes cells to escape from the arrested condition and cancer recurrence (Figure 1B). However, more direct evidence about the stemness recovery of senescent tumor cells remains to be investigated in the future.
Senescence-Associated Signaling Crosstalk Between Tumor Cells and Their Niche Cells
Senescent cells can secrete a complex mixture of proinflammatory cytokines, chemokines, growth factors and other cytokines, which is also called as SASP.10 These secretomes might be an important mediator of the cross-talk between senescent cells and nonsenescent cells (Figure 1C).
The Effects of Senescent Cancer Cells on Neighboring Cells
At the early stage of tumors, SASPs contribute to increased tumor growth control by reinforcing and/or “spreading” senescence-associated cell cycle exit via autocrine and paracrine signaling. SASPs might be required for stable cell cycle arrest. For example, IL-6 and IL-8, the major components of the SASP, have been shown to restrain the proliferation of malignant cells by reinforcing the senescence growth arrest in an autocrine fashion.59 PAI-1 was also found to be essential for the induction of senescence even in the absence of p53.60 In other studies, senescent MCF-7 breast cancer cells triggered by adriamycin were observed to have an antiproliferative effect in which they induced a senescent state of cell cycle arrest in naive MCF-7 cells by secreting factors in vitro.61 Paracrine-induced senescence by SASPs might be important for the inhibition of tumor progression. Moreover, secreted SASP factors can directly modulate the immune response by signaling to the immune system. As described above, senescent cancer cells recruit immune cells to remove the senescent tumor cells, consequently limiting tumor progression via engagement of the innate and adaptive immune systems.
Other than frequently acting as an anticancer mechanism, senescent cells have recently been considered beneficial to cancer progression and relapse in some settings.10,18 Their contribution includes creating an immunosuppressive and protumor tissue microenvironment that protects some cancer cells from being cleared and permits tumor cell residues, thus positively favoring cancer relapse and progression. One of the functions of SASPs is that they can suppress the tumor immune response, which contributes to cancer promotion. For example, a recent study found that chemotherapy-induced senescent cells were able to circumvent immune clearance through concerted MMP-mediated shedding of NKG2D ligands and paracrine suppression of NKG2D–dependent immunosurveillance.8 A study conducted by Toso et al showed that Pten-null senescent prostate tumors released a subset of immunosuppressive SASP cytokines. These SASP factors could block both the proliferation and activity of immune cells, which promoted tumor progression and chemoresistance.52 Through analyzing proteome profiling of senescent melanoma cells induced by doxorubicin, the senescent melanoma cells were revealed to secret a panoply of proteins to contribute to tumor immunosuppression or tumor immune evasion.62 In summary, SASP factors are not always the same, and tissue-specific.
Other reports have revealed that the SASPs secreted by senescent cancer cells can also impact the metastatic and invasion potential of cancer cells.12,63–65 The SASP factors from malignant tumor cells can alter the tissue microenvironment, thus creating a local region of the tumor-promoting microenvironment that stimulates nearby premalignant and malignant cells metastasis and invasion.12 Chemotherapy-triggered senescent mesothelioma cells strongly enhanced epithelial-to-mesenchymal (EMT) induction and chemoresistance mediated by SASP.63 In a mouse model of breast cancer, p95HER2-induced senescent breast cancer cells promoted metastasis in a noncell-autonomous manner.64 Across the types of thyroid cancer, senescent cells were found to be capable of promoting neighboring nonsenescent cancer cell invasion and metastasis via CXCL12-CXCR4 signaling.65
In addition to these functions, SASPs have been suggested to contribute to the proliferation and stemness of cancer cells. Indeed, cisplatin treatment-induced senescent melanoma cells stimulated the growth of nonsenescent cancer cells mediated by IL-1α and IL-8.66 Similarly, after exposure to senescence-inducing doxorubicin or irradiation, the emergence and maintenance of CSCs was promoted in multiple myeloma, which was correlated with the upregulation of the SASPs secreted by non-CSCs, particularly with RANTES expression.67 To date, this has yet to be specifically studied between TIS cancer cells and the tumor microenvironment. In summary, the SASPs have dual effects of tumor suppression and tumor promotion. Therefore, patients may obtain beneficial outcomes from the elimination of senescent cells.
Effects of Senescent Stromal Cells on Neighboring Cells
Obviously, chemotherapeutic drugs not only induce cancer cell senescence but also contribute to stromal cells entering senescence. Similar to senescent cancer cells, senescent stromal cells have a dual role in tumor development. On the one hand, senescence is widely accepted as a firm tumor barrier because it can prevent tumorigenesis and limit the tumor proliferative life span. SASPs from senescent mesenchymal stromal cells (MSCs) could block the proliferation of immortalized prostate cells (PNT2) by inducing senescence, which acted as a weapon effective against pre-tumorigenesis.68 Lujambio et al found that senescent hepatic stellate cells could stimulate the recruitment and M1 polarization of macrophages to suppress tumorigenesis and to form an antitumor microenvironment.69 On the other hand, contrary to the originally proposed tumor suppressor function of senescent cells, SASPs secreted by senescent stromal cells may play a protumor role in tumor development. For example, a recent study suggested that conventional chemotherapies induced senescence of the prostate stromal cell line PSC27. As a result, amphiregulin (AREG) derived from senescent stromal cells enhanced cancer cell resistance to chemotherapeutic agents and increased programmed cell death 1 ligand (PD-L1)-mediated immunosuppression.9 In a breast cancer mouse model, bleomycin-induced senescent human fibroblasts stimulated the growth of cotransplanted breast cancer cells (MDA-MB-231) by secreting metalloproteases.70 In another study in lung cancer, the antitumor drug boanmycin could induce fibroblast senescence, and the long-term treatment-induced senescence could alter the lung tumor microenvironment by increasing the expression of IL-6, thus contributing to tumor relapse and metastasis.71
Targeting Senescence Cancer Cells
As discussed above, cellular senescence has beneficial and detrimental effects on cancer development.2,18 Senescent cells initially inhibit tumor development, while accumulating senescent cells promotes tumor development. For instance, palbociclib initially blocked tumor cell proliferation by inducing cancer cell senescence. However, with prolonged treatment with palbociclib, a tumor-permissive environment that promoted tumor growth would be established.21 Given the detrimental effect of senescent cells on cancer, removing existing senescent cells may increase therapeutic efficacy. The Campisi group demonstrated that the removal of senescent cells after doxorubicin treatment had clinical potential because it could reduce the incidence of cancer relapse and metastasis, as well as side effects of chemotherapy.16 Some researchers established “INK-ATTAC” transgenic mice in which p16Ink4a-positive cells were depleted by the dimerizer AP20187 (AP).23 The mouse model suggested that the removal of senescent cells reduced spontaneous tumor formation. Similarly, the p16-3MR transgenic mouse model in which tracking and eliminating senescent cells could be permitted indicated that targeting senescent cells could prevent or delay cancer relapse as well as reduce chemotherapy side effects.24 Altogether, these data highlight the benefits of targeting these senescent cells in cancer treatment. Senotherapy as an antitumor strategy targeting senescent cells has been proposed. Current senotherapy is focused on: i) specific removal of senescent cells by senolytics; ii) SASP neutralization (anti-SASP strategy); and iii) improving immune-mediated senescent cell elimination.72
Senolytics
Among senotherapies targeting senescent cells, senolytics are the most developed and well-studied, and show promising results in cancer treatment (Table 1). According to studies, senolytics mainly interfere with the pathway involved in apoptosis to achieve selective elimination and target of senescent cells, which are also called senescent-cell anti-apoptotic pathways (SCAPs), such as the BCL-2/BCL-XL, PI3K/AKT, p53/p21, Ephirins/dependence receptors/tyrosine kinases and the hypoxia inducible factor (HIF-1α) pathway.73 In recent years, another HSP-90 SCAP has been reported.74 For instance, dasatinib + quercetin (D+Q) treatment, which targets several of these SCAPs, was effective in eliminating senescent human umbilical vein cells (HUVECs) rather than control cells in vitro.73 ABT737, which targets BCL-2, BCL-W and BCL-XL, was found specifically to induce apoptosis in senescent cells and efficiently to clear them in two independent senescent mouse models.75 Similarly, navitoclax (ABT-263, targets BCL-XL, BCL-2, and BCL-W) was demonstrated that it to eliminate senescent cells effectively by inducing apoptosis in a cell type- and species-independent manner.76 In another study, Wang et al found that ABT-263 could selectively kill senescent melanoma and lung cancer cells, providing a potential strategy for sequential using after a pro-senescence drug for cancer.25
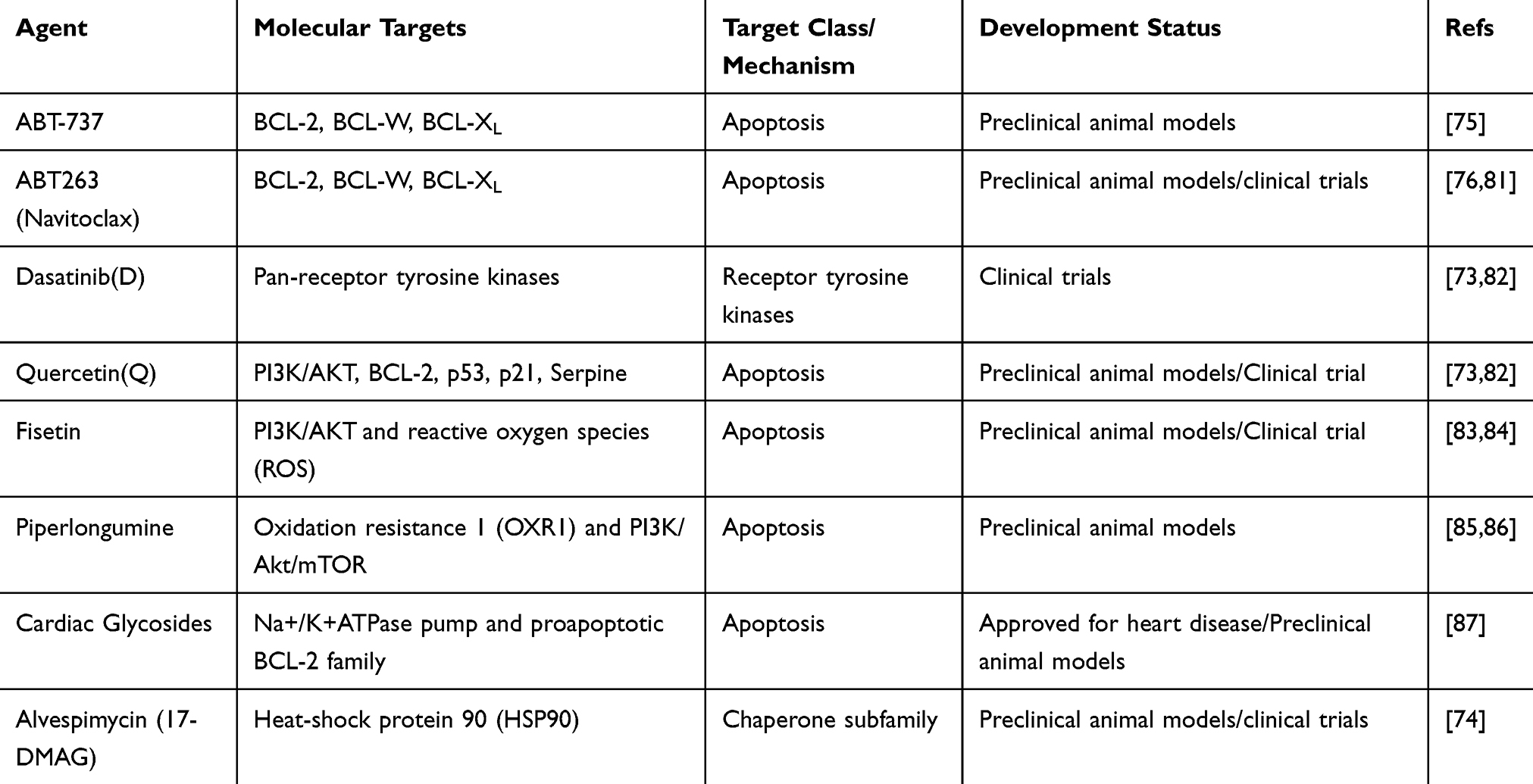 |
Table 1 Candidate Senolytics |
Although numerous reports have shown the benefits of senolytics in age-related diseases including cancer, there are still unwanted damage/side effects. Some evidence suggests that systemic administration of BCL-2 family inhibitors such as navitoclax (ABT-263) results in thrombocytopenia and neutropenia.77–79 It would be desirable to control the abundance of senolytics by intermittent dosing, without affecting effectiveness. In addition, different senescent cell types may rely on different antiapoptotic mechanisms. Therefore, it is difficult to contend that these senolytic drugs are generally effective for all types of senescent cells. In the future, senolytics need to be more specific or combined with other drugs to solve this problem. A new strategy has been described that could overcome the problem of specificity of senolytics using galactooligosaccharides–coated nanoparticles containing senolytics when administered in a chronic manner.80
SASP Inhibitors
Blocking the SASP is another potential means to inhibit the functions of senescent cells as they relate to tumor progression (Table 2). SASP inhibitors can target pathways such as p38 MAPK, NF-κB, and mTOR, to suppress SASP production. Alternatively, inhibition of specific SASP factors could also be achieved, as is the case for IL-6, IL-8 and TNFα. Rapamycin (selective MTORC1 complex inhibitor) can repress the ability of senescent fibroblast cells to stimulate cell proliferation and tumorigenesis in vivo by modulating SASPs by selectively suppressing IL1A translation.88 As our own work has showed, rapamycin promoted hematopoietic stem cell (HSC) survival by inhibiting senescence.89 Another mTOR inhibitor, AZD8055, efficiently eliminated TIS cells and synergized with prosenescence drug to reduce tumor burden and increase survival in mice model of aggressive liver cancer.26 Metformin has proven efficient in decreasing the secretion of inflammatory cytokines in senescent cells by inhibiting the phosphorylation of IκB and IKKα/β and the activation of NF-κB.90 The AK1 inhibitor 5Z-7 weaken the SASP phenotype of senescent cells treated with chemotherapeutic agents by inhibiting both the p38 MAPK and PI3K-Akt-mTOR pathways. Thus, 5Z-7 substantially sensitizes cancer cells to chemotherapy and promotes tumor regression by affecting the expression of SASP factors but not senescence.91 Anakinra, an IL-1 receptor (IL-1R) antagonist, markedly reduced the invasiveness of metastatic cancer cells by decreasing senescence-associated IL-6/IL-8 secretion.92 Notably, the SASP has a complex background dependency and particular components have effects on tumor development.10,14 In addition, if signaling cascades upstream of SASPs, such as p38 MAPK or PI3K–AKT–mTOR were blocked, unwanted side effects would appear such as promoting tumorigenesis and relapse, because SASP development is orchestrated by multiple signaling pathways and these signaling pathways are not exclusive to senescence.41,72 Thus, to enhance the therapeutic efficacy and to make SASP inhibitors more specific for senescent cells, it is important to identify which SASP compounds have tumor-promoting functions and which ones possess antitumorigenic functions.
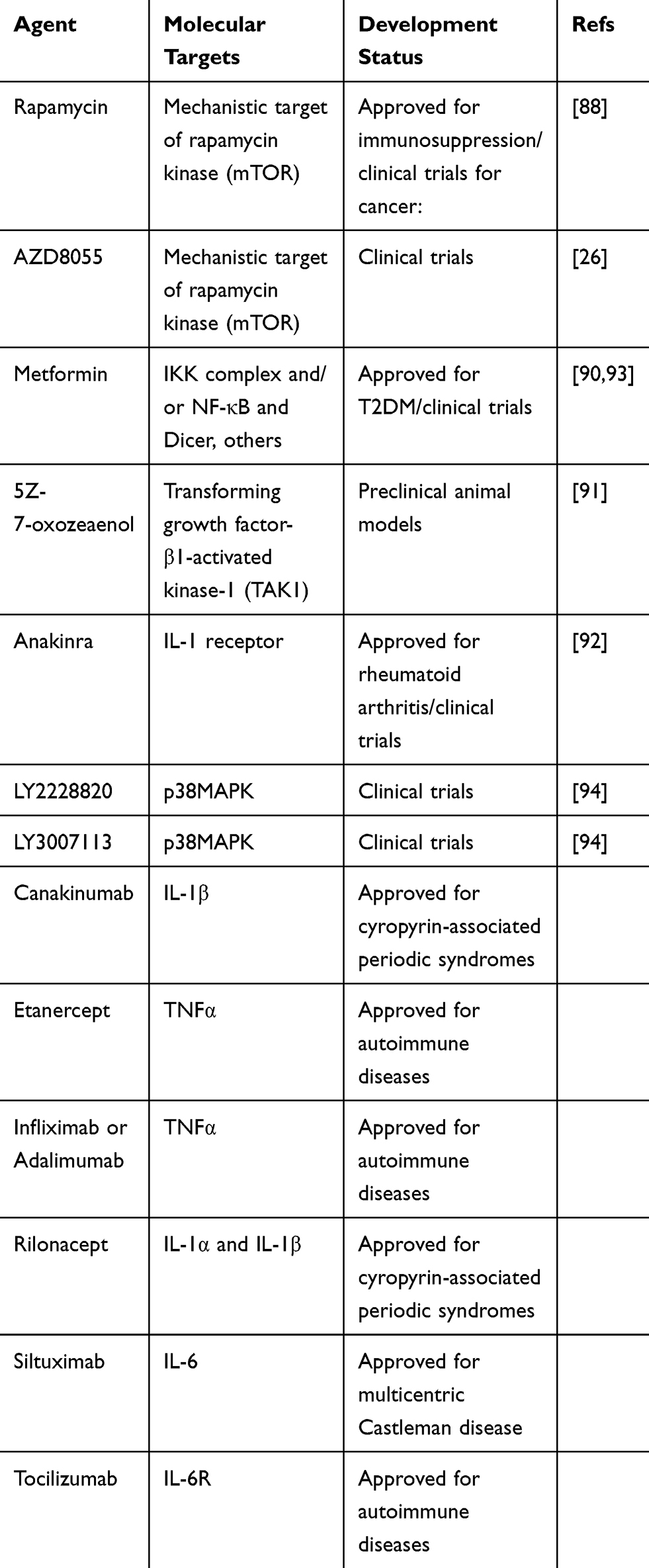 |
Table 2 Candidate SASP Inhibitors |
Improving Immune System Function
Senotherapy to improve the immune system-mediated clearance of senescent cells could reduce the senescent cell burden and limit tumor proliferation. This could be achieved through chimeric antigen receptor T cells (CAR-Ts) that could target specific senescence-related molecules or increase immune activity to enhance surveillance.18 Of note, a recent study suggests that PD-1 checkpoint blockade reawakens exhausted CD8+ T cells following senescence induction, thus triggering antitumor immunity and producing tumor cell death.95 As a consequence, the addition of anti-PD-1 therapy to the prosenescence regimen promoted tumor regression. To date, this therapy has not been identified with enough experiments.
Targeting Senescent Cancer cells – Two-Step Cancer Therapies
Numerous studies have demonstrated that cancer-associated senescent cells are initially helpful for the tumor-suppressive response. Therefore, the induction of cellular senescence is considered an effective and powerful strategy for cancer therapy with fewer severe side effects of cytotoxicity.5 RA and/or arsenic trioxide can activate pro-senescence Trp53 signaling to eradicate leukemia-initiating cells, ultimately definitively curing acute promyelocytic leukemia.7 In preclinical mouse models of KRAS-mutant pancreatic ductal adenocarcinoma (PDAC), a combination of trametinib and palbociclib (T/P) could drive PDAC cells into senescence, which resulted in vascular remodeling that in turn facilitated chemotherapeutic drug delivery and CD8+ cell infiltration.95 However, as mentioned above, the unwanted accumulation of senescent cells can be harmful, which could turn into a potential threat such as stemness-reprogrammed and immune suppression. To reconcile this controversy, “one–two punch” approach to cancer therapy has been proposed, in which pro-senescence treatments are followed by senotherapy.72 This method may achieve a better purpose of extending both the short-term and long-term survival of cancer patients. In this regard, the use of senotherapy can reduce the side effects of pro-senescence therapies by killing senescent cells from the tumor as well as removing the SASP source permanently and reducing side effects from cellular senescence. For example, limiting therapy-induced SASPs by inhibiting the p38 MAPK-MK2 pathway could rescue chemotherapy-induced bone loss in doxorubicin-treated mouse models.17 Wang et al found that TIS cells became vulnerable to apoptosis induction by ABT263 in vitro, and demonstrated the feasibility of the sequential therapy for cancer in which senotherapy could be administered concomitantly or after senescence-inducing treatment.25 In preclinical mouse models of ovarian and breast cancer, concurrent PARPi and ABT263 treatment was observed to have a significant potentiation of cell killing. ABT263 enhanced the limitation of resistance by inducing PARPi-induced senescent apoptosis.96 Researchers have found that panobinostat could kill persistent senescent cancer cells that arose post-Taxol therapy by decreasing the expression of BCL-XL and increasing acetylated H3. Under this combination treatment, the tumor burden could be significantly reduced.97 A recent study suggests that pro-senescence therapy with a CDC7 inhibitor, combined with mTOR inhibitor that can selectively eliminate TIS cells, may elicit a more-effective inhibition of growth in liver cancer without causing damage to normal cells.26 A later study on a mouse model of aggressive breast cancer showed that the use of the nano-encapsulated senolytic agent navitoclax could selectively eliminate palbociclib-induced senescence, which contributed to the limitation of tumor growth and metastases.98 Although the application of prosenescence therapy represents a successful cancer treatment strategy, an increasing number of studies have shown that some patients still relapse, perhaps because of the existence of long-term senescent cells that promote tumor recurrence. These studies suggest that the “one–two punch” approach of cancer therapy may provide a new therapeutic opportunity to improve antitumor efficacy and outcomes in cancer patients.
Conclusion and Future Perspectives
Over the past decades, our understanding of the cellular and molecular mechanisms that drive senescence has exploded. Both conventional and targeted antitumor drugs could induce cancer cell senescence as a primary response. The induction of senescence is a potential means of cancer treatment because it leads to a persistent cytostatic state in various cancer types. Interestingly, recent studies suggest that cellular senescence is a dynamic process rather than a binary choice in which senescence-associated secretomes display highly dynamic compositional changes. Long-term senescent cells have a deleterious impact on their neighbors, which is correlated with SASP.2,11 Of note, chemotherapy-induced senescent cancer cells were found to have the potential to escape from the growth arrest and re-entry into the cell cycle with high aggressivity,20 which argued against the therapeutics of driving tumor cells into senescence. However, to date there are no reliable markers to isolate true senescent cells. In the future, more sufficient evidence is needed to prove senescence-associated reprogramming.
Perhaps we could develop a newer agent that drives the cancer cells into a “deep” senescent state in which the arrest state is irreversible; thus, the senescent cells would not be able to escape from senescence and decrease the possibility of stemness. This agent may be combined with other compounds that mediate senescent cell emulations, such as senolytic drugs and immune-boosting drugs (Figure 2). This combination also called “one–two punch” approach, which first applies pro-senescence treatments to induce senescence and then to eliminate senescent cancer cells by senotherapy.1,25,26 This combination therapy could address the dual behavior of senescence and provide a therapeutic benefit in cancer treatment. Nevertheless, studies have shown that senolytic cause some side effects. For example, navitoclax results in thrombocytopenia and neutropenia.73 As such, we can expect that there are side and off-target effects, which should be carefully taken into consideration. A major challenge for combination therapies involving senotherapy will be the question of how to target the right cells at the right time. Therefore, it is essential to identify more reliable senescence markers to assess the therapeutic effects of pro-senescence and senolytic drugs. In the future, we could use single-cell profiling technologies to characterize the phenotypic spectrum of senescent cells as well as unravel to the heterogeneity of senescence and the SASP and the role senescence plays in cancer. Further studies addressing these questions could bring a great breakthrough for the precise treatment of cancer.
 |
Figure 2 The dynamic process of chemotherapy-induced senescence. Cancer cells undergo senescence after exposure to chemotherapeutic agents in response to DNA damage, and then activate the p53/p21CIP1 and/or p16INK4a/Rb tumor suppressor networks. On the one hand, the senescent cancer cells are cleared by activating the immune system. On the other hand, failure to clear CIS cells can last in the long-term and even escape from the senescent state. The accumulating senescent cells are able to shape their surroundings toward a tumor-permissive and chronic inflammatory microenvironment by SASP. The escaping senescent cells could reprogram with stemness recovery which contributes to tumor relapse. Using senotherapy after senescence-inducing chemotherapy could mitigate unnecessary side effects of the senescent cells. |
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81170524, 30871097,81770192, 81370660, 81973999, 81300412).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128(4):1238–1246. doi:10.1172/JCI95148
2. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol. 2019;21(1):94–101. doi:10.1038/s41556-018-0249-2
3. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi:10.1016/0014-4827(61)90192-6
4. Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20(2):150–155. doi:10.1016/j.ceb.2008.01.007
5. Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102(20):1536–1546. doi:10.1093/jnci/djq364
6. Chang BD, Broude EV, Dokmanovic M, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59(15):3761–3767.
7. Ablain J, Rice K, Soilihi H, de Reynies A, Minucci S, de The H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med. 2014;20(2):167–174. doi:10.1038/nm.3441
8. Munoz DP, Yannone SM, Daemen A, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight. 2019;5. doi:10.1172/jci.insight.124716
9. Xu Q, Long Q, Zhu D, et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD‐L1)‐mediated immunosuppression. Aging Cell. 2019;18(6). doi:10.1111/acel.13027
10. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. 2019;19(8):439–453. doi:10.1038/s41568-019-0156-2
11. Ritschka B, Storer M, Mas A, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31(2):172–183. doi:10.1101/gad.290635.116
12. Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi:10.1371/journal.pbio.0060301
13. Coppé J-P, Rodier F, Patil CK, Freund A, Desprez P-Y, Campisi J. Tumor suppressor and aging biomarker p16INK4a induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286(42):36396–36403. doi:10.1074/jbc.M111.257071
14. Basisty N, Kale A, Jeon OH, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18(1):e3000599. doi:10.1371/journal.pbio.3000599
15. Shao L, Feng W, Li H, et al. Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arf-independent manner. Blood. 2014;123(20):3105–3115. doi:10.1182/blood-2013-07-515619
16. Demaria M, O’Leary MN, Chang J, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7(2):165–176. doi:10.1158/2159-8290.CD-16-0241
17. Yao Z, Murali B, Ren Q, et al. Therapy-induced senescence drives bone loss. Cancer Res. 2020;80(5):1171–1182. doi:10.1158/0008-5472.CAN-19-2348
18. Burton DGA, Stolzing A. Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res Rev. 2018;43:17–25. doi:10.1016/j.arr.2018.02.001
19. Milanovic M, Fan DNY, Belenki D, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553(7686):96–100. doi:10.1038/nature25167
20. Saleh T, Tyutyunyk-Massey L, Murray GF, et al. Tumor cell escape from therapy-induced senescence. Biochem Pharmacol. 2019;162:202–212. doi:10.1016/j.bcp.2018.12.013
21. Guan X, LaPak KM, Hennessey RC, et al. Stromal senescence by prolonged CDK4/6 inhibition potentiates tumor growth. Mol Cancer Res. 2017;15(3):237–249. doi:10.1158/1541-7786.MCR-16-0319
22. Saleh T, Tyutyunyk-Massey L, Gewirtz DA. Tumor cell escape from therapy-induced senescence as a model of disease recurrence after dormancy. Cancer Res. 2019;79(6):1044–1046. doi:10.1158/0008-5472.CAN-18-3437
23. Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–189. doi:10.1038/nature16932
24. Jeon OH, Kim C, Laberge RM, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–781. doi:10.1038/nm.4324
25. Wang L, Leite de Oliveira R, Wang C, et al. High-throughput functional genetic and compound screens identify targets for senescence induction in cancer. Cell Rep. 2017;21(3):773–783. doi:10.1016/j.celrep.2017.09.085
26. Wang C, Vegna S, Jin H, et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature. 2019;574(7777):268–272. doi:10.1038/s41586-019-1607-3
27. Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell. 2019;179(4):813–827. doi:10.1016/j.cell.2019.10.005
28. Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005;65(7):2795–2803. doi:10.1158/0008-5472.CAN-04-1270
29. Chang BD, Xuan Y, Broude EV, et al. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18(34):4808–4818. doi:10.1038/sj.onc.1203078
30. Te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62(6):1876–1883.
31. Schmitt CA, Fridman JS, Yang M, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335–346. doi:10.1016/s0092-8674(02)00734-1
32. Ewald JA, Peters N, Desotelle JA, Hoffmann FM, Jarrard DF. A high-throughput method to identify novel senescence-inducing compounds. J Biomol Screen. 2009;14(7):853–858. doi:10.1177/1087057109340314
33. Schwarze SR, Fu VX, Desotelle JA, Kenowski ML, Jarrard DF. The identification of senescence-specific genes during the induction of senescence in prostate cancer cells. Neoplasia. 2005;7(9):816–823. doi:10.1593/neo.05250
34. Zhou B, Elledge S. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408(6811):433–439. doi:10.1038/35044005
35. Aasland D, Gotzinger L, Hauck L, et al. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019;79(1):99–113. doi:10.1158/0008-5472.CAN-18-1733
36. Peng D, Wang H, Li L, et al. miR-34c-5p promotes eradication of acute myeloid leukemia stem cells by inducing senescence through selective RAB27B targeting to inhibit exosome shedding. Leukemia. 2018;32(5):1180–1188. doi:10.1038/s41375-018-0015-2
37. Qu K, Lin T, Wang Z, et al. Reactive oxygen species generation is essential for cisplatin-induced accelerated senescence in hepatocellular carcinoma. Front Med. 2014;8(2):227–235. doi:10.1007/s11684-014-0327-1
38. You R, Dai J, Zhang P, Barding GA, Raftery D. Dynamic metabolic response to adriamycin-induced senescence in breast cancer cells. Metabolites. 2018;8(4):95. doi:10.3390/metabo8040095
39. Saleh T, Bloukh S, Carpenter VJ, et al. Therapy-induced senescence: an “old” friend becomes the enemy. Cancers (Basel). 2020;12(4):822. doi:10.3390/cancers12040822
40. Lin HK, Chen Z, Wang G, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464(7287):374–379. doi:10.1038/nature08815
41. Alimonti A, Nardella C, Chen Z, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest. 2010;120(3):681–693. doi:10.1172/JCI40535
42. Lodygin D, Menssen A, Hermeking H. Induction of the Cdk inhibitor p21 by LY83583 inhibits tumor cell proliferation in a p53-independent manner. J Clin Invest. 2002;110(11):1717–1727. doi:10.1172/jci0216588
43. McDermott MSJ, Conlon N, Browne BC, et al. HER2-targeted tyrosine kinase inhibitors cause therapy-induced-senescence in breast cancer cells. Cancers (Basel). 2019;11(2):197. doi:10.3390/cancers11020197
44. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi:10.1038/nature05529
45. Chien Y, Scuoppo C, Wang X, et al. Control of the senescence-associated secretory phenotype by NF- B promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25(20):2125–2136. doi:10.1101/gad.17276711
46. Soriani A, Zingoni A, Cerboni C, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113(15):3503–3511. doi:10.1182/blood-2008-08-173914
47. Liu Y, Hawkins OE, Su Y, et al. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-kappaB impairs this drug-induced senescence. EMBO Mol Med. 2013;5(1):149–166. doi:10.1002/emmm.201201378
48. Vilgelm AE, Johnson CA, Prasad N, et al. Connecting the dots: therapy-induced senescence and a tumor-suppressive immune microenvironment. J Natl Cancer Inst. 2016;108(6):djv406. doi:10.1093/jnci/djv406
49. Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–551. doi:10.1038/nature10599
50. Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720–724. doi:10.1038/nature03890
51. Ewald J, Desotelle J, Almassi N, Jarrard D. Drug-induced senescence bystander proliferation in prostate cancer cells in vitro and in vivo. Br J Cancer. 2008;98(7):1244–1249. doi:10.1038/sj.bjc.6604288
52. Toso A, Revandkar A, Di Mitri D, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9(1):75–89. doi:10.1016/j.celrep.2014.08.044
53. Was H, Barszcz K, Czarnecka J, et al. Bafilomycin A1 triggers proliferative potential of senescent cancer cells in vitro and in NOD/SCID mice. Oncotarget. 2017;8(6):9303. doi:10.18632/oncotarget.14066
54. Was H, Czarnecka J, Kominek A, et al. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol Ther. 2018;19(1):63–75. doi:10.1080/15384047.2017.1385675
55. Leikam C, Hufnagel AL, Otto C, et al. In vitro evidence for senescent multinucleated melanocytes as a source for tumor-initiating cells. Cell Death Dis. 2015;6:e1711. doi:10.1038/cddis.2015.71
56. Nacarelli T, Fukumoto T, Zundell JA, et al. NAMPT inhibition suppresses cancer stem-like cells associated with therapy-induced senescence in ovarian cancer. Cancer Res. 2020;80(4):890–900. doi:10.1158/0008-5472.CAN-19-2830
57. Li Y, Zhao H, Huang X, et al. Embryonic senescent cells re-enter cell cycle and contribute to tissues after birth. Cell Res. 2018;28(7):775–778. doi:10.1038/s41422-018-0050-6
58. Yu Y, Schleich K, Yue B, et al. Targeting the senescence-overriding cooperative activity of structurally unrelated H3K9 demethylases in melanoma. Cancer Cell. 2018;33(2):322–336e8. doi:10.1016/j.ccell.2018.01.002
59. Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. doi:10.1016/j.cell.2008.03.039
60. Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877–884. doi:10.1038/ncb1448
61. Di X, Bright AT, Bellott R, et al. A chemotherapy-associated senescence bystander effect in breast cancer cells. Cancer Biol Ther. 2008;7(6):864–872. doi:10.4161/cbt.7.6.5861
62. Chavanet A, Hill KR, Jimenez-Andrade Y, Choo MK, White K, Park JM. Intracellular signaling modules linking DNA damage to secretome changes in senescent melanoma cells. Melanoma Res. 2020;30(4):336–347. doi:10.1097/CMR.0000000000000671
63. Canino C, Mori F, Cambria A, et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene. 2012;31(26):3148–3163. doi:10.1038/onc.2011.485
64. Angelini PD, Zacarias Fluck MF, Pedersen K, et al. Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Res. 2013;73(1):450–458. doi:10.1158/0008-5472.CAN-12-2301
65. Kim YH, Choi YW, Lee J, Soh EY, Kim J-H, Park TJ. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun. 2017;8(1). doi:10.1038/ncomms15208
66. Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012;3:e446. doi:10.1038/cddis.2012.183
67. Sun X, Shi B, Zheng H, et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018;9(3):260. doi:10.1038/s41419-018-0303-9
68. Alessio N, Aprile D, Squillaro T, et al. The senescence-associated secretory phenotype (SASP) from mesenchymal stromal cells impairs growth of immortalized prostate cells but has no effect on metastatic prostatic cancer cells. Aging (Albany NY). 2019;11(15):5817–5828. doi:10.18632/aging.102172
69. Lujambio A, Akkari L, Simon J, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153(2):449–460. doi:10.1016/j.cell.2013.03.020
70. Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67(7):3117–3126. doi:10.1158/0008-5472.can-06-3452
71. Chen P, Guo H, Chen J, Fu Y. The chemotherapeutic drug boanmycin induces cell senescence and senescence-associated secretory phenotype factors, thus acquiring the potential to remodel the tumor microenvironment. Anticancer Drugs. 2016;27(2):84–88. doi:10.1097/CAD.0000000000000304
72. Sieben CJ, Sturmlechner I, van de Sluis B, van Deursen JM. Two-step senescence-focused cancer therapies. Trends Cell Biol. 2018;28(9):723–737. doi:10.1016/j.tcb.2018.04.006
73. Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–658. doi:10.1111/acel.12344
74. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8(1):422. doi:10.1038/s41467-017-00314-z
75. Yosef R, Pilpel N, Tokarsky-Amiel R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190. doi:10.1038/ncomms11190
76. Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78–83. doi:10.1038/nm.4010
77. Rudin CM, Hann CL, Garon EB, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18(11):3163–3169. doi:10.1158/1078-0432.CCR-11-3090
78. Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. doi:10.1158/0008-5472.CAN-07-5836
79. Qi B, Hardwick JM. A Bcl-xL timer sets platelet life span. Cell. 2007;128(6):1035–1036. doi:10.1016/j.cell.2007.03.002
80. Munoz-Espin D, Rovira M, Galiana I, et al. A versatile drug delivery system targeting senescent cells. EMBO Mol Med. 2018;10(9). doi:10.15252/emmm.201809355
81. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15(3):428–435. doi:10.1111/acel.12445
82. Xu M, Pirtskhalava T, Farr JN, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24(8):1246–1256. doi:10.1038/s41591-018-0092-9
83. Zhu Y, Doornebal EJ, Pirtskhalava T, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY). 2017;9(3):955–963. doi:10.18632/aging.101202
84. Yousefzadeh MJ, Zhu Y, McGowan SJ, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018;36:18–28. doi:10.1016/j.ebiom.2018.09.015
85. Wang Y, Chang J, Liu X, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY). 2016;8(11):2915–2926. doi:10.18632/aging.101100
86. Wang F, Mao Y, You Q, Hua D, Cai D. Piperlongumine induces apoptosis and autophagy in human lung cancer cells through inhibition of PI3K/Akt/mTOR pathway. Int J Immunopathol Pharmacol. 2015;28(3):362–373. doi:10.1177/0394632015598849
87. Triana-Martinez F, Picallos-Rabina P, Da Silva-alvarez S, et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat Commun. 2019;10(1):4731. doi:10.1038/s41467-019-12888-x
88. Laberge RM, Sun Y, Orjalo AV, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–1061. doi:10.1038/ncb3195
89. Luo Y, Li L, Zou P, et al. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014;97(1):20–29. doi:10.1097/TP.0b013e3182a7fcf8
90. Moiseeva O, Deschenes-Simard X, St-Germain E, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell. 2013;12(3):489–498. doi:10.1111/acel.12075
91. Zhang B, Fu D, Xu Q, et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat Commun. 2018;9(1). doi:10.1038/s41467-018-04010-4
92. Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106(40):17031–17036. doi:10.1073/pnas.0905299106
93. Noren Hooten N, Martin-Montalvo A, Dluzen DF, et al. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell. 2016;15(3):572–581. doi:10.1111/acel.12469
94. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30(8):1536–1548. doi:10.1038/emboj.2011.69
95. Ruscetti M, Morris JP, Mezzadra R, et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell. 2020;181(2):424–441.e21. doi:10.1016/j.cell.2020.03.008
96. Fleury H, Malaquin N, Tu V, et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat Commun. 2019;10(1):2556. doi:10.1038/s41467-019-10460-1
97. Samaraweera L, Adomako A, Rodriguez-Gabin A, McDaid HM, Novel A. Indication for panobinostat as a senolytic drug in NSCLC and HNSCC. Sci Rep. 2017;7(1):1900. doi:10.1038/s41598-017-01964-1
98. Galiana I, Lozano-Torres B, Sancho M, et al. Preclinical antitumor efficacy of senescence-inducing chemotherapy combined with a nanoSenolytic. J Control Release. 2020;323:624–634. doi:10.1016/j.jconrel.2020.04.045
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.