Back to Journals » Clinical Interventions in Aging » Volume 9
The current research status of normal tension glaucoma
Received 6 May 2014
Accepted for publication 20 June 2014
Published 16 September 2014 Volume 2014:9 Pages 1563—1571
DOI https://doi.org/10.2147/CIA.S67263
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Xue-Song Mi,1,2 Ti-Fei Yuan,3,4 Kwok-Fai So2,4,5
1Department of Ophthalmology, The First Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China; 2Department of Anatomy, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, People’s Republic of China; 3School of Psychology, Nanjing Normal University, Nanjing, People’s Republic of China; 4Department of Ophthalmology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, People’s Republic of China; 5GHM Institute of CNS Regeneration, Jinan University, Guangzhou, People’s Republic of China
Abstract: Normal tension glaucoma (NTG) is a progressive optic neuropathy that mimics primary open-angle glaucoma, but lacks the findings of elevated intraocular pressure or other mitigating factors that can lead to optic neuropathy. The present review summarized the causes, genetics, and mechanisms underlying NTG in both animal models and human patients. We also proposed that the neurovascular unit is a therapeutic target for NTG management.
Keywords: aging, genetics, neurovascular unit, primary open-angle glaucoma, treatment
Clinical aspects
Normal tension glaucoma (NTG) is a progressive optic neuropathy that mimics primary open-angle glaucoma (POAG), but lacks the findings of elevated intraocular pressure (IOP) or other mitigating factors that can lead to optic neuropathy.1 The main clinical features include normal angles on gonioscopy, cupping of the optic nerve, and visual field loss correlated with cupping that shows the progressive damage of the optic nerve.1–3
Epidemiologically, several risk factors have been shown to be related to NTG, the first of which is age. The mean age in years of patients with NTG reported in many studies is in the 60s.1,4 The second risk factor is sex. Some studies have suggested that there is a greater population of women with NTG than men;4 however, this phenomenon may be due to the longer life span of females compared to males.5 The third risk factor is race. The incidence of NTG in different populations is not the same. It has been reported that there is a higher incidence of NTG in Asian populations, such as Japanese, compared to European populations. As many as two-thirds of glaucoma instances in Japan may be NTG.6 Although the relationship between NTG and other diseases is not clear, some systemic diseases have been reported to concur with NTG, such as vascular disease, migraine, vasospasm, and immune-related diseases.2 The diagnosis of NTG is a kind of exclusion, which means that the diagnosis is used to exclude other possible etiologies of optic nerve cupping with or without visual field loss when NTG is suspected. The first step in diagnosis is to rule out chronic anemia, cardiopathies, acute blood loss, episodes of systemic hypotension, decreased cerebral blood flow, blood dysplasias, neurosyphilis, etc, from the medical history. The second step in diagnosis is to rule out other glaucomas through a reliable IOP reading, angle examination by gonioscopy, and the status of fundus. The third step in diagnosis is to perform visual field (VF) testing to confirm whether there are specific glaucomatous VF defects.7
For the management of NTG, the initial approach is to observe if there is any documented progression of the disease, which includes the following indications: 1) signs of change of retinal nerve fiber layer, optic peri-papilla, and visual fields; 2) family history of NTG with rapid progression; 3) visual symptoms suspected to have had any progression; and 4) recurrent optic disc hemorrhages.4 Although there is no elevation of IOP in NTG, IOP reduction is considered the essential treatment. It has been reported that a 25%–30% reduction of IOP from baseline is the initial target to slow the disease.4 However, another report suggested that, even with a 30% reduction of IOP, progress still occurred in a significant proportion of cases (40% after 4 years).8 Therefore, other IOP-independent treatments are needed. Calcium-channel blockers have been used in a clinical study to increase optic nerve head perfusion. Recently, the neuroprotection approach has been introduced to rescue the survival of neurons in neurodegenerative diseases of the eye and brain, including glaucoma. Many agents are considered promising in animal studies, although there is not yet reliable evidence to show that they are beneficial to human patients.9
The basic research and clinical investigations of NTG
Genetics of NTG
Etiologically, the genetic background of most types of glaucoma is complex. However, many ongoing studies have shown the inherited trait with some gene mutation in NTG patients. Scanning for gene mutation and association with NTG will benefit the understanding of the specific function of the genes involved in the mechanism of NTG. Below are listed some of the identified genes and their related functions in the development of NTG.
A polymorphism of the endothelin receptor A gene has been found to be associated with NTG,10,11 which suggests the involvement of endothelin-1 (ET-1) signaling pathways in the development of NTG. Optic atrophy type 1 (OPA1) gene is reported to be related to NTG.12 It has been suggested that the mitochondrial OPA1 could provide defense of retinal ganglion cells (RGCs) from pressure-mediated retinal damage.13 Altered OPA1 gene expression could directly induce apoptotic cell death in cultured RGC-5 cells.14 Mutation in the optineurin (OPTN) gene was reported to be associated with NTG in the hereditary POAG family.15 The functional role of OPTN, as described in the literature, included nuclear factor kappa B regulation, vesicular trafficking, immune response, etc. The mutated OPTN gene could induce mutation of E50K and H486R genes, resulting in death of RGC-5 cells by weakness of antioxidation.16 There have been reports of increasing candidate-gene alterations in the NTG population worldwide, such as p53 gene,17 TLR4,18 MFN1,19 MFN2,19 PARL,19 GLC1F,20 SRBD1,21 and ELOVL5.21 These reports indicate that all of the gene alterations should be due to IOP-independent factors that are involved in some specific signaling pathway to target the degenerating events of RGCs or their axons in the development of NTG.
As of now, some of these genes have already been investigated, and clear gene functions, which have been confirmed with studies using tissue culture and animal models, have been identified; others have been recently discovered and do not yet have a clearly identified gene function. However, for many of the genes, these is still a lack of large demographic data that would support their classification as major risk factors for developing NTG. Future investigations will continue to identify more new genes, and will clarify the roles of the mutated genes and the relationship that they have with the outside environment.
Animal models related to the development of NTG
As many gene mutations have already been reported to be associated with NTG in demographic studies, increasing reports on transgenic mice with the phenotypes related to human glaucoma have been published (Table 1).22–24 On the other hand, there are still many published NTG mouse models without confirmed genetic relation to human NTG.25–28,30 However, all studies are helpful in understanding the pathogenesis of NTG. Recent discoveries from studies of these transgenic mice are listed below.
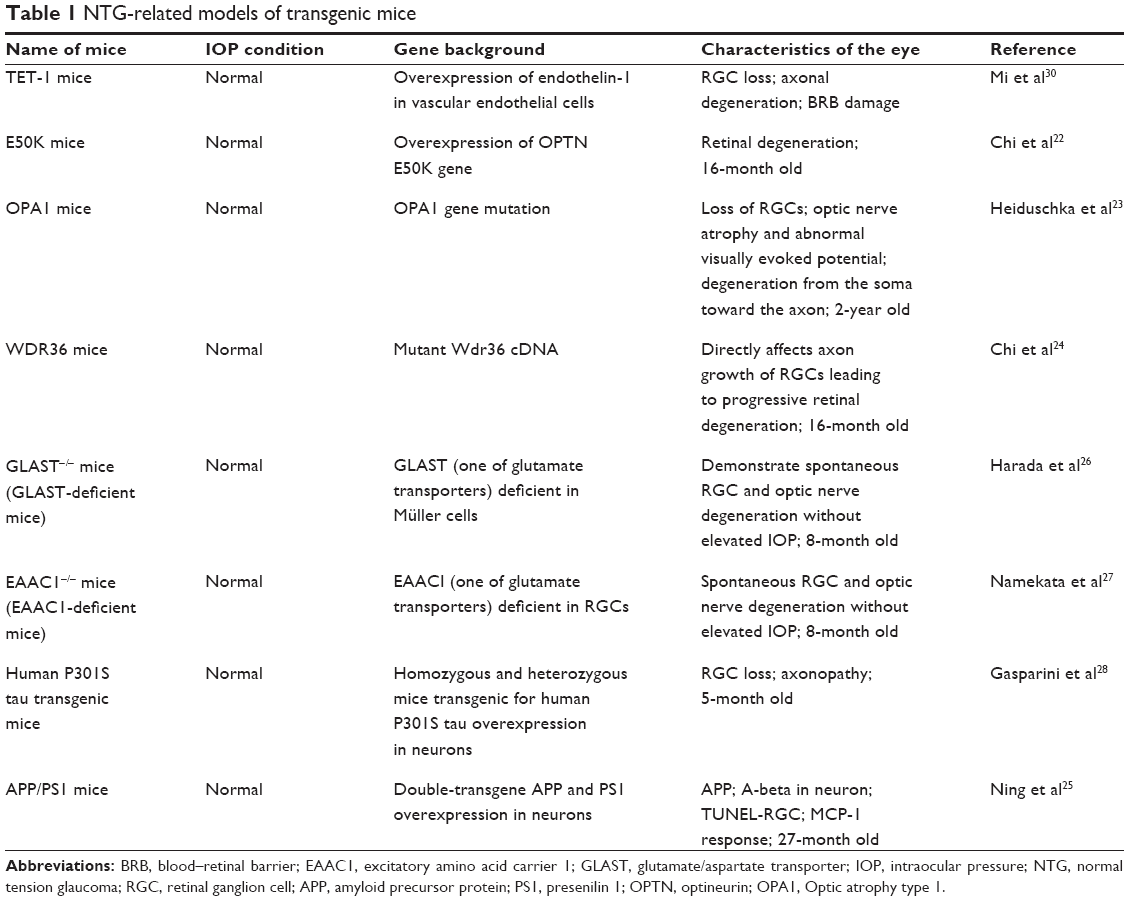 | Table 1 NTG-related models of transgenic mice |
OPTN gene mutation was one of the reported NTG-associated genes.16 Correspondingly, the transgenic mice with over-expression of OPTN (containing E50K point mutants) showed significant retinal degeneration.22 OPA1 is another reported human NTG-causing gene. OPA1 gene-mutated mice were showed with loss of RGCs, optic nerve atrophy, and abnormal visually evoked potential.23 WD repeat-containing protein 36 (WDR36)24 has been identified as being associated with POAG.29 It was reported that the mutant WDR36 could directly affect the axon growth of RGCs, leading to progressive retinal degeneration in mice.24
There are some transgenic mice for which the gene mutations have not been reported to be related to human NTG, but that hold the similar phenotypes. These include glutamate/aspartate transporter-deficient mice, which lack glutamate transporters glutamate/aspartate transporter,26 and excitatory amino acid carrier 1-deficient mice.27 More recently, some Alzheimer’s disease (AD)-related transgenic mice, such as P301 mice28 and APP/PS1 (amyloid precursor protein/ presenilin 1) mice,25 have been reported to have RGC loss and axonopathy without elevated IOP. These mice will be useful in understanding the AD-like pathogenic mechanisms in glaucoma and NTG.
Due to the large amount of clinical evidence for the association of ischemic optic neuropathy with NTG,4,30 there is an over-expression of ET-1 experimental induced animal model related to the development of NTG that is mainly focused on the ischemic-associated mechanisms. Utilizing microapplication of ET-1, Cioffi et al32 developed a chronic optic nerve ischemia model in monkeys. Optic neuropathy with associated optic nerve cupping was produced using a similar method in rabbit eyes that could resemble the clinical findings in NTG. To date, extraneous application of ET-1 to induce ischemic optic neuropathy has been reported in many kinds of animals, such as Rhesus monkeys, rabbits, and SD rats, with administration of different dosages and through different methods of administration.32–35 However, extraneous delivery of ET-1 does not fully mimic the real effect of the stable and persistent endogenous functions produced by animals. After all, NTG is a kind of chronic disease, which affects the elderly more than the young. Investigation on the long-term effect of overexpression of ET-1 may provide insight into the mechanism of NTG. Our previous study reported that NTG-like retinal degeneration appeared progressively more with age in transgenic mice, with over-expression of ET-1 in vascular endothelial cells (TET-1 mice). The phenotype of TET-1 mice involves major morphological changes, including the loss of RGCs, degeneration of optic axons, blood vessel changes, and retinal gliosis, which was helpful in understanding the effect of long-term exposure of endothelial ET-1 in glaucoma and other vascular-related retinal degeneration.30
The role of ocular blood flow (OBF) in NTG
The relationship of OBF, blood pressure (BP), and NTG is an important topic in this area of research. High BP is a risk factor in high-tension glaucoma (HTG), as is evidenced by the positive correlation between systolic BP and IOP.37 On the other hand, low BP, especially the nocturnal drop of BP, has been shown to have a greater prevalence in those with NTG.38,39 Low BP will decrease ocular perfusion pressure by the dynamic formula of pressure,40 where the corresponding result is the reduction of OBF, and thus increases the risk of development or progression of NTG.
Assessing OBF is important in evaluating its role in NTG; however, because the blood supply to the back segment of the eye comes from two sets of vascular systems (the retinal and the choroidal system), measurement of OBF is difficult. To date, there is not a single comprehensive technique to assess OBF. Different measurements provide different details of vascular parameters, and can be interpreted differently. Thus, accuracy and multifunction are the trends of the prospective application of current OBF assessment approaches. Flammer et al41 summarized the OBF measuring techniques that applied in clinic and animal research. Below are listed some updated OBF tools and their applications.
The most popular method in clinical settings is color Doppler imaging. It is used to assess the blood flow velocity of retrobulbar vessels using resistivity index as the parameter. A higher value of resistivity index represents greater resistance of vasculature, indicating deleterious significance.42 Lack of quantification of vessel diameter in this method limits the application in terms of assessing OBF directly. Another device is the Langham OBF system, which measures pulsatile OBF of choroidal perfusion by monitoring the rhythmic change of IOP during the cardiac cycle.43 Combining the technique of confocal scanning laser tomography with laser Doppler flowmetry, the method of confocal scanning laser Doppler flowmetry, or Heidelberg retinal flowmetry, can assess the capillary blood flow of inner retinal layers and the optic disk.44 Angiography, including fluorescein fundus angiography and indocyanine green, can provide information on velocity of blood flow in the retina and choroid by visualizing perfusion of ocular vessels.45 The Canon laser Doppler blood flowmeter (Canon Inc., Tokyo, Japan) provides assessments of both vessel diameter and blood speed of retinal blood flow.46 Color Doppler optical coherence tomography couples the protocols of color Doppler with those of optical coherence tomography and provides analysis of the speed of OBF within the visualized eye tissue.47 The updated retinal oximeters provide convenient assessment of retinal vessel oxygenation associated with a pseudocolor fundus image and are easier to perform, however, not all oximeters can measure OBF.48
There are many other approaches still used in clinic settings to assess different parts of ocular vasculature and to indicate particular information related to OBF (eg, the laser speckle phenomenon49 and the blue field entoptic technique).50 The general limitation of these techniques is their single function, which cannot provide as much information as the other multifunctional instruments described above.
Experimental methods are important in investigating the role of OBF in the mechanism of glaucoma in animals. The above assessments can also be used as nonintrusive techniques on living animals; however, in some small-sized animals, such as rodents, many of the methods are not validated. The methods used currently for rodents include the microsphere method51 to detect the relative intensity of OBF, vascular casting techniques,52 and the ink-perfusion method.53 Vascular casting techniques and the ink-perfusion method are aimed at visualizing the alteration of retinal vasculature in order to reflect the change of OBF. Overall, even if there has been no uniform standard to date, different methods could still provide much detailed information related to the alteration of OBF in NTG.43
In summary, OBF has various roles in NTG: 1) OBF is an indicator for evaluating vascular dysregulation; 2) reduction of OBF suggests the presence of ischemic mechanism in NTG patients; and 3) assessment of OBF can be used as an indicator in vascular protective therapy in NTG.
IOP-independent mechanisms in NTG
The damaging factors involved in NTG
Although IOP plays a predominant role in glaucoma, either in HTG or NTG, IOP-independent factors (such as oxidative stress, glutamate toxicity, vascular factors, etc) still play important roles in the development of NTG. Among IOP-independent, vascular factors have been most attended to.54 Further, these vascular factors have been classified as either systemic vascular risk factors, such as nocturnal hypotension and vasospasm, or local risk factors, such as disc hemorrhages, peripapillary atrophy, and choroidal sclerosis.55,56 The latter classification is considered to be more important than the former, for a more direct relationship with the glaucomatous process.57 The presence of decreased OBF in NTG patients3 and the prevalent incidence of NTG among those with vasospasm55,56 provide overwhelming evidences to indicate the significance of vascular factors in the development or progression of NTG. Flammer58 uses the term “vascular dysregulation” in reference to the mechanism of glaucoma, and suggested that vascular dysregulation as an initial damage in the mechanism of NTG,59 which led to the unstable OBF in NTG.59 Vascular dysregulation increases the impact of ischemia, affecting not only retinal tissue but also the optic nerve, and contributes to the degeneration of RGCs and their axons. Under the conditions of ischemia, many vascular-related factors have been shown to be involved in the pathogenesis of NTG, eg, oxidative stress, ET-1, glutamine, vasogenic cytokines, etc.60
Aging is another important risk factor in demographic studies of glaucoma,2 especially in NTG.30,61 With regard to senescence, glaucoma has been considered an age-related disease. With regard to cytology, aging has been proposed to play a role in contributing to the damage of nuclear and mitochondrial DNA in mammals.62 Many clinical and experimental findings have shown that aging-related factors are involved in the mechanism of glaucoma. Amyloid-β (Aβ), the major neuronal toxic factor in AD, is one of the molecular links between glaucoma and neurodegenerative disease of central nervous system. Recently, expression of Aβ and amyloid precursor protein (APP) was observed in the retina and optic nerve in experiment-induced chronic ocular hypertension rats,63 chronic ocular hypertension mice,64 and old-DBA/2J mice.65 In a mouse model of AD using mutated APP transgenic mice, apoptosis of RGCs was also found to be associated with over-expression of Aβ.25 These data suggest that Aβ plays a role in apoptosis of RGCs, with or without elevated IOP. In addition, some studies suggest that ischemia could promote the deposition of Aβ in tissue by increasing the production from APP and through decreased cleaning via the blood vessels,66 indicating that similar mechanism might be involved in NTG. In retinal samples from patients with POAG or NTG, the immunoreactivity of advanced glycation end products and their receptor, RAGE, have been found, which suggested the process of aging was accelerated in POAG and NTG.67
Flammer and Mozaffarieh summarized the pathogenesis of glaucomatous optic neuropathy.59 The causative factors in the mechanism of NTG in relation to neurodegenerative disease using the concept of neurovascular dysfunction are summarized below. The term neurovascular unit refers to a collection of different cells around blood vessels, including neurons, glia cells, and vascular cells. These cells come together to form a structural and functional integrated unit to maintain the balance of the microenvironment outside of the cells (Figure 1).60,66,68,69 In normal conditions, the neurovascular unit has the function of regulating OBF via the end-feet activity of astrocytes,70 the autoregulating blood vessels,59 and regulating some special molecules, such as nitric oxide (NO), ET-1, etc.71 Other functions include controlling the exchange across the blood–brain barrier, immune surveillance, hemostatic balance, and trophic function.66 Neurovascular dysfunction is characterized by multiple structural alterations associated with disruption of neurovascular unit functions.
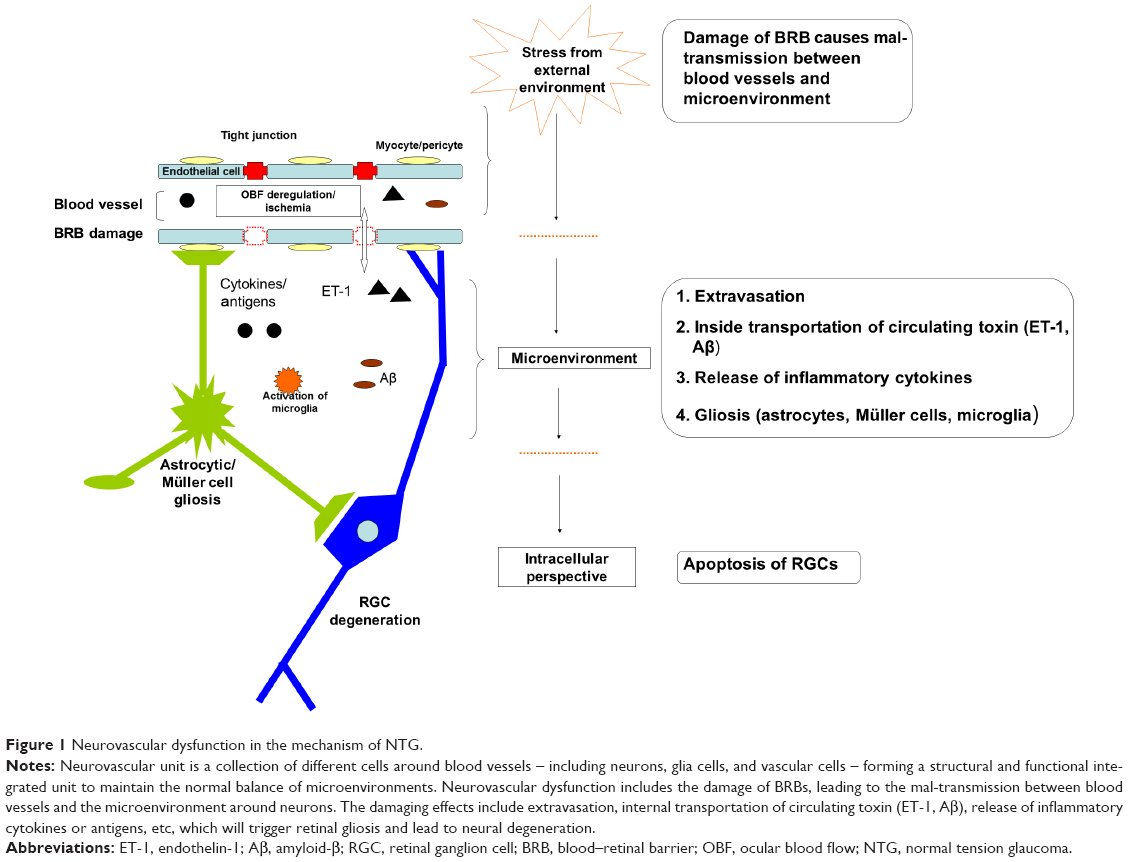 | Figure 1 Neurovascular dysfunction in the mechanism of NTG. |
The first course or process of neurovascular dysregulation is vascular dysfunction, consisting of vascular oxidative stress and inflammation, which reduces the OBF and thereby aggravates ischemia.60,72 The second course or process of neurovascular dysregulation is damage of blood–retinal barriers (BRB), including protein extravasation,73 the transportation of Aβ across the blood vessel to tissue,74 and release of ET-1 and matrix metalloproteinase 9.75,76 All of these factors could contribute to the degeneration of RGCs and to the release of inflammatory cytokines or antigens, which might trigger the immune response and could be the cause of the involvement of the autoimmune mechanism.77 There is increasing evidence that indicates the presence of BRB damage in glaucoma, including NTG,78–81 where the BRB damage is also suggested to be a cause of optic disc hemorrhage.82 The third course or process of neurovascular dysregulation is gliosis. Reactivated glial cells, such as astrocyte, can release NO and TNF-α, provoking many deleterious signaling pathways that are involved in the neurodegeneration of glaucoma, which has been discussed in previous studies.77,83,84 The last course or process of neurovascular dysregulation is loss of trophic support. The condition of neurovascular dysfunction disrupts the bioactivity and signaling of growth factors, such as vascular endothelial growth factor and brain-derived neurotrophic factor, which has an impact on the normal function and survival of the cells in the neurovascular unit.85–88
Apoptosis of RGCs from the extrinsic and intracellular signaling pathways
It is now generally accepted that apoptosis is the major pathway of RGC death in glaucoma.89 Both the extrinsic and intracellular pathways of apoptosis are involved in RGC death. The former pathway can be mediated via the death receptor of the TNF super-family on the cell membrane,89 and the latter is mediated by mitochondrion.90 The NTG-related models of transgenic mice are listed in Table 1. Some of the mutated genes or abnormally expressed proteins could affect the apoptosis of RGCs, and would therefore be involved in the mechanisms of NTG,26,27 although proof is needed.
Thus, in regard to vascular dysfunction, one possible mechanism in NTG could be that which is summarized below and in Figure 1. The risk factors, such as vascular factors, aging-related factors, etc, disrupt the BRB leading to neurovascular dysfunction, which activates the response of endothelial cells and glial cells, and induces direct insult to neurons; then, more damaging factors could be released from the BRB into the microenvironment of the neurovascular unit to provoke an intracellular event, such as apoptosis, resulting in the degeneration of RGCs. A previous study91 summarized that neurodegeneration in glaucoma goes through two phases: direct damage to RGC and axons, and secondary damage by responses of nonneuronal cells. The secondary damage is considered to be the major cause of RGC loss in glaucoma.91 Thus, it is reasonable to target the rescue of the vascular from dysfunction and control of gliosis. Doing so would maintain the microenvironment of the neurovascular unit, which could be the essential therapeutic mechanism for NTG.
The current concepts regarding the relationship between NTG and HTG
The commonly held categorization of NTG is as a subtype of POAG.1 However, there is evidence to show the distinction between NTG and POAG.3,30,92 Results for current investigations have offered both similarity and difference between NTG and POAG.3,30,92
Clinically, NTG was initially described from clinical features of glaucoma. Thus, similar to POAG, the main management for NTG is to reduce IOP,93 which has been shown to slow the progress of the disease. Some studies on the cornea showed the corneal thickness of NTG patients is thinner than normal, resulting in the lower IOP reading, which suggested that relatively high IOP status may present in NTG patients. Thus, the condition in NTG is similar to the conditions in POAG after adjusting the IOP reading according to the relative corneal thickness.94,95 However, there is other evidence observed in NTG patients that differs from POAG patients, eg, the incidence of optic disk hemorrhage is higher, the decrease of retinal nerve fiber layer is earlier, and the shape of defect of VF is different.96 Although, these observations were controversial,71 it still hints at the significance of the distinction of NTG under its phenotype. Most researchers believe that the causative factor is the alteration of OBF.3,41,97 Strong evidence showed the presence of vascular problems or abnormal perfusion in NTG patients.93 Other evidence suggested a populational relationship of systemic vascular diseases with NTG patients,98,99 which was not shown in POAG patients.
With regard to pathophysiological mechanisms, the similarity and difference between NTG and POAG still coexist. Based on the vascular theory of glaucoma, if there is no elevation of IOP, other causative factors related to vascular abnormality could lead to glaucomatous change.41,54 IOP-independent factors play an important role in the mechanism of NTG, which is different from the dominant role of high IOP for the mechanical mechanism in the development of POAG.59 However, basic research regards both of them as neurodegenerative diseases, where there is a similar signaling pathway of apoptosis resulting in the loss of RGCs.59,100 In addition, similar cellular or protein responses, such as gliosis, ET-1 increase, glutamate toxicity, oxidative stress, etc, are involved in the mechanism.101,102 Even similar gene mutation is found both in NTG and POAG based on the epidemiological evidence.103–105 Both NTG and POAG are prevalent in senile populations.61,93 Some studies showed evidence that the anti-Alzheimer agents exhibit benefit to POAG patients.106,107 Thus, presently, more and more researchers consider glaucomas as a group of aging-related disease similar to neurodegenerative disease in the central nervous system.108,109
Up to now, because the definite mechanism of POAG is still unclear, it is not the appropriate time to separate completely NTG from POAG. Maybe the initial causative factor is different. It is likely that NTG is due to vascular abnormality and POAG is due to increased IOP. However, neurodegeneration in the retina and optic axons is the common final pathway in both diseases. After all, the mechanism and the therapy for both diseases are clearly related to each other.
From the above depiction, in the research field of NTG, there are many issues that need to be further investigated, for example, gene-scanning works of demography in different races aiming to discover more genetic characteristics of NTG patients. Then, more lines of transgenic mice should be used to mimic the disease course of NTG to provide more chances to discover some specific mechanisms and therapeutic interventions in vivo. Clarification of the mechanism is proposed to find some special biomarkers, which will be beneficial to the early diagnosis of NTG in the clinical setting. In addition, more attention should be paid to the treatments concerning antiaging and blood vessel protection as a kind of neuroprotection in NTG.
Acknowledgments
Our study was supported by the Natural Science Foundation of China 2013 (No. 81300766), the National Program on Key Basic Research Project of China (973 Program: 2011CB707501), the Cultivation and Innovation Fund from Jinan University (No. 21613311), and the Cultivation and Innovation Fund from the First Affiliated Hospital of Jinan University (No. 2013203).
Disclosure
The authors report no conflicts of interest in this work.
References
Cantor LB, WuDunn D. 11 Normal-tension glaucoma. In: Color Atlas of Glaucoma. 1998:155. | ||
Shields MB. Textbook of glaucoma. Vol 3. The University of Michigan: Williams & Wilkins; 1998. | ||
Shields MB. Normal-tension glaucoma: is it different from primary open-angle glaucoma? Curr Opin Ophthalmol. 2008;19(2):85–88. | ||
GLAUCOMA RTOOA. Normal Tension Glaucoma. Primary care of the glaucomas. 2000:113. | ||
Klein BE, Klein R, Sponsel WE, et al. Prevalence of glaucoma. The Beaver Dam Eye Study. Ophthalmology. 1992;99(10):1499–1504. | ||
Levene RZ. Low tension glaucoma: a critical review and new material. Surv Ophthalmol. 1980;24(6):621–664. | ||
Orgül S, Zawinka C, Gugleta K, Flammer J. Therapeutic strategies for normal-tension glaucoma. Ophthalmologica. 2000;219(6):317–323. | ||
Membrey WL, Bunce C, Poinoosawmy DP, Fitzke FW, Hitchings RA. Glaucoma surgery with or without adjunctive antiproliferatives in normal tension glaucoma: 2 Visual field progression. Br J Ophthalmol. 2001;85(6):696–701. | ||
Weinreb RN, Levin LA. Is neuroprotection a viable therapy for glaucoma? Arch Ophthalmol. 1999;117(11):1540–1544. | ||
Ishikawa K, Funayama T, Ohtake Y, et al. Association between glaucoma and gene polymorphism of endothelin type A receptor. Mol Vis. 2005;11:431–437. | ||
Kim SH, Kim JY, Kim DM, et al. Investigations on the association between normal tension glaucoma and single nucleotide polymorphisms of the endothelin-1 and endothelin receptor genes. Mol Vis. 2006;12: 1016–1021. | ||
Mabuchi F, Tang S, Kashiwagi K, Yamagata Z, Iijima H, Tsukahara S. The OPA1 gene polymorphism is associated with normal tension and high tension glaucoma. Am J Ophthalmol. 2007;143(1):125–130. | ||
Dai Y, Weinreb RN, Kim KY, et al. Inducible nitric oxide synthase-mediated alteration of mitochondrial OPA1 expression in ocular hypertensive rats. Invest Ophthalmol Vis Sci. 2011;52(5): 2468–2476. | ||
Ju WK, Kim KY, Lindsey JD, et al. Elevated hydrostatic pressure triggers release of OPA1 and cytochrome C, and induces apoptotic cell death in differentiated RGC-5 cells. Mol Vis. 2009;15:120–134. | ||
Alward WL, Kwon YH, Kawase K, et al. Evaluation of optineurin sequence variations in 1,048 patients with open-angle glaucoma. Am J Ophthalmol. 2003;136(5):904–910. | ||
Chalasani ML, Swarup G, Balasubramanian D. Optineurin and its mutants: molecules associated with some forms of glaucoma. Ophthalmic Res. 2009;42:176–184. | ||
Wiggs JL. Genetic etiologies of glaucoma. Arch Ophthalmol. 2007; 125(1):30–37. | ||
Shibuya E, Meguro A, Ota M, et al. Association of Toll-like receptor 4 gene polymorphisms with normal tension glaucoma. Invest Ophthalmol Vis Sci. 2008;49(10):4453–4457. | ||
Wolf C, Gramer E, Müller-Myhsok B, et al. Evaluation of nine candidate genes in patients with normal tension glaucoma: a case control study. BMC Med Genet. 2009;10:91. | ||
Murakami K, Meguro A, Ota M, et al. Analysis of microsatellite polymorphisms within the GLC1F locus in Japanese patients with normal tension glaucoma. Mol Vis. 2010;16:462–466. | ||
Writing Committee for the Normal Tension Glaucoma Genetic Study Group of Japan Glaucoma Society1; Meguro A, Inoko H, Ota M, Mizuki N, Bahram S. Genome-wide association study of normal tension glaucoma: common variants in SRBD1 and ELOVL5 contribute to disease susceptibility. Ophthalmology. 2010;117:1331–1338. | ||
Chi ZL, Akahori M, Obazawa M, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet. 2010;19(13):2606–2615. | ||
Heiduschka P, Schnichels S, Fuhrmann N, et al. Electrophysiological and histologic assessment of retinal ganglion cell fate in a mouse model for OPA1-associated autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci. 2010;51(3):1424–1431. | ||
Chi ZL, Yasumoto F, Sergeev Y, et al. Mutant WDR36 directly affects axon growth of retinal ganglion cells leading to progressive retinal degeneration in mice. Hum Mol Genet. 2010;19(19):3806–3815. | ||
Ning A, Cui J, To E, Ashe KH, Matsubara J. Amyloid-beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci. 2008;49(11):5136–5143. | ||
Harada T, Harada C, Nakamura K, et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J Clin Invest. 2007;117(7):1763–1770. | ||
Namekata K, Harada C, Guo X, et al. Interleukin-1 attenuates normal tension glaucoma-like retinal degeneration in EAAC1-deficient mice. Neurosci Lett. 2009;465(2):160–164. | ||
Gasparini L, Crowther RA, Martin KR, et al. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging. 2011;32:419–433. | ||
Stone EM, Fingert JH, Alward WL, et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275(5300):668–670. | ||
Mi XS, Zhang X, Feng Q, Lo AC, Chung SK, So KF. Progressive retinal degeneration in transgenic mice with overexpression of endothelin-1 in vascular endothelial cells. Invest Ophthalmol Vis Sci. 2012;53(8):4842–4851. | ||
Buono LM, Foroozan R, Sergott RC, Savino PJ. Is normal tension glaucoma actually an unrecognized hereditary optic neuropathy? New evidence from genetic analysis. Curr Opin Ophthalmol. 2002; 13(6):362–370. | ||
Cioffi GA, Orgül S, Onda E, Bacon DR, Van Buskirk EM. An in vivo model of chronic optic nerve ischemia: The dose-dependent effects of endothelin-l on the optic nerve microvasculature. Curr Eye Res. 1995; 14(12):1147–1153. | ||
Masuzawa K, Jesmin S, Maeda S, et al. A model of retinal ischemia-reperfusion injury in rats by subconjunctival injection of endothelin-1. Exp Biol Med (Maywood). 2006;231(6):1085–1089. | ||
Sugiyama T, Mashima Y, Yoshioka Y, Oku H, Ikeda T. Effect of unoprostone on topographic and blood flow changes in the ischemic optic nerve head of rabbits. Arch Ophthalmol. 2009;127(4):454–459. | ||
Nagano H, Wei PZ, Wen CQ, et al. Effects of kallidinogenase on ischemic changes induced by repeated intravitreal injections of endothelin-1 in rabbit retina. Curr Eye Res. 2007;32(2):113–122. | ||
Taniguchi T, Shimazawa M, Sasaoka M, Shimazaki A, Hara H. Endothelin-1 impairs retrograde axonal transport and leads to axonal injury in rat optic nerve. Curr Neurovasc Res. 2006;3(2):81–88. | ||
Dielemans I, Vingerling JR, Algra D, Hofman A, Grobbee DE, de Jong PT. Primary open-angle glaucoma, intraocular pressure, and systemic blood pressure in the general elderly population. Ophthalmology. 1995;102(1):54–60. | ||
Hayreh SS, Zimmerman MB, Podhajsky P, Alward WL. Nocturnal arterial hypotension and its role in optic nerve head and ocular ischemic disorders. Am J Ophthalmol. 1994;117(5):603–624. | ||
Meyer JH, Brandi-Dohrn J, Funk J. Twenty four hour blood pressure monitoring in normal tension glaucoma. Br J Ophthalmol. 1996;80(10): 864–867. | ||
Deokule S, Weinreb RN. Relationships among systemic blood pressure, intraocular pressure, and open-angle glaucoma. Can J Ophthalmol. 2008;43(3):302–307. | ||
Flammer J, Orgül S, Costa VP, et al. The impact of ocular blood flow in glaucoma. Prog Retin Eye Res. 2002;21(4):359–393. | ||
Ehrlich R, Harris A, Siesky BA, et al. Repeatability of retrobulbar blood flow velocity measured using color Doppler imaging in the Indianapolis Glaucoma Progression Study. J Glaucoma. 2011;20:540–547. | ||
Harris A, Kagemann L, Ehrlich R, Rospigliosi C, Moore D, Siesky B. Measuring and interpreting ocular blood flow and metabolism in glaucoma. Can J Ophthalmol. 2008;43:328–336. | ||
Chauhan BC, Smith FM. Confocal scanning laser Doppler flowmetry: experiments in a model flow system. J Glaucoma. 1997;6(4): 237–245. | ||
Arend O, Harris A, Martin BJ, Remky A. Scanning laser ophthalmoscopy-based evaluation of epipapillary velocities:: method and physiologic variability. Surv Ophthalmol. 1999;44(Suppl 1):S3–S9. | ||
Feke GT, Goger DG, Tagawa H, Delori FC. Laser Doppler technique for absolute measurement of blood speed in retinal vessels. IEEE Trans Biomed Eng. 1987;34(9):673–680. | ||
Ben-nun J. Comparative flow velocity of erythrocytes and leukocytes in feline retinal capillaries. Invest Ophthalmol Vis Sci. 1996;37(9): 1854–1859. | ||
Hardarson SH, Harris A, Karlsson RA, et al. Automatic retinal oximetry. Invest Ophthalmol Vis Sci. 2006;47(11):5011–5016. | ||
Tamaki Y, Araie M, Hasegawa T, Nagahara M. Optic nerve head circulation after intraocular pressure reduction achieved by trabeculectomy. Ophthalmology. 2001;108(3):627–632. | ||
Riva CE, Petrig B. Blue field entoptic phenomenon and blood velocity in the retinal capillaries. J Opt Soc Am. 1980;70(10):1234–1238. | ||
Wang L, Fortune B, Cull G, McElwain KM, Cioffi GA. Microspheres method for ocular blood flow measurement in rats: size and dose optimization. Exp Eye Res. 2007;84(1):108–117. | ||
Morrison JC, Cepurna WO, Johnson EC. Modeling glaucomatous optic nerve damage. Int Ophthalmol Clin. 1999;39(3):29–41. | ||
Jianbin T, Liang H, Jufang H, et al. Improved method of ink-gelatin perfusion for visualising rat retinal microvessels. Acta Histochem Cytochem. 2008;41(5):127–133. | ||
Yamamoto T, Kitazawa Y. Vascular pathogenesis of normal-tension glaucoma: a possible pathogenetic factor, other than intraocular pressure, of glaucomatous optic neuropathy. Prog Retin Eye Res. 1998; 17(1):127–143. | ||
Gasser P, Flammer J, Guthauser U, Mahler F. Do vasospasms provoke ocular diseases? Angiology. 1990;41(3):213–220. | ||
Pache M, Dubler B, Flammer J. Peripheral vasospasm and nocturnal blood pressure dipping – two distinct risk factors for glaucomatous damage? Eur J Ophthalmol. 2003;13(3):260–265. | ||
Béchetoille A. Vascular risk factors in glaucoma. Curr Opin Ophthalmol. 1996;7(2):39–43. | ||
Flammer J. The vascular concept of glaucoma. Surv Ophthalmol. 1994;38 Suppl:S3–S6. | ||
Flammer J, Mozaffarieh M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Surv Ophthalmol. 2007;52(Suppl 2):S162–S173. | ||
Carvey PM, Hendey B, Monahan AJ. The blood–brain barrier in neurodegenerative disease: a rhetorical perspective. J Neurochem. 2009; 111(2):291–314. | ||
Fournier AV, Damji KF, Epstein DL, Pollock SC. Disc excavation in dominant optic atrophy: differentiation from normal tension glaucoma. Ophthalmology. 2001;108(9):1595–1602. | ||
Vijg J. Somatic mutations and aging: a re-evaluation. Mutat Res. 2000; 447(1):117–135. | ||
McKinnon SJ, Lehman DM, Kerrigan-Baumrind LA, et al. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Invest Ophthalmol Vis Sci. 2002;43(4):1077–1087. | ||
Kipfer-Kauer A, McKinnon SJ, Frueh BE, Goldblum D. Distribution of amyloid precursor protein and amyloid-beta in ocular hypertensive C57BL/6 mouse eyes. Curr Eye Res. 2010;35(9):828–834. | ||
Goldblum D, Kipfer-Kauer A, Sarra GM, Wolf S, Frueh BE. Distribution of amyloid precursor protein and amyloid-beta immunoreactivity in DBA/2J glaucomatous mouse retinas. Invest Ophthalmol Vis Sci. 2007;48(11):5085–5090. | ||
Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010; 120:287–296. | ||
Tezel G, Luo C, Yang X. Accelerated aging in glaucoma: immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Invest Ophthalmol Vis Sci. 2007; 48(3):1201–1211. | ||
Pournaras CJ, Rungger-Brändle E, Riva CE, Hardarson SH, Stefansson E. Regulation of retinal blood flow in health and disease. Prog Retin Eye Res. 2008;27(3):284–330. | ||
Popescu BO, Toescu EC, Popescu LM, et al. Blood–brain barrier alterations in ageing and dementia. J Neurol Sci. 2009;283(1–2):99–106. | ||
Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10(11):1369–1376. | ||
Geijssen HC, Greve EL. Vascular concepts in glaucoma. Curr Opin Ophthalmol. 1995;6(2):71–77. | ||
Faraci FM. Reactive oxygen species: influence on cerebral vascular tone. J Appl Physiol (1985). 2006;100(2):739–743. | ||
Farrall AJ, Wardlaw JM. Blood–brain barrier: ageing and microvascular disease – systematic review and meta-analysis. Neurobiol Aging. 2009; 30(3):337–352. | ||
Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med. 2003;9(7):907–913. | ||
Cleary C, Buckley CH, Henry E, McLoughlin P, O’Brien C, Hadoke PW. Enhanced endothelium derived hyperpolarising factor activity in resistance arteries from normal pressure glaucoma patients: implications for vascular function in the eye. Br J Ophthalmol. 2005;89(2):223–228. | ||
Golubnitschaja O, Yeghiazaryan K, Liu R, et al. Increased expression of matrix metalloproteinases in mononuclear blood cells of normal-tension glaucoma patients. J Glaucoma. 2004;13(1):66–72. | ||
Tezel G, Wax MB. The immune system and glaucoma. Curr Opin Ophthalmol. 2004;15(2):80–84. | ||
Schwartz B, Rieser JC, Fishbein SL. Fluorescein angiographic defects of the optic disc in glaucoma. Arch Ophthalmol. 1977;95(11):1961–1974. | ||
Tsukahara S. Hyperpermeable disc capillaries in glaucoma. Adv Ophthalmol. 1978;35:65–72. | ||
Arend O, Remky A, Plange N, Kaup M, Schwartz B. Fluorescein leakage of the optic disc in glaucomatous optic neuropathy. Graefes Arch Clin Exp Ophthalmol. 2005;243(7):659–664. | ||
Schwartz B. Circulatory defects of the optic disk and retina in ocular hypertension and high pressure open-angle glaucoma. Surv Ophthalmol. 1994;38 Suppl:S23–S34. | ||
Grieshaber MC, Flammer J. Does the blood–brain barrier play a role in Glaucoma? Surv Ophthalmol. 2007;52(Suppl 2):S115–S121. | ||
Tezel G, Wax MB. Glaucoma. Chem Immunol Allergy. 2007;92: 221–227. | ||
Wax MB, Tezel G. Immunoregulation of retinal ganglion cell fate in glaucoma. Exp Eye Res. 2009;88(4):825–830. | ||
Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464–472. | ||
Savva GM, Wharton SB, Ince PG, et al. Age, neuropathology, and dementia. N Engl J Med. 2009;360(22):2302–2309. | ||
Tong L, Balazs R, Soiampornkul R, Thangnipon W, Cotman CW. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging. 2008;29(9):1380–1393. | ||
Venters HD, Dantzer R, Kelley KW. A new concept in neurodegeneration: TNF alpha is a silencer of survival signals. Trends Neurosci. 2000;23(4):175–180. | ||
Chowdhury I, Tharakan B, Bhat GK. Current concepts in apoptosis: the physiological suicide program revisited. Cell Mol Biol Lett. 2006; 11(4):506–525. | ||
Tatton WG, Chalmers-Redman RM, Tatton NA. Apoptosis and anti-apoptosis signalling in glaucomatous retinopathy. Eur J Ophthalmol. 2001;11(Suppl 2):S12–S22. | ||
Tombran-Tink J, Barnstable CJ, Shields B, editors. Optic neuropathy and ganglion cell degeneration in glaucoma. In: Mechanisms of the glaucomas: disease processes and therapeutic modalities. Totowa: Humana Press; 2008:393–424. | ||
Anderson DR; Normal Tension Glaucoma Study. Collaborative normal tension glaucoma study. Curr Opin Ophthalmol. 2003;14(2):86–90. | ||
Anderson DR. Normal-tension glaucoma (Low-tension glaucoma). Indian J Ophthalmol. 2011;59 Suppl:S97–S101. | ||
Kniestedt C, Lin S, Choe J, et al. Correlation between intraocular pressure, central corneal thickness, stage of glaucoma, and demographic patient data: prospective analysis of biophysical parameters in tertiary glaucoma practice populations. J Glaucoma. 2006;15(2):91–97. | ||
Shah H, Kniestedt C, Bostrom A, Stamper R, Lin S. Role of central corneal thickness on baseline parameters and progression of visual fields in open angle glaucoma. Eur J Ophthalmol. 2007:17(4):545–549. | ||
Barry CJ, Cooper RL, Eikelboom RH. Optic disc haemorrhages and vascular abnormalities in a glaucoma population. Aust N Z J Ophthalmol. 1997;25(2):137–144. | ||
Caprioli J, Coleman AL; Blood Flow in Glaucoma Discussion. Blood pressure, perfusion pressure, and glaucoma. Am J Ophthalmol. 2010; 149(5):704–712. | ||
Leung DY, Tham CC, Li FC, Kwong YY, Chi SC, Lam DS. Silent cerebral infarct and visual field progression in newly diagnosed normal-tension glaucoma: a cohort study. Ophthalmology. 2009;116(7):1250–1256. | ||
Suzuki J, Tomidokoro A, Araie M, et al. Visual field damage in normal-tension glaucoma patients with or without ischemic changes in cerebral magnetic resonance imaging. Jpn J Ophthalmol. 2004;48(4):340–344. | ||
Crish SD, Calkins DJ. Neurodegeneration in glaucoma: progression and calcium-dependent intracellular mechanisms. Neuroscience. 2011;176:1–11. | ||
Saccà SC, Izzotti A. Oxidative stress and glaucoma: injury in the anterior segment of the eye. Prog Brain Res. 2008;173:385–407. | ||
Delaney Y, Walshe TE, O’Brien C. Vasospasm in glaucoma: clinical and laboratory aspects. Optom Vis Sci. 2006;83(7):406–414. | ||
Sripriya S, Uthra S, Sangeetha R, et al. Low frequency of myocilin mutations in Indian primary open-angle glaucoma patients. Clin Genet. 2004;65(4):333–337. | ||
Mukhopadhyay A, Acharya M, Mukherjee S, et al. Mutations in MYOC gene of Indian primary open angle glaucoma patients. Mol Vis. 2002;8:442–448. | ||
Rezaie T, Child A, Hitchings R, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295(5557):1077–1079. | ||
Normando EM, Coxon KM, Guo L, Cordeiro MF. Focus on: amyloid beta. Exp Eye Res. 2009;89(4):446–447. | ||
Guo L, Duggan J, Cordeiro MF. Alzheimer’s disease and retinal neurodegeneration. Curr Alzheimer Res. 2010;7(1):3–14. | ||
McKinnon SJ. Glaucoma: ocular Alzheimer’s disease. Front Biosci. 2003;8:s1140–s1156. | ||
Wostyn P, Audenaert K, De Deyn PP. Alzheimer’s disease: cerebral glaucoma? Med Hypotheses. 2010;74(6):973–977. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.